

Abstract
Mutations in the α-crystallin domain of 4 of the small heat shock proteins (sHsp) (Hsp27/HspB1, αA-crystallin/ HspB4, αB-crystallin/HspB5, and HspB8) are responsible for dominant inherited diseases in humans. One such mutation at a highly conserved arginine residue was shown to cause major conformational defects and intracellular aggregation of αA- and αB-crystallins and HspB8. Here, we studied the effect of this Arg mutation on the structure and function of Hsp27. Chinese hamster Hsp27 with Arg148 replaced by Gly (Hsp27R148G) formed dimers in vitro and in vivo, which contrasted with the 12- or 24-subunit oligomers formed by the wild-type protein (Hsp27WT). Despite these alterations, Hsp27R148G had a chaperone activity almost as high as Hsp27WT. The dimers of Hsp27R148G did not further deoligomerize on phosphorylation and like the dimers formed by phosphorylated Hsp27WT were not affected by the deletion of the N-terminal WD/EPF (single letter amino acid code) motif, suggesting that mutation of Arg148, deletion of the N-terminal WD/EPF motif, and phosphorylation of Ser90 may produce similar structural perturbations. Nevertheless, the structure of Hsp27R148G appeared unstable, and the mutated protein accumulated as aggregates in many cells. Both a lower basal level of phosphorylation of Hsp27R148G and the coexpression of Hsp27WT could reduce the frequency of formation of these aggregates, suggesting possible mechanisms regulating the onset of the sHsp-mediated inherited diseases.
INTRODUCTION
Heat shock protein (Hsp)27/HspB1 belongs to the family of small heat shock proteins (sHsp), which is composed of 10 members in mammals, including the well-known αA- and αB-crystallin proteins, also named HspB4 and HspB5, respectively. Most of these proteins form oligomeric structures with a molecular mass of 150 to 800 kDa (Arrigo and Landry 1994; Kappé et al 2003). The structural hallmark of this family of proteins is a conserved sequence of some 80–100 residues known as the α-crystallin domain in their carboxyl terminus (de Jong et al 1988, 1993; Ehrnsperger et al 1998). According to recent crystallographic data of a bacterial and plant sHsp, the oligomerization results from multiple inter- and intramolecular interactions within the α-crystallin domain. This domain is structured as a sandwich of 2 beta sheets, which, depending on the sHsp, may or may not require stabilization by sequences located at the N-terminus (Kim et al 1998; van Montfort et al 2001; Koteiche and McHaourab 2002). Such a requirement for N-terminal sequences has been demonstrated for Hsp27 and for αA- and αB-crystallins, all of which form dimers when their N-terminus is truncated (Merck et al 1992; Lambert et al 1999; Feil et al 2001). In the case of Hsp27, a WD/EPF motif was identified as the sequence at the N-terminus that is essential for oligomerization. A model based on the structure of wheat Hsp16.5 suggested that the WD/ EPF motif of Hsp27 mediates intramolecular interaction with a hydrophobic surface left exposed in the organized α-crystallin domain (Kim et al 1998; van Montfort et al 2001; Thériault et al 2004). This interaction was proposed to be modulated by phosphorylation, explaining how phosphorylation causes major structural remodeling and deoligomerization of Hsp27 into dimers (Lavoie et al 1995; Lambert et al 1999; Thériault et al 2004). Phosphorylation is induced during stress downstream of the stress-activated protein kinase p38 and is essential in many protective activities of the protein against stress (Rouse et al 1994; Lavoie et al 1995; Guay et al 1997; Thériault et al 2004).
A point mutation changing Arg116 to cysteine in the α-crystallin domain of αA-crystallin (αAR116C) is responsible for a dominant congenital cataract disease in humans (Litt et al 1998). A similar mutation in αB-crystallin replacing Arg120 to glycine (αBR120G) was found in a French family with an autosomal dominant desmin-related myopathy (Vicart et al 1998). Equivalent missense mutations replacing Lys141 of HspB8 to glutamate or asparagine was found in distal hereditary motor neuropathy (Irobi et al 2004). Arginine or lysine are found at this residue in almost all sHsp across the phylogeny and are likely to play an important structural role. In fact, analyses of αAR116C and αBR120G indicated that the proteins were severely modified in their supramolecular organization and ended up in large oligomers of over 1000 kDa, which, in cells, collapsed into inclusion bodies resembling aggresome (Bova et al 1999; Kumar et al 1999; Perng et al 1999; Chávez Zobel et al 2003). αA- and αB-crystallin are expressed at high levels in the lens, where they form heterooligomers. A loss of chaperone activity by αAR116C or a loss of its capacity to polymerize with αB-crystallin is thought to be responsible for the observed insolubilization of αB-crystallin and for the increase in lens opacity. In the muscle, the loss of a specific chaperone function of αB-crystallin at the level of the intermediate filament resulting in the aggregation of desmin has been suggested as being the cause of the αBR120G-mediated disease (Bova et al 1999; Kumar et al 1999; Perng et al 1999; Wang et al 2001). However, a gain of toxic function by the aggregated αBR120G mutant is also a possible cause of the disease, particularly because stress activates the transcription of the αB-crystallin gene (Chá vez Zobel et al 2003).
In this study, we analyzed the effects of a mutation equivalent to that found in αAR116C, αBR120G, and HspB8K141E or HspB8K141E in Chinese hamster Hsp27 (Hsp27R148G). We found that changing Arg148 by glycine destabilized the proteins in a manner very similar to phosphorylation or deletion of the WD/EPF motif and provoked the deoligomerization of the protein into dimers. The mutant protein showed little alteration in its chaperone activity; however, similar to αBR120G, Hsp27R148G partly aggregated in the cells suggesting a possible toxic effect of this mutation in vivo.
