

RESUME
La dyslipidémie familiale chez les nourrissons est une pathologie rare caractérisée par des taux anormaux de lipides dans le sang, tels que le cholestérol et les triglycérides. Le diagnostic précoce pose un défi en raison de symptômes peu spécifiques et de critères lipidiques différents de ceux observés chez les adultes. À travers l'étude de deux cas cliniques de dyslipidémie familiale (Hypercholestérolémie familiale de type 1 et Hyperlipidémie familiale combinée de type 2b), nous mettons en lumière les difficultés diagnostiques et thérapeutiques rencontrées chez les nourrissons. Nous soulignons l'importance cruciale d'une approche multidisciplinaire dans la prise en charge et le dépistage précoce. Dans le premier cas, un garçon âgé de 3 mois, dont la famille avait des antécédents de dyslipidémie, a été diagnostiqué lors d'une bronchiolite. Les investigations ont montré un sérum lactescent, une pseudo hyponatrémie, et un profil lipidique anormal. Son hyperlipidémie familiale de type 1 a été confirmé par électrophorèse des lipides. Malgré une gestion alimentaire et l'allaitement maternel, il a développé une pancréatite sévère jugulée par des soins intensifs. Le deuxième cas concernait une fille qui a présenté à l'âge de 3 mois des vomissements et de l'irritabilité. Les tests de laboratoire ont révélé une pseudo hyponatrémie, des anomalies hématologiques et des troubles lipidiques. Son hyperlipidémie familiale de type 2b a été confirmée par électrophorèse des lipides. Elle a bien évolué sous régime spécialisé, présentant quelques épisodes de pancréatite sans atteindre les critères de gravité clinique ou radiologique.
ABSTRACT
Dyslipidemia in infants is a rare condition characterized by abnormal levels of lipids in the blood, such as cholesterol and triglycerides. Early diagnosis poses a challenge due to nonspecific symptoms and lipid criteria differing from adults. Through two clinical cases of familial dyslipidemia (Type 1 Familial Hypercholesterolemia and Type 2b Combined Familial Hyperlipidemia), we highlight the diagnostic and therapeutic challenges encountered in infants, emphasizing the importance of a multidisciplinary approach in care and early screening. In the first case, a 3-month-old boy with a family history of dyslipidemia was diagnosed during bronchiolitis, revealing milky serum, pseudohyponatremia, and abnormal lipid profile. His Type 1 familial hyperlipidemia was confirmed by lipid electrophoresis. Despite dietary management and breastfeeding, he developed severe pancreatitis, successfully treated with intensive care. The second case involved a girl who presented at 3 months with vomiting and irritability. Laboratory tests indicated pseudohyponatremia, hematologic abnormalities, and lipid disturbances. Her Type 2b familial hyperlipidemia was confirmed by lipid electrophoresis. She responded well to a specialized diet, experiencing few pancreatitis episodes without meeting clinical or radiological severity criteria.
Introduction
Dyslipidemia is rare in children and infants (1-2).
Early diagnosis of dyslipidemia in infants poses complexity due to nonspecific symptoms and lipid criteria differing from adults (3).
Dyslipidemia is classified into different types: First one is hypercholesterolemia where total cholesterol levels, low-density lipoprotein cholesterol, or nonhigh-density lipoprotein cholesterol levels elevated above values appropriate for age.
The second one is hypertriglyceridemia where fasting plasma triglyceride level above values appropriate for age.
And the final one is combined or mixed dyslipidemia which is a combination of abnormal lipid levels, most commonly elevated triglycerides and decreased high-density lipoprotein cholesterol(2).
Given its rarity, a treatment protocol is poorly elucidated.
Treating dyslipidemia in infants is challenging due to the lack of age-appropriate therapeutic options.
Additionally, complications like acute pancreatitis face the absence of clear management protocols specific to infants.
Through the clinical and evolutionary description of two cases of familial hypercholesterolemia diagnosed in two infants, and a literature review, this article highlights the diagnostic and therapeutic challenges encountered in infants, emphasizing the importance of a multidisciplinary approach in care and early screening.
