

Abstract
A subset of genes in mammals are subject to genomic imprinting. The mouse H19 gene, for example, is active only when maternally inherited and the neighboring Igf2 gene is paternally expressed. This imprinted expression pattern is regulated by the imprinting control region (ICR) upstream of the H19 gene. A maternally inherited H19 ICR inhibits Igf2 gene activation by the downstream enhancer due to its insulator function while it suppresses H19 gene transcription by promoter DNA methylation when paternally inherited. These parent-of-origin specific functions depend on the allele-specific methylation of the ICR DNA, which is established during gametogenesis. Therefore, the ICR may also function as a landmark for epigenetic modifications. To examine whether the ICR confers these activities autonomously, we introduced a 2.9-kbp ICR-containing DNA fragment into a human β-globin yeast artificial chromosome at the 3′ end of the locus control region and established transgenic mouse lines. Expression of all of the β-like globin genes was higher when the transgene was paternally inherited. In accord with this result, transgenic ICR DNA from nucleated erythrocytes was more heavily methylated when paternally transmitted. Chromatin immunoprecipitation assays confirmed that CCCTC binding factor is preferentially recruited to the maternal transgenic ICR in vivo. Surprisingly however, the parent-of-origin specific methylation pattern was not observed in germ cell DNA in testis, demonstrating that methylation was established after fertilization. Thus, the ICR autonomously recapitulated imprinting within the normally nonimprinted transgenic β-globin gene locus, but the temporal establishment of imprinting methylation differs from that at the endogenous Igf2/H19 locus.
Keywords: H19, transgenic mouse, yeast artificial chromosome
Differentially methylated domains, which are characterized by CpG-rich sequences, are frequently found within imprinted loci and are involved in parent-of-origin specific gene expression (1). For example, the imprinting control region (ICR) in the Igf2/H19 locus is differentially methylated depending on its parent of origin. The ICR, located 2-4 kb upstream of the H19 gene (H19 ICR, Fig. 1A), controls several aspects of the imprinting mechanism, part of which is governed by the enhancer blocking protein CTCF and its four recognition motifs within the ICR (2-5). Because CTCF binding is sensitive to DNA methylation, insulator activity can be regulated through methylation of CpG motifs in the CTCF binding site (6). It was demonstrated that a hypomethylated maternal ICR allows CTCF binding and insulator function, which prevents the shared enhancer, located 3′ to H19 (7), from interacting with the distal Igf2 gene (enhancer blocking). In contrast, a hypermethylated paternal ICR attenuates H19 transcription by direct promoter methylation (silencing, ref. 8) and prevents CTCF from binding to the ICR, thereby allowing the distal Igf2 gene to be activated by the H19 3′ enhancer. Thus, the establishment of differential methylation of the H19 ICR in germ cells constitutes the central imprinting mechanism in this locus.
Fig. 1.

Experimental procedure. (A) Genomic structure of the Igf2/H19 locus. The Igf2 and H19 genes (open boxes) are ≈90 kbp apart, and the expression of both genes depends on the shared 3′ enhancer (gray boxes). The ICR is located within a SacI-BamHI fragment lying approximately -2.0 to -5.0 kbp relative to the transcription initiation site of H19 gene. The positions of the CTCF binding sites are shown as black rectangles in the enlarged map. (B) Structure of the human β-globin YAC (13). The positions of the β-like globin genes are shown relative to the LCR. SfiI restriction enzyme sites are located 5′ to HS5, between HS4 and HS3, and in the right arm of the YAC. Probes used for long-range structural analysis in D are shown as solid rectangles. The whole β-globin locus is contained within two SfiI fragments (10 and 100 kbp). (C) Structure of the mutant loci. The ICR (inverted orientation) fragment was floxed (filled triangles) and introduced between HS1 and the ε-globin gene in the human β-globin YAC. Subsequent in vivo cre-loxP recombination enabled generation of pseudo wild-type TgM (loxP). (D) Long-range structural analysis of transgenes. DNA from thymus cells was digested with SfiI in agarose plugs, separated by pulsed-field gel electrophoresis, and hybridized separately to probes in B.
Another powerful vertebrate insulator, the chicken β-globin LCR-HS4 (ChβGI), also bears a CpG dinucleotide in its CTCF binding site, but otherwise lacks homology to the H19 ICR. Hence, it could theoretically function as a methylation-sensitive ICR. To test this hypothesis, Szabo et al. (9) replaced the endogenous H19 ICR with ChβGI and established a knock-in mouse. In this mutant mouse, ChβGI was constitutively active regardless of its parent of origin, and the H19 gene was biallelically expressed. Thus, although the presence of a CTCF binding site is sufficient to confer insulator activity, the parent-of-origin-specific methylation pattern is regulated by other sequences present within or around the H19 ICR.