MATERIALS AND METHODS
Plasmids
pSVHa27WT, pSVHa27AA, pSVHa27EE, and pSVHa27Δ5–23 code for Chinese hamster wild type (Hsp27WT), phosphorylation mutants with both Ser15 and Ser90 changed to Alanine or Glutamate (Hsp27AA or Hsp27EE) or a deletant of the residues 5 to 23 (Hsp27Δ5–23), respectively (Lambert et al 1999). The same plasmids were used as template to produce by polymerase chain reaction (PCR) the plasmids pSVHaR148G, pSVHaR148GAA, pSVHaR148GEE, and pSVHaR148GΔ5–23, which contain, in addition, a codon change from AGG to GGG corresponding to an Arg to Gly mutation at position 148, yielding the mutants Hsp27R148G, Hsp27R148G-AA, Hsp27R148G-EE, and Hsp27R148GΔ5–23, respectively. The pHu27WT plasmid was made by inserting the complementary DNA of human wild-type Hsp27 at the EcoRI restriction site of the expression vector pCIneo (Promega, Madison, WI, USA). The pSVHa27WT and pSVHaR148G plasmids were used as templates to make plasmids coding for the Glutathione S-Transferase (GST)-Chinese hamster Hsp27 fusion proteins GST-Hsp27WT and GST-Hsp27R148G. These plasmids were amplified by PCR with 28-bp primers containing floating ends corresponding to the restrictions sites of BamHI and EcoRI. The PCR products were digested and inserted in phase with the carboxy terminus of GST protein into the pGEX-4T3 plasmid (Amersham Biosciences, Baie d'Urfé, QC, Canada), yielding pGEXHsp27WT and pGEXHsp27R148G.
Cell culture and transfection
Mouse NIH3T3 and Chinese hamster CCL39 cells were grown at 37°C in a humidified atmosphere containing 5% CO2 in Dulbecco's modified Eagle medium (High glucose, Invitrogen, Burlington, ON, Canada) supplemented with 5% fetal bovine serum (CCL39) or 10% calf serum (NIH3T3). Twenty-four hours after plating, the cells were transfected with 2.5 to 9 μg of plasmid DNA using the calcium phosphate precipitation method in the presence of chloroquine at 100 μM (NIH3T3) or 50 μM (CCL39). Five hours later, the cells were washed twice with their culture medium and used 1 to 3 days later. They were either prepared for immunofluorescence microcopy or lysed by sonication in buffer containing 10 mM Tris-HCl, pH 7.4, 3.33% glycerol, 1 mM ethylene diamine tetraacetic acid (EDTA), and 3 mM dithiothreitol (DTT). The cell lysates were centrifuged at 18 000 × g for 5 minutes at 4°C and the supernatant used for glycerol gradient or glutaraldehyde cross-linking analyses.
Immunofluorescence analysis
The cells were fixed at 24 or 48 hours after the beginning of the transfection with 3.7% (wt/vol) formaldehyde in phosphate-buffered saline (PBS) (10 mM sodium phosphate pH 7.5, 150 mM sodium chloride) during 20 minutes at room temperature. Thereafter they were permeabilized for 15 minutes in a saponin buffer (0.1% [wt/vol] saponin in PBS) and blocked 30 minutes with 5% (wt/ vol) free fatty acid milk in the saponin buffer. They were then incubated for 1 hour with the anti-Chinese hamster Hsp27 antibody L2R3 diluted 1:100 in blocking buffer and incubated with biotin-conjugated anti-rabbit immunoglobulin G (IgG) antibodies (Jackson Immunoresearch Lab, West Grove, PA, USA) for 1 hour. The antibody-antigen complexes were revealed for 30 minutes with a solution of streptavidin Texas Red diluted 1:100 in blocking buffer (Amersham Biosciences). The images were collated with a Confocal Microscope MRC-600 (Biorad Laboratories, Mississauga, ON, Canada) equipped with a Diaphot-TDM microscope (Nikon Canada, Montreal, QC, Canada).
Glycerol gradient profile
Glycerol gradient analyses were made essentially as before (Lambert et al 1999). The cell extracts were prepared 48 hours after transfection and loaded onto a 12.0-mL linear glycerol gradient (10–40%) made in gradient buffer (25 mM N-2-hydroxythylpiperazine-N′-2-ethane-sulfonic acid [HEPES], 1 mM EDTA, and 1 mM DTT, pH 7.4). After 18 hours of centrifugation at 30 000 rpm in an SW-40 rotor (Beckman Coulter, Mississauga, ON, Canada) at 4°C, the gradient was fractionated and aliquots were analyzed by Western blot using the L2R3 antibody to detect Hsp27 (Lavoie et al 1995; Lambert et al 1999). The antigen-antibody complexes were detected using a goat anti-rabbit IgG conjugated to horseradish peroxidase and the chemiluminescence substrate Super Signal Ultra (Pierce Co., Rockford, IL, USA). The luminescence reaction was captured on radiological BML-1 film (Kodak Co., Toronto, ON, Canada). The digitalized images were analyzed and quantified using the softwares NIH-Image and Microsoft Excel.
Cross-linking with glutaraldehyde
The extracts were diluted with 1 volume of 0% to 0.8% (wt/vol) glutaraldehyde in water and incubated for 30 minutes at 30°C. The reaction was stopped with 1 volume of Tris-HCL 1 M, pH 7.6, containing sodium dodecyl sulfate (SDS) 10% (wt/vol) and 10 mM EDTA. The extracts were analyzed by 10% SDS polyacrylamide gel electrophoresis and the content of cross-linked Hsp27 species detected after blotting and staining with the Hsp27 L2R3 antibody and 125I-labeled goat anti-rabbit IgG. The radioactivity was measured using a Storm imaging system (Amersham Biosciences, Baie d'Urfé, QC, Canada).
Expression and purification of recombinant proteins
Recombinant Hsp27WT (rHsp27) and Hsp27R148G (rHsp27R148G) were produced in Escherichia coli BL21 Codon+ RIL (Stratagene, Cedar Creek, TX, USA) transformed with plasmids pGEXHsp27WT and pGEXHsp27R148G and purified to homogeneity, essentially as described before (Thériault et al 2004). Digestion with thrombin to dissociate GST from the Hsp27 proteins yielded a final purified proteins containing 2 additional aminoacids (Ser and Gly) before the initial methionine residue.