Indeed, the first case underscores an incidental diagnosis, and the diverse complications associated with the condition, whereas the second case highlights an uncommon complication linked to combined familial hyperlipidemia type 2b.
Observations
Case A
At 3 months of age, a boy born to consanguineous parents presented with acute febrile dyspnea attributed to acute bronchiolitis.
The infant had a family history of dyslipidemia and vascular events on both the paternal and maternal sides.
Laboratory examinations
Initial laboratory investigations were hindered by lactescent serum, showing hemoglobin at 20.4 g/ dL and unmeasurable mean corpuscular hemoglobin concentration (MCHC).
The ionogram indicated unmeasurable values and a false hyponatremia at 120 mmol/L.
Based on this presentation, dyslipidemia was strongly suspected.
A lipid profile revealed triglycerides at 16.4 g/L (reference range: 0.57-1.71 g/L), total cholesterol at 1.38 g/L (0.7-1.75 g/L), and HDL-cholesterol at 0.14 g/L (0.35-0.65 g/L).
Secondary dyslipidemia investigation was negative: TSH=4uIU/L (1-4 uIU/L), and renal and hepatic tests were normal.
Electrophoresis confirmed the diagnosis of Type I hyperlipoproteinemia: high chylomicrons at 44.7% (0.00%), LDL at 24% (34-59%), VLDL at 27.5% (2-34%), and HDL at 3.8% (20-49%).
Management
The child was kept on exclusive breastfeeding until the age of complementary feeding.
The parents received several therapeutic education sessions on hypolipidemic dietary measures, including reducing simple sugar intake, replacing simple sugars with complex sugars, reducing caloric intake from fats, and increasing omega-3 fatty acid consumption.
The mother followed the dietary advice during breastfeeding.
However, these recommendations were challenging to implement during the infant's introduction to solid foods.
Evolution
The course was marked by the onset of multiple acute pancreatitis with abdominal computed tomography (CT) showing Stage E acute pancreatitis according to the Balthazar classification, with a severity index of 8 (Figure 1).
The concomitant triglyceride level was 92 g/L.
Management of severe infantile pancreatitis involved 48- hour parenteral nutrition, analgesia with paracetamol, third-generation cephalosporin-based antibiotic therapy in combination with metronidazole and a macrolide, and gastric dressing.
The course was marked by good clinical improvement.
Figure 1. Case(a) Abdominal CT scan stage E acute pancreatitis.

At the age of 10 months, the patient presented with gastrointestinal bleeding secondary to well-managed intestinal giardiasis with metronidazole.
Currently, patient did not show cutaneous xanthomas, pancreatic insufficiency (exocrine or endocrine), or cardiovascular impairment.
A residual pseudocyst was observed on ultrasound without complications.
Case B
She was the daughter of consanguineous parents with a family history of complicated dyslipidemia.
She was admitted during the neonatal period due to prematurity resulting from chorioamnionitis with neonatal meningitis treated for 21 days.
She had a mixed diet, consisting of both breast milk and formula.
At the age of 3 months, she experienced vomiting, abdominal pain, and irritability.
Laboratory examinations
Initials laboratory tests were difficult to interpret, showing a hemoglobin level of 24.4 g/dL, a MCV of 69.3fL, a MCHC of 60 pg, and an ionogram indicating hyponatremia at 108 mEq/L, potassium at 4.2 mEq/L, and chloride at 80 mEq/L.
Considering this pseudohyponatremia, protein levels returned to normal at 65 g/L, and a lipid profile revealed hypertriglyceridemia at 13.87 mmol/L (reference range: 0.7-2 mmol/L) and cholesterol levels at 3.29 mmol/L (3.8-5.2 mmol/L).
As part of the etiological investigation, liver and renal function tests were normal, and thyroidstimulating hormone (TSH) was within the normal range at 1.62 uIU/mL (0.25-5 uIU/mL), with free thyroxine (FT4) at 10 pmol/L (9-19 pmol/L).
Primary dyslipidemia was suspected and confirmed by lipid electrophoresis, which showed chylomicrons at 0% (0.00%), LDL at 48.1% (34- 59%), VLDL at 39.4% (2-34%), and HDL at 5.8% (20-49%).