Size exclusion chromatography
Size exclusion chromatography was performed on a Superose 12 column (Amersham Biosciences) equilibrated in a 150-mM sodium chloride solution containing 25 mM Tris-HCl, pH 7.9, 10% (wt/vol) glycerol, and 0.5 mM DTT. The flow was maintained at 0.5 mL/min. The elution profile was determined by measuring the absorbance at 280 nm. The molecular weight markers were from Sigma Chemical Co., Mississauga, ON, Canada.
In vitro thermoaggregation assays
Denaturation of citrate synthase (CS) was measured at 43°C at a concentration of 75 nM (dimer) in a solution of 40 mM HEPES, pH 7.5, containing varying concentrations of Hsp27WT or rHsp27R148G. Thermal aggregation was determined by measuring light scattering at 320 nm at 30-second intervals using an Ultraviolet/Visible Spectrophotometer Cary 1-Bio (Varian Canada, Mississauga, ON, Canada) equipped with a temperature-controlled cell holder as described before (Thériault et al 2004).
RESULTS
Supramolecular organization of Hsp27R148G
The effect of the R148G mutation on the supramolecular organization of Hsp27 was first evaluated in vitro by looking at the bacterially produced recombinant Hsp27 (rHsp27) and Hsp27R148G (rHsp27R148G) by size exclusion chromatography. As reported previously (Thériault et al 2004), wild-type rHsp27 formed in vitro molecules with size corresponding approximately to dodecamers. The R148G mutation induced important modification in the structure because rHsp27R148G was found mainly as dimers (Fig 1A). This effect of the R148G mutation was confirmed in vivo. NIH3T3 cells were transfected with pSVHa27WT (Fig 1B) or pSVHa27R148G (Fig 1C) and extracts were prepared and fractionated by ultracentrifugation on glycerol gradients. As shown previously (Lambert et al 1999), wild-type Hsp27 was expressed mainly as an oligomeric complex of 700 kDa corresponding to 24-mers. In contrast, Hsp27R148G sedimented at a size corresponding to dimers or tetramers. We next looked at the effect of phosphorylation of Hsp27 on the size of the protein by treating the transfected cells for 2 hours with 200 μM sodium arsenite. As described previously, such treatment induced the phosphorylation and the dissociation of the Hsp27WT molecules into species of the size predicted for dimers. No or little size change was observed in the case of Hsp27R148G.
Fig 1.

Size distribution of the heat shock protein (Hsp)27 and Hsp27R148G oligomers. (A) In vitro: Recombinant Hsp27WT (shaded area) and Hsp27R148G (empty area) were prepared as described in Materials and Methods and analyzed by size exclusion chromatography on Superose 12 HR column. Molecular weight standards are shown on the top axis: dimers of thyroglobulin (340 kDa), β-galactosidase (116 kDa), phosphorylase b (97 kDa), albumin (66 kDa), ovalbumin (45 kDa), carbonic anhydrase (31 kDa). (B, C) In vivo: NIH3T3 cells were transfected with plasmids to yield expression of Hsp27WT (B) or Hsp27R148G (C) and treated (□) or not (▪) with arsenite (200 mM, 2 hours) to induce the phosphorylation of Hsp27. The cell extracts were fractionated by centrifugation on glycerol gradient, and Hsp27 was detected in each fraction by immunoblot. Molecular mass markers are indicated at the top axis: 20 S proteasome (700 kDa), β-galactosidase (540 kDa), 15S proteasome (350 kDa), firefly luciferase (62 kDa), p38 (38 kDa)
Glutaraldehyde cross-linking
To better characterize the nature of the Hsp27 complexes, cellular extracts of NIH3T3 cells transfected with pSVHa27WT or pSVHa27R148G were treated with increasing concentrations of glutaraldehyde, and the cross-linked products were analyzed by Western blot using anti-Hsp27 antibodies. Cross-linking wild-type Hsp27 in control cells with increasing concentrations of glutaraldehyde results in the formation of a family of regularly spaced bands corresponding to multimers made of a varying number of 27-kDa subunits. Cross-linked species with a maximal size much larger than 200 kDa (the biggest molecular weight standard used) were obtained at the highest concentration of glutaraldehyde (Fig 2A). In contrast, even at the highest concentration of the cross-linker, wild-type Hsp27 extracted from arsenite-treated cells was cross-linked into dimers (Fig 2B). The cross-linking of Hsp27R148G extracts confirmed the sedimentation analyses. Most Hsp27R148G from both control and arsenite-treated cells could not be cross-linked in species larger than dimers (Fig 2C, D).
Fig 2.

Size distribution of heat shock protein (Hsp)27 mutants as evaluated by glutaraldehyde cross-linking. NIH3T3 cells were transfected with the appropriate plasmid to yield expression of Hsp27WT (A, B), Hsp27R148G (C, D), Hsp27R148G-EE (E), Hsp27R148G-AA (G), or Hsp27R148GD5–23 (F), and treated (B, D) or not (A, C, E–G) with arsenite (200 mM, 2 hours) to induce phosphorylation of Hsp27. The cell extracts were incubated with increasing concentrations of glutaraldehyde (Glu; from left to right: 0%, 0.002%, 0.005%, 0.01%, 0.02%, 0.04%, 0.08%, 0.1%, 0.2%, 0.4% wt/vol), the cross-linked proteins were separated on sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and Hsp27 was detected by immunoblot. Molecular weight markers are shown on the left: myosin (200 kDa), β-galactosidase (116 kDa), phosphorylase b (97 kDa), albumin (66 kDa), ovalbumin (45 kDa), carbonic anhydrase (31 kDa)
In contrast to its effect on the structure of the wild-type Hsp27, arsenite treatment had no effect on the oligomeric properties of Hsp27R148G. Hsp27R148G could be constitutively phosphorylated in vivo. To determine whether a change in the basal level of phosphorylation might have influenced the size of the Hsp27R148G molecule, NIH3T3 cells were transfected with plasmids coding for Hsp27R148G mutants in which the 2 sites of phosphorylation of Chinese hamster Hsp27, Ser15 and Ser90, were replaced by either Alanine (Hsp27R148G-AA) to prevent phosphorylation or Glutamate (Hsp27R148G-EE) to mimic phosphorylation. In the case of Hsp27WT, replacement of Ser15 and Ser90 by a nonphosphorylatable residue (Hsp27AA) stabilizes the molecule in a highly polymerized state and results in a distribution of cross-linked species identical to unphosphorylated wild-type Hsp27, whereas replacement by an acidic residue mimicking phosphorylation (Hsp27EE) yields exclusively dimers (Lambert et al 1999). We found that neither the presence of nonphosphorylatable nor of pseudophosphorylated residues had an effect on the size of the Hsp27R148G molecules. Hsp27R148G-AA was not stabilized to higher molecular species, and Hsp27R148G-EE was not dissociated in species smaller than those formed by Hsp27R148G (Fig 2E, F).
We found previously that wild-type Hsp27 deleted of Arg5 to Tyr23 (Hsp27Δ5–23) also forms phosphorylation-insensitive dimers in vivo (Lambert et al 1999; Thériault et al 2004). The residues 5 to 23 of Hsp27 contain a WD/ EPF motif that is essential for the maintenance of the oligomeric structure of the protein. To study the resemblance between the 2 alterations in Hsp27, we constructed the plasmid pSVHaR148GΔ5–23 coding for a Δ5–23 deletant of Hsp27R148G. Cross-linking assays with the protein expressed in NIH3T3 cells indicated that deletion of the WD/EPF motif did not caused further changes in the oligomerization of the protein, suggesting that mutation at Arg148 and deletion of the WD/EPF motif produce similar structural alterations (Fig 2G).
Molecular chaperone activity
An equivalent mutation replacing Arg120 in αB-crystallin by Gly (αBR120G) yields a reduction of the molecular chaperone activity of αB-crystallin (Bova et al 1999; Kumar et al 1999; Perng et al 1999). We therefore tested the ability of rHsp27R128G, as compared with rHsp27, to protect CS from thermal denaturation at 43°C (Fig 3). rHsp27R148G prevented the denaturation of CS in a dose-dependent manner and almost as efficiently as rHsp27. In comparison, the 50% protection level was attained at an rHsp27:CS molar ratio of about 0.5 for the wild-type protein as compared to 0.8 for the mutant. For this calculation, all molecules were considered as dimers.
Fig 3.

Chaperone activities of the heat shock protein (Hsp)27 proteins. (A) Kinetics of thermal aggregation of citrate synthase (CS) at 43°C alone (•) or in the presence of Hsp27WT at concentration of 80 nM (▪) or Hsp27R148G at concentrations of 14.2 (▴), 28.5 (♦), 57 (○), or 114 nM (□). All concentrations are calculated for dimeric molecules. Light scattering at 320 nm is indicated in arbitrary units. (B) Dose-dependent protection of denaturation as shown by the relative light scattering at 40 minutes as a function of the molar ratio of Hsp27 to CS. Concentrations were calculated for dimeric citrate synthase and dimeric Hsp27WT (▪) or Hsp27R148G (▴) molecules, or dimeric CS and dodecameric Hsp27WT (□)
Formation of inclusion bodies
The intracellular distribution of Hsp27WT and Hsp27R148G was analyzed by immunofluorescence after transfection of the appropriate plasmids in NIH3T3 or in CCL39 cell lines. In both cell lines, Hsp27 wild type had a uniform cytoplasmic distribution, in agreement with previously published works (Arrigo and Landry 1994; Lavoie et al 1995). Hsp27R148G similarly had a uniform distribution in most cells; however, inclusion bodies were seen in many of the transfected cells (Fig 4). In general, small aggregates of Hsp27R148G were seen scattered over the cytoplasm at 24 hours after transfection. By 48 hours, the small aggregates had coalesced into large amorphous aggregates mostly at perinuclear locations in 40–50% of the cells (Fig 5A).
Fig 4.

Intracellular aggregation of Hsp27R148G. Confocal immunofluorescence microcopy revealed the presence of Hsp27-positive inclusion bodies in NIH3T3 (a, b) or CCL39 (c) transfected with Hsp27R148G. Examples of small aggregates scattered in the cytoplasm (arrows) and large perinuclear aggregates are presented. The cells were fixed 24 hours (a) or 48 hours (b, c) after transfection. In b, the cells were also treated with arsenite as described in the legend of Figure 5. Hsp, heat shock protein
Fig 5.

Role of phosphorylation on the intracellular aggregation of Hsp27R148G mutants. (A) NIH3T3 cells were transfected to express Hsp27R148G (HaR) and treated when indicated (Ars) 48 hours later with arsenite (200 mM, 2 hours) to induce phosphorylation of the protein or were transfected with a pseudophosphorylated (Hsp27R148G-EE, HaREE) or nonphosphorylatable Hsp27R148G-AA (HaRAA) mutants. (B) The cells were transfected to express Hsp27R148G and exposed to arsenite in the presence of SB203580 (SB) at 5 mM added 1 hour before to block the basal and induced level of phosphorylation. (C) NIH3T3 cells were transfected with vectors coding for Hsp27R148G, Hsp27R148G-EE, or Hsp27R148G-AA, alone (open bars and “−”) or together with a vector coding for the human wild-type Hsp27 (close bars). The insert shows in the same order as the bar graph a Western blot illustrating the constant level of expression of the Chinese hamster Hsp27 mutants in the presence (+) or not (−) of the human wild-type protein (Hu). In all cases, the cells were fixed with formaldehyde and processed for immunofluorescence to count the percentage of Chinese hamster Hsp27-expressing cells in which aggregates were seen. Hsp, heat shock protein
We evaluated the effect of phosphorylation on the frequency of in vivo aggregation by Hsp27R148G. NIH3T3 cells were transfected with the expression vectors pSVHa27R148G and treated with arsenite to induce phosphorylation or with SB203580 to block phosphorylation, and the number of cells with cytoplasmic aggregates were counted at 48 hours and compared with cells transfected with pSVHa27R148GAA or pSVHa27R148GEE. SB203580 is an inhibitor of the MAP kinase p38, the Hsp27 kinase kinase, and was used to reduce the basal level of phosphorylation of the proteins. In 3 experiments, arsenite induced a slight but not significant increase in the number of cells with aggregates of Hsp27R148G. A similar minor increase was also observed with Hsp27R148G-EE. In contrast, only 15% of the cells expressing Hsp27R148G-AA, the nonphosphorylable mutant, formed aggregates. SB203580 added to the cell culture 1 hour before the treatment also reduced the frequency of aggregation down to 15% (Fig 5B). We concluded that phosphorylation higher than the basal level cannot further destabilize the mutant protein. However, the basal Hsp27 phosphorylation activity is probably already an important destabilizing factor.