The diagnosis of combined familial hyperlipidemia (Type IIb) was confirmed.
Management
The patient was placed on a special hypolipidemic diet with therapeutic education from the nutritionist.
Evolution
The course was marked by the occurrence of multiple episodes of abdominal pain and vomiting, without pancreatic involvement on biological and radiological examination.
At the age of two years, she presented with abdominal pain and vomiting.
The diagnosis of acute pancreatitis was made, classified as Stage D according to Balthazar on abdominal computed tomography, with concurrent hypertriglyceridemia at 29.7 mmol/L.
Symptomatic treatment was administered, and there was good progress and ultrasound control after three months without anomalies.
Currently, the patient is 4 years old and follows a hypolipidemic diet with normal triglyceride and cholesterol levels.
On examination, her weight is 16 kg (+1SD), height is 100 cm (average), with a body mass index of 16 (+1SD) and normal blood pressure.
She does not present gerontoxon or xanthelasma.
Cardiovascular evaluation is normal, with normal fasting blood glucose and an HbA1c level of 5.5%.
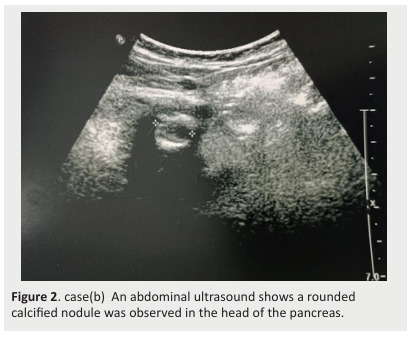
A recent ultrasound revealed a rounded calcified nodule in the head of the pancreas, with a normal-sized liver and regular contour (Figure 2).
Figure 2. case(b) An abdominal ultrasound shows a rounded calcified nodule was observed in the head of the pancreas.
Discussions
We studied two cases involving infants born to consanguineous parents with early-onset dyslipidemias.
The first case concerned a 3-month-old boy with a family history of dyslipidemia, where suspicion arose during bronchiolitis, revealing milky serum, pseudohyponatremia, and disturbed lipid profile.
His Type 1 familial hyperlipidemia was confirmed by lipid electrophoresis.
Despite dietary management and breastfeeding, he developed severe pancreatitis, successfully managed with intensive care.
The second case involved a girl with a family history of dyslipidemia, who presented at 3 months with vomiting and irritability.
Laboratory tests revealed pseudohyponatremia, hematologic, and lipid disturbances.
Her Type 2b familial hyperlipidemia was confirmed by lipid electrophoresis.
She responded well to a specialized diet, experiencing few episodes of pancreatitis without clinical or radiological severity criteria.
Dyslipidemia in children is a medical condition characterized by abnormal levels of lipids in the blood, such as cholesterol and triglycerides (4).
It can be diagnosed following complications caused by dyslipidemia or incidentally during routine blood tests or evaluations for other health issues, as in our two cases where dyslipidemia was diagnosed following hospitalization for acute bronchiolitis and acute gastroenteritis.
Dyslipidemias can be classified into different types, including familial hypercholesterolemia, hypertriglyceridemia, hyperlipoproteinemia, and other less common forms according to Fredrickson's classification (5 ).
The severity of the clinical presentation depends on the class of dyslipidemia.
Family history plays an important role in establishing the diagnosis.
A thorough evaluation of family members may be necessary to detect other cases of dyslipidemia and implement appropriate prevention and treatment measures (3).
Hence, the importance of dyslipidemia screening in children, recommended between 9 and 11 years of age for those without family history, and starting from the age of 2 years if cases are described in the family(6).
However, severe forms complicated at an early age before 24 months of life advocate for prenatal screening in couples with a family dyslipidemia history, especially autosomal dominant transmission.
Prenatal diagnosis could help these children start early nutritional therapy to prevent severe pancreatic complications as diagnosed in our patients(6).
Children with dyslipidemia may present symptoms such as cutaneous xanthomas, eruptive xanthomas, and retinal lipemia observed in familial exogenous hypertriglyceridemia (type I) (5), or xanthelasmas and corneal arcus in combined familial hyperlipidemia type IIb.