NIH3T3 cells express undetectable amounts of endogenous Hsp27. We investigated the effect of expressing the wild-type protein on the aggregation of the mutants. The cells were transfected with appropriate vectors to express Hsp27R148G, Hsp27R148G-AA, or Hsp27R148G-EE, alone or together with the vector pSVHu27WT directing the expression of human Hsp27WT. A human Hsp27 protein was used because of the availability of antibodies that recognize specifically the human from the Chinese hamster Hsp27. Forty-eight hours after the beginning of the transfection, the cells were fixed and the percentage of transfected cells in which aggregates of mutant proteins were found was determined (Fig 5C). The cotransfection of wild-type Hsp27 decreased 5, 6, and 9 times the frequency of formation of aggregates by the mutants Hsp27R148G, Hsp27R148G-AA, and Hsp27R148G-EE, respectively. Western blot analyses indicated that the expression of the human protein had no influence on the expression of mutant Chinese hamster proteins (Fig 5C, insert). This suggested that under the physiological situation, the production of a wild-type protein from the normal allele could to some extent rescue the stability of the mutant protein.
DISCUSSION
Mutations of Arg116 in human αA-crystallin or Arg120 in human αB-crystallin lead to the development of cataracts and myopathy, respectively. A mutation at the corresponding residue in HspB8, Lys141, causes a neuromuscular disorder. This study indicates that an equivalent mutation at Arg148 of Chinese hamster Hsp27 produces major structural perturbations and hence suggests that this residue, which is among the best conserved within the crystallin domain of sHsp, has also a well-conserved structural role. We found that the R148G mutation destabilizes the oligomeric structure of the protein resulting in its dissociation into dimers. This was confirmed by 3 different techniques: glycerol gradient sedimentation, glutaraldehyde cross-linking of the protein expressed in the cells, and size exclusion chromatography of the bacterially produced protein. The R148G mutation produced a structural phenotype apparently identical to that induced by either phosphorylation of Ser90 or deletion of the WD/ EPF motif. However, the effect was not due to a spontaneously induced phosphorylation because a nonphosphorylatable Hsp27R148G mutant (Hsp27R148G-AA) also was expressed as dimers. Finally, combinations of 2 alterations, that is, pseudophosphorylation or WD/EPF deletion together with the R148G mutation, also yielded dimers, suggesting that the 3 alterations produced a destabilizing effect through similar mechanisms.
Although sHsp from distant species such as plant, bacteria, and mammals have evolved considerably, crystallographic studies have indicated that the structure of the monomers and their organization into dimers, the building block of the oligomer, is highly conserved. In Methanococcus jannaschii Hsp16.5 and wheat Hsp16.9, the α-crystallin core is organized as 2 sheets of β-strands that face each other in a sandwich configuration. There are some differences in the expected role of the conserved arginine in these 2 species, but in both cases a stabilizing role is proposed. Arg107 of Hsp16.5 (equivalent to Arg148 in Hsp27) connects through hydrogen bonds the strands β1 and β2 on the opposing sheets of the same molecule (Kim et al 1998). In wheat Hsp16.9, β1 is missing and Arg108 makes a salt-bridge with Glu100 located on the loop between β5 and β7 of the opposing molecule of the dimer (van Montfort et al 2001). In Hsp27, as well as αB-crystallin, β1 is also missing and the connecting loop between β5 and β7 is considerably shorter and lacks position equivalent to Glu100, making it unlikely that R148 interacts with this loop (Guruprasad and Kumari 2003; Thériault et al 2004). We showed that in Hsp27, like in Hsp16.9, a small hydrophobic sequence corresponding to the WD/EPF motif in the N-terminus of the protein plays the role of the missing β1 strand of Hsp16.5, patching a hydrophobic surface made by the residues Ile96, Gln98, Tyr150, Thr151, and Pro153 of β2 and β7, at the interface of the 2 sheets. It is interesting that in this model of Hsp27 obtained based on its homology with wheat Hsp16.9, Arg148 is also located at the top of the sandwich, also at the interface between the β2 and β7 strands (Fig 6, cover version where Arg148 is shown in red). Arg148 is very close to the hydrophobic surface and is in fact likely making H-bond with Tyr150 on β7 and the nearby Ser94 (data not shown). It is therefore conceivable that, as suggested by the results shown here, mutation of Arg148 produces perturbations similar to those resulting from the deletion of the WD/EPF motif, both resulting in the exposure of hydrophobic surface. In fact, phosphorylation of Ser90 may cause similar perturbation. Ser90 is also located on top of the sandwich between the 2 β-sheets. Phosphorylation of Ser90 might similarly interfere with the intramolecular bonds between the WD/ EPF motif and the hydrophobic surface (Thériault et al 2004).
Fig 6.
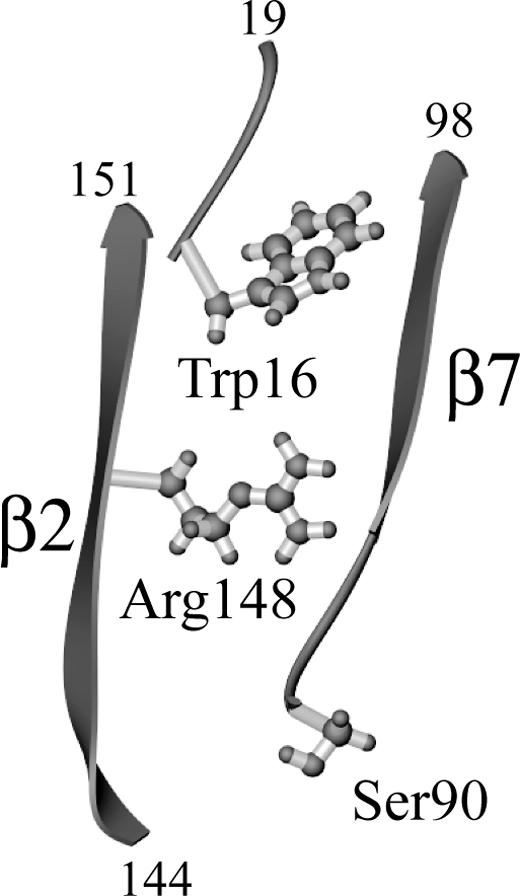
Drawing of the 3-dimensional structure of heat shock protein (Hsp)27 illustrating the relative position of Ser90, Arg148, and the N-terminal WD/EPF motif. Only residues 16 to 19, 90 to 98, and 144 to 151 are shown. The α-crystallin domain of Hsp27 is composed of β-strands organized as a sandwich of 2 β-sheets (Thériault et al 2004). Only 2 β-strands on top of each of these sheets, β2 and β7, are shown. Arg148 on β2 emerges at the interior of the sandwich where it faces and establishes H-bonds with residues on β2 and β7. Trp16 within the WD/EPF motif patches the hydrophobic surface formed by residues at the interface of β2 and β7. The position of Ser90 on top of the sandwich is also illustrated. The figure was generated from the coordinates of Hsp27 (Thériault et al 2004), using Ribbons (Carson 1997)
How can such perturbations at the top interface of the β-sheets result in the dissociation of the oligomer into dimers? It is possible that, in order to patch the hydrophobic surface generated by the β2 and β7 strands, the R148G mutant, the ΔWD/EPF deletion, as well as the phosphorylated protein, must adopt a new dimeric structure that is incompatible with oligomerization. In fact, there appears to exist considerable differences in the behavior of different sHsp once the oligomer is destabilized. Mutation of the conserved arginine in αB- and αA-crystallins causes further aggregation rather than the dissociation of the oligomer into dimers (Kumar et al 1999; Chávez Zobel et al 2003). It is possible that in these cases, the resulting protein is unable to adopt a new stable dimeric configuration and hence unfolds and aggregates. This is in line with the previously reported observation that whereas Hsp27 is found in cells in equilibrium between dimers and 24-mers, αB-crystallin is mainly found in equilibrium between 24-mers and very large species of molecular weight larger than 2000 KDa (Lambert et al 1999; Chávez Zobel et al 2003; Thériault et al 2004). Hence, even the wild-type αB-crystallin may be less stable in a dimeric configuration than Hsp27.
Despite major structural alterations, the R148G mutant still retains considerable chaperone activity. In fact, the chaperone activity of the R148G mutant is almost unchanged (less than 2-fold reduction) as compared with the wild-type protein. Of course, this small difference in activity becomes more important when the true oligomeric nature of the proteins is taken into account. On a molar basis, the dodecameric wild-type protein would be at least 6 times more efficient than the dimeric mutant. This change in activity is very similar to what we observed before for the pseudophosphorylated mutant of Hsp27WT, Hsp27EE, which had also a chaperone activity per mole (of dimers) reduced by about 90% when compared with the dodecamer of Hsp27WT. This suggests that the dissociated dimers, whether produced by phosphorylation or by the R148G mutation, can bind denatured proteins and protect them as well as the dimers within the oligomer. This is consistent with the situation in yeast where Hsp26 binds the substrates as dimers and then oligomerizes with other dimers and their substrate to form large oligomers with the denatured proteins kept attached onto their surface (Haslbeck et al 1999). Studies with mouse Hsp27, αB-crystallin, and bacterial Hsp16.6 also have shown that both large and small oligomers of sHsp can bind and protect denatured substrates from aggregation (Ehrnsperger et al 1999; Feil et al 2001; Giese and Vierling 2002).
The identification of a number of human genetic diseases associated with mutations in proteins of the sHsp family highlights the essential role of these proteins in normal cell physiology. Indeed, in addition to the aforementioned mutations at sites corresponding to the R148 residue of Hsp27 in αA-crystallin, αB-crystallin, and HspB8 (Litt et al 1998; Vicart et al 1998; Irobi et al 2004), several other disease-causing mutations have been identified in αB-crystallin (Selcen and Engel 2003) and also Hsp27 (Evgrafov et al 2004). In all cases investigated, although the exact cause of the disease is not totally clear, formation of aggresome-like structures that become by themselves toxic to the cells seems an important aspect in the progression of the disease (Litt et al 1998; Vicart et al 1998). Our immunofluorescence confocal analyses showed that Hsp27R148G similarly forms in a large proportion of the cells cytoplasmic aggregates which, with time, accumulates in amorphous perinuclear structures. We have identified 2 factors that could potentially affect the development of these aggregates and which can probably also be important in the formation of aggregates by other small Hsp mutants. The first is the homeostatic role of the normal allele. Coexpression of Hsp27WT together with Hsp27R148G can stabilize the latter, preventing the accumulation of intracellular aggregates. Similar results were described before in the case of the αBR120G mutant, which was found to be stabilized by copolymerizing with the wild-type αB-crystallin protein (Chávez Zobel et al 2003). This factor is likely to be particularly important for Hsp27 because its expression is highly inducible by stress, including the stress associated with the accumulation of denatured proteins. Under these circumstances, the increased expression of the wild-type gene that would normally accompany the increased expression of the mutated gene is likely to contribute in a major manner to reduce the toxicity of the mutated proteins. A second factor, which is also highly modulated during stress, is phosphorylation. We found that blocking the basal phosphorylation of Hsp27 (which may be higher under transfection conditions) by inhibiting the activity of the stress-activated p38 pathway reduced the formation of Hsp27R148G aggregates. This was due to a direct effect of phosphorylation on the protein because the nonphosphorylatable Hsp27R148G-AA mutant also had a reduced capacity to aggregate in the cells. Conversely, under more normal physiological conditions where basal phosphorylation may be lower, stresses that cause increased phosphorylation might be expected to exacerbate the toxicity of the mutations, thereby accelerating the expression of the disease. These predictions probably apply to other small Hsps as well because at least αB-crystallin is also phosphorylated and changes its oligomeric organization on phosphorylation (Ito et al 1997, 2001).