Other complications include acute pancreatitis, mainly for type 1 dyslipidemia(7).
Type IIb hyperlipidemia is characterized by phenotypic variation within the same family and fluctuation within an individual over time, with fluctuating hypercholesterolemia and hypertriglyceridemia, explaining the acute pancreatitis in our second case(8) and the ultrasound image showing past episodes of silent acute pancreatitis.
Dietary intervention for children and adolescents with familial hyperlipidemia (FH) is a complex task that needs to be individualized and adapted, taking into consideration, first and foremost, the nutritional adequacy for growth and development.
Additionally, it should consider multiple aspects related to the child/adolescent, such as age, tastes, preferences, family background, socioeconomic context, and country of residence(9).
The treatment of dyslipidemia in children usually involves lifestyle modifications, such as adopting a balanced diet and regular physical activity.
Statins and fibrates can only be prescribed from the age of 8 years due to their side effects and lack of market authorization before this age.
Therapeutic management of infants remains complex due to the lack of specific options for this age group.
Establishing protocols for the optimal management of pancreatic complications in infants is essential(10).
Dyslipidemia in infants requires a multidisciplinary approach involving pediatricians, lipidology specialists, and nutritionists.
Protocols for managing pancreatic complications in infants should be established for optimal care.
Long-term regular follow-up is essential to prevent long-term complications in these patients.
FH can manifest as either homozygous (or compound heterozygous) or heterozygous, with a gene dosage effect.
Homozygous FH is exceptionally rare, occurring at a frequency of 1 in 1,000,000, while heterozygous FH affects 1 in 250-500 individuals. (11 )
The decision for primary prevention therapy is based on LDL levels, family history (early cardiovascular events), and often on non-invasive measurement of atherosclerosis.
It is therefore imperative to identify candidates for familial hypercholesterolemia as early as possible, a task facilitated by genetic analysis today.
In this regard, clinicians should consider FH in the presence of a family history of early atherosclerosis (occurrence of myocardial infarction before age 55 in men and 65 in women) or hyperlipidemia in a first-degree relative.
On clinical examination, adults may exhibit tendon xanthomas or corneal arcus before the age of 45.
The biological diagnosis is based on repeated measurements of LDL cholesterol > 5 mmol/L in adults and > 3.35 mmol/L in children, after excluding secondary hypercholesterolemia due to chronic diseases (liver disease, thyroid dysfunction, diabetes, nephrotic syndrome, etc.).
Considering the heterogeneity of clinical presentation and severity of different known mutations, confirmation of diagnosis through molecular analysis is strongly recommended(12).
A cascade screening approach is preferred over universal screening, and family history remains the cornerstone for identifying individuals at risk.
Proteins encoded by genes involved in this pathology play a role in the elimination of LDL cholesterol: the LDL receptor (LDLR), apolipoprotein B (APOB), proprotein convertase subtilisin/kexin type 9 (PCSK9), and apolipoprotein E (APOE).
To date, over 1000 different mutations in the LDLR gene have been associated with FH. (12) (11)
Given the complicated nature of genetic testing, there is significant role of genetic counseling for professionals treating hypercholesterolemic patients.
Genetic counseling should begin when the proband is suspected to have diagnosis of FH.
The discussion should include an explanation of inheritance patterns, information about genetic testing, including potential benefits, risks, and potential for incidental or uncertain findings. (11)
The estimated prevalence of FCH is approximately 0.5% to 4%.
Despite its prevalence and potential health consequences, familial combined hyperlipidemia (FCH) is often underdiagnosed and undertreated. (11)
This has led to ongoing research efforts aimed at better understanding its underlying genetic and metabolic mechanisms and developing effective treatment strategies(13).
The genetic component of familial combined hyperlipidemia (FCH) has not been fully elucidated to date.
The pattern of inheritance of FCH was initially reported as autosomal dominant in 1973 by Goldstein et al.
Subsequent research has specified and proposed that FCH can be familial or nonfamilial, indicating a multigenic mode and complex inheritance ( 13) (11).