Acknowledgments
The authors thank Gabriel Charest and Pierre Lavigne from Université de Sherbrooke for helpful discussions and providing the drawing of Fig 6. This work was supported by the Canadian Institutes of Health Research, Grant MT-7088, and the Canada Research Chair in Cellular Stress-signal Transduction. J.R.T. received studentships from the Société de recherche sur le cancer, Inc., and the Fonds de la recherche en Santé du Québec/Fonds Québécois de la recherche sur la nature et les technologies. A.T.C.-Z. was supported by the Fundación Gran Mariscal de Ayacucho and the Universidad Centroccidental Lisandro Alvarado, Venezuela.
REFERENCES
- Arrigo AP, Landry J 1994 Expression and function of the low-molecular-weight heat shock proteins. In: The Biology of Heat Shock Proteins and Molecular Chaperones, ed Morimoto RI, Tissières A, Georgopoulos C. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 335–373. [Google Scholar]
- Bova MP, Yaron O, Huang Q, Ding L, Haley DA, Stewart PL, Horwitz J. Mutation R120G in alphaB-crystallin, which is linked to a desmin-related myopathy, results in an irregular structure and defective chaperone-like function. Proc Natl Acad Sci U S A. 1999;96:6137–6142. doi: 10.1073/pnas.96.11.6137.0027-8424(1999)096[6137:MRIAWI]2.0.CO;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson M. Ribbons. Methods Enzymol. 1997;277:493–505.0076-6879(1997)277[0493:R]2.0.CO;2 [PubMed] [Google Scholar]
- Chávez Zobel AT, Loranger A, Marceau N, Thériault JR, Lambert H, Landry J. Distinct chaperone mechanisms can delay the formation of aggresomes by the myopathy-causing R120G alphaB-crystallin mutant. Hum Mol Genet. 2003;12:1609–1620. doi: 10.1093/hmg/ddg173.0964-6906(2003)012[1609:DCMCDT]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- de Jong WW, Leunissen JA, Leenen PJ, Zweers A, Versteeg M. Dogfish alpha-crystallin sequences. Comparison with small heat shock proteins and Schistosoma egg antigen. J Biol Chem. 1988;263:5141–5149.0021-9258(1988)263[5141:DASCWS]2.0.CO;2 [PubMed] [Google Scholar]
- de Jong WW, Leunissen JAM, Voorter CE. Evolution of the alpha-crystallin/small heat-shock protein family. Mol Biol Evol. 1993;10:103–126. doi: 10.1093/oxfordjournals.molbev.a039992.0737-4038(1993)010[0103:EOTSHP]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Ehrnsperger M, Buchner J, and Gaestel M 1998 Structure and function of small heat-shock proteins. In: Molecular Chaperones in the Life Cycle of Proteins—Structure, Function and Mode of Action, ed Fink AL, Goto Y. Marcel Dekker, NY, 533–575. [DOI] [PubMed] [Google Scholar]
- Ehrnsperger M, Lilie H, Gaestel M, Buchner J. The dynamics of hsp25 quaternary structure. Structure and function of different oligomeric species. J Biol Chem. 1999;274:14867–14874. doi: 10.1074/jbc.274.21.14867.0021-9258(1999)274[14867:TDOHQS]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Evgrafov OV, Mersiyanova I, and Irobi J. et al. 2004 Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat Genet. 36:602–606. [DOI] [PubMed] [Google Scholar]
- Feil IK, Malfois M, Hendle J, van Der Zandt H, Svergun DI. A novel quaternary structure of the dimeric alpha-crystallin domain with chaperone-like activity. J Biol Chem. 2001;276:12024–12029. doi: 10.1074/jbc.M010856200.0021-9258(2001)276[12024:ANQSOT]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Giese KC, Vierling E. Changes in oligomerization are essential for the chaperone activity of a small heat shock protein in vivo and in vitro. J Biol Chem. 2002;277:46310–46318. doi: 10.1074/jbc.M208926200.0021-9258(2002)277[46310:CIOAEF]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Guay J, Lambert H, Gingras-Breton G, Lavoie JN, Huot J, Landry J. Regulation of actin filament dynamics by p38 map kinase-mediated phosphorylation of heat shock protein 27. J Cell Sci. 1997;110:357–368. doi: 10.1242/jcs.110.3.357.0021-9533(1997)110[0357:ROAFDB]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Guruprasad K, Kumari K. Three-dimensional models corresponding to the C-terminal domain of human alphaA- and alphaB-crystallins based on the crystal structure of the small heat-shock protein HSP16.9 from wheat. Int J Biol Macromol. 2003;33:107–112. doi: 10.1016/s0141-8130(03)00074-6.0141-8130(2003)033[0107:TMCTTC]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Haslbeck M, Walke S, Stromer T, Ehrnsperger M, White HE, Chen S, Saibil HR, Buchner J. Hsp26: a temperature-regulated chaperone. EMBO J. 1999;18:6744–6751. doi: 10.1093/emboj/18.23.6744.0261-4189(1999)018[6744:HATC]2.0.CO;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irobi J, Van Impe K, and Seeman P. et al. 2004 Hot-spot residue in small heat-shock protein 22 causes distal motor neuropathy. Nat Genet. 36:597–601. [DOI] [PubMed] [Google Scholar]
- Ito H, Kamei K, Iwamoto II, Inaguma Y, Nohara D, Kato K. Phosphorylation-induced change of the oligomerization state of alphaB-crystallin. J Biol Chem. 2001;276:5346–5352. doi: 10.1074/jbc.M009004200.0021-9258(2001)276[5346:PCOTOS]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Ito H, Okamoto K, Nakayama H, Isobe T, Kato K. Phosphorylation of αB-crystallin in response to various types of stress. J Biol Chem. 1997;272:29934–29941. doi: 10.1074/jbc.272.47.29934.0021-9258(1997)272[29934:POBIRT]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Kappé G, Frank E, Verschuure P, Boelens WC, Leunissen JAM, de Jong WW. The human genome encodes 10 α-crystallin– related small heat shock proteins: HspB1–10. Cell Stress Chaperones. 2003;8:53–61. doi: 10.1379/1466-1268(2003)8<53:thgecs>2.0.co;2.1466-1268(2003)008[0053:THGECR]2.0.CO;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KK, Kim R, Kim SH. Crystal structure of a small heat-shock protein. Nature. 1998;394:595–599. doi: 10.1038/29106.0028-0836(1998)394[0595:CSOASH]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Koteiche HA, McHaourab HS. The determinants of the oligomeric structure in Hsp16.5 are encoded in the alpha-crystallin domain. FEBS Lett. 2002;519:16–22. doi: 10.1016/s0014-5793(02)02688-1.0014-5793(2002)519[0016:TDOTOS]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Kumar LV, Ramakrishna T, Rao CM. Structural and functional consequences of the mutation of a conserved arginine residue in alphaA and alphaB crystallins. J Biol Chem. 1999;274:24137–24141. doi: 10.1074/jbc.274.34.24137.0021-9258(1999)274[24137:SAFCOT]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Lambert H, Charette SJ, Bernier AF, Guimond A, Landry J. HSP27 multimerization mediated by phosphorylation-sensitive intermolecular interactions at the amino terminus. J Biol Chem. 1999;274:9378–9385. doi: 10.1074/jbc.274.14.9378.0021-9258(1999)274[9378:HMMBPI]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Lavoie JN, Lambert H, Hickey E, Weber LA, Landry J. Modulation of cellular thermoresistance and actin filament stability accompanies phosphorylation-induced changes in the oligomeric structure of heat shock protein 27. Mol Cell Biol. 1995;15:505–516. doi: 10.1128/mcb.15.1.505.0270-7306(1995)015[0505:MOCTAA]2.0.CO;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litt M, Kramer P, LaMorticella DM, Murphey W, Lovrien EW, Weleber RG. Autosomal dominant congenital cataract associated with a missense mutation in the human alpha crystallin gene CRYAA. Hum Mol Genet. 1998;7:471–474. doi: 10.1093/hmg/7.3.471.0964-6906(1998)007[0471:ADCCAW]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Merck KB, De Haard-Hoekman WA, Oude Essink BB, Bloemendal H, De Jong WW. Expression and aggregation of recombinant alpha A-crystallin and its two domains. Biochim Biophys Acta. 1992;1130:267–276. doi: 10.1016/0167-4781(92)90439-7.0006-3002(1992)1130[0267:EAAORA]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Perng MD, Muchowski PJ, van Den IP, Wu GJ, Hutcheson AM, Clark JI, Quinlan RA. The cardiomyopathy and lens cataract mutation in alphaB-crystallin alters its protein structure, chaperone activity, and interaction with intermediate filaments in vitro. J Biol Chem. 1999;274:33235–33243. doi: 10.1074/jbc.274.47.33235.0021-9258(1999)274[33235:TCALCM]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Rouse J, Cohen P, Trigon S, Morange M, Alonso-Llamazares A, Zamanillo D, Hunt T, Nebreda AR. A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell. 1994;78:1027–1037. doi: 10.1016/0092-8674(94)90277-1.0092-8674(1994)078[1027:ANKCTB]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Selcen D, Engel AG. Myofibrillar myopathy caused by novel dominant negative alpha B-crystallin mutations. Ann Neurol. 2003;54:804–810. doi: 10.1002/ana.10767.0364-5134(2003)054[0804:MMCBND]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Thériault JR, Lambert H, Chávez-Zobel AT, Charest G, Lavigne P, Landry J. Essential role of the N-terminal WD/EPF motif in the phosphorylation-activated protective function of mammalian Hsp27. J Biol Chem. 2004;279:23463–23471. doi: 10.1074/jbc.M402325200.0021-9258(2004)279[23463:EROTNE]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- van Montfort RL, Basha E, Friedrich KL, Slingsby C, Vierling E. Crystal structure and assembly of a eukaryotic small heat shock protein. Nat Struct Biol. 2001;8:1025–1030. doi: 10.1038/nsb722.1072-8368(2001)008[1025:CSAAOA]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Vicart P, Caron A, and Guicheney P. et al. 1998 A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy. Nat Genet. 20:92–95. [DOI] [PubMed] [Google Scholar]
- Wang X, Osinska H, Klevitsky R, Gerdes AM, Nieman M, Lorenz J, Hewett T, Robbins J. Expression of R120G-alphaB-crystallin causes aberrant desmin and alphaB- crystallin aggregation and cardiomyopathy in mice. Circ Res. 2001;89:84–91. doi: 10.1161/hh1301.092688.0009-7330(2001)089[0084:EORCAD]2.0.CO;2 [DOI] [PubMed] [Google Scholar]