Figure 3. Evolution of lipid profile under dietry regimen Case A : HF type 1, Case B: HF type 2b .

Conclusion
A proactive and preventive approach is crucial to ensure a better quality of life for these young patients.
These two cases highlight the crucial importance of screening for dyslipidemia in infants under two years old.
Early diagnosis is essential to initiate preventive measures and avoid long-term complications.
Family history is a key factor in guiding the diagnosis and preventive management, especially in families at risk of familial dyslipidemia.
References
- Jayaram S, Meera S, Kadi S, Sreenivasa N. An Interesting Case of Familial Homozygous Hypercholesterolemia—A Brief Review. Indian J Clin Biochem. 2012 Jul;27(3):309–313. doi: 10.1007/s12291-011-0165-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Management of Dyslipidemias in Children - DynaMed. 2024. https://www.dynamed.com/management/management-of-dyslipidemias-in-children#GUID78466972-8724-470E-B126-3E38E7547B95 https://www.dynamed.com/management/management-of-dyslipidemias-in-children#GUID78466972-8724-470E-B126-3E38E7547B95
- Filippo MD. Aspects génotypiques et phénotypiques des dyslipidémies primitives rares affectant le métabolisme des lipoprotéines riches en triglycérides
- Singh S, Bittner V. Familial hypercholesterolemia--epidemiology, diagnosis, and screening. Curr Atheroscler Rep. 2015;17(2):482. doi: 10.1007/s11883-014-0482-5. [DOI] [PubMed] [Google Scholar]
- Brun N, Rodondi N. [How to deal with familial dyslipidemia in clinical practice?] Rev Med Suisse. 2012 Mar;8(331):494–500. [PubMed] [Google Scholar]
- US Preventive Services Task Force, Bibbins-Domingo K, Grossman DC, Curry SJ, et al. Screening for Lipid Disorders in Children and Adolescents: US Preventive Services Task Force Recommendation Statement. JAMA. 2016 Aug;316(6):625–633. doi: 10.1001/jama.2016.9852. [DOI] [PubMed] [Google Scholar]
- Defesche JC, Gidding SS, Harada-Shiba M, Hegele RA, et al. Familial hypercholesterolaemia. Nat Rev Dis Primer. 2017 Dec;3:17093. doi: 10.1038/nrdp.2017.93. [DOI] [PubMed] [Google Scholar]
- Chora JR, Iacocca MA, Tichý L, Wand H, et al. The Clinical Genome Resource (ClinGen) Familial Hypercholesterolemia Variant Curation Expert Panel consensus guidelines for LDLR variant classification. Genet Med Off J Am Coll Med Genet. 2022 Feb;24(2):293–306. doi: 10.1016/j.gim.2021.09.012. [DOI] [PubMed] [Google Scholar]
- Capra ME, Biasucci G, Crivellaro E, Banderali G, et al. Dietary intervention for children and adolescents with familial hypercholesterolaemia. Ital J Pediatr. 2023 Jun;49(1):77. doi: 10.1186/s13052-023-01479-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen RZ, Freeman AJ. Pancreatitis in Children. Pediatr Clin North Am. 2021 Dec;68(6):1273–1291. doi: 10.1016/j.pcl.2021.07.012. [DOI] [PubMed] [Google Scholar]
- Patni N, Ahmad Z, Wilson DP, et al. Endotext. MDText.com, Inc.; 2000. Genetics and Dyslipidemia. [Google Scholar]
- Hyperlipidemies_-_Dr_Peretti.pdf. https://www.gfhgnp.org/wordpress/wp-content/themes/kTheme-v2-gfhgnp/pdf/publications/lyon-2015/Hyperlipidemies_-_Dr_Peretti.pdf. 2023. https://www.gfhgnp.org/wordpress/wp-content/themes/kTheme-v2-gfhgnp/pdf/publications/lyon-2015/Hyperlipidemies_-_Dr_Peretti.pdf
- Padda IS, Fabian D, Johal GS. StatPearls. StatPearls Publishing; 2024. Familial Combined Hyperlipidemia. [PubMed] [Google Scholar]