The methylation imprinting activity of the H19 ICR has been tested in heterologous loci. In the RSVIgmyc transgene locus (10), both maternal and paternal transgenic ICRs (from 1182 to 3411 nt, GenBank accession no. AF049091) were highly and equally methylated, demonstrating that the H19 ICR alone was not capable of marking parental origin in this transgenic mouse (TgM). However, a slightly different H19 ICR (from 1126 to 3508 nt), when knocked-in to the α-fetoprotein (afp) locus, was differentially methylated, with the paternal allele being more heavily methylated (11). Furthermore, the afp paternal allele was less abundantly expressed than the maternal allele, probably due to promoter methylation mediated by the transgenic ICR. Thus, the H19 ICR conferred both autonomous methylation imprinting and silencing activities in this study. Because of these contradictory results, the autonomy of ICR marking by methylation imprinting has not yet been firmly established.
The human β-globin locus contains five developmentally regulated β-like globin genes (5′-ε, Gγ, Aγ, δ, and β-3′, ref. 12). Abundant expression of all five genes depends on the locus control region (LCR), a powerful enhancer, located at the 5′ end of the locus (Fig. 1B). In TgM, the human ε- and γ-globin genes are both expressed in primitive erythroid cells of the yolk sac (at 9.5 days postcoitum). Abundant β- and diminishing γ-globin gene expression is observed in definitive erythroid cells of the fetal liver (≈14.5 days postcoitum), and the two γ-globin genes are completely silenced in adult erythroid cells.
To evaluate the enhancer blocking activity of human LCR-HS5 (HS, hypersensitive site), we previously reported experiments in which an HS5 fragment was inserted 3′ to the LCR within a human β-globin yeast artificial chromosome (YAC) and generated TgM lines (HS5, ref. 14). Analysis of globin gene transcription revealed that human HS5 carried a CTCF site-dependent enhancer blocking activity, which was most pronounced in definitive erythroid cells. This observation suggested that this experimental system might be amenable to examination of insulator-dependent genomic imprinting mechanisms.
Here, we asked whether the H19 ICR could recapitulate genomic imprinting in a transgenic human β-globin locus. To this end, we introduced a 2.9-kbp H19 ICR fragment 3′ to HS1 in the β-globin YAC and established four intact, single-copy TgM lines. In germ cells of the testis, the transgenic H19 ICR was almost completely devoid of methylation, which was in striking contrast to the hypermethylated endogenous H19 ICR in the same samples, demonstrating that no dominant methylation imprinting activity was conferred by the ICR fragment in the germ cell environment. However, in adult erythroid cells, differential CpG methylation between paternal and maternal H19 ICRs was observed. As a consequence, the insulator protein CTCF was enriched preferentially on the maternal H19 ICR, and the paternal allele generated significantly higher expression levels of all of the β-like globin genes. Importantly, this parent-of-origin-dependent expression was reprogrammable over generations, one of the fundamental features of genomic imprinting. These results demonstrate that the establishment of methylation imprinting, which is critical for controlling imprinted gene expression, can occur during the postfertilization period and that a 2.9-kbp H19 ICR fragment is sufficient for autonomously recapitulating this process.
Materials and Methods
Yeast Targeting Vector and Transgenic Mice. The mouse ICR fragment (a generous gift of Shirley Tilghman, Princeton University, Princeton) was excised as a 2.9-kbp SacI-BamHI fragment (from 833 to 3696 nt, GenBank accession no. AF049091). After converting the SacI site into a BamHI site with linker oligonucleotides, the fragment was subcloned into BamHI-digested pHS1/loxPw+ (14) in reverse orientation to generate pHS1/loxPw+/ICR. This plasmid DNA was linearized with SpeI and used for YAC mutagenesis (14). Generation and structural analysis of human β-globin YAC TgM has been described in ref. 14. Removal of the ectopic ICR copy was conducted by mating β-globin YAC TgM with cre recombinase TgM (15).
Semiquantitative RT-PCR. Total RNA was extracted from yolk sacs (9.5 days postcoitum), fetal livers (14.5 days postcoitum), or phenylhydrazine-induced anemic adult (1-2 month old) spleens by using ISOGEN (Nippon Gene) and converted to cDNA with reverse transcriptase (ReverTra Ace, Toyobo, Osaka). The PCR condition has been described in ref. 14. An aliquot of each PCR reaction was electrophoresed on 8% polyacrylamide gels, dried, and subjected to x-ray autoradiography and phosphor-imaging for quantitative analysis.
Bisulfite DNA Modification. Genomic DNA was extracted from anemic adult spleens or testes from 1- to 2-month-old TgM by using standard procedures. After XbaI digestion, 1 μg of DNA was treated with sodium bisulfite according to the manufacturer's recommendation (Zymo Research, Orange, CA).
PCR Amplification, Cloning, and Sequence Analysis of Bisulfite-Treated DNA. Each subregion of the ICR was amplified by nested PCR with LA Taq polymerase (Takara Bio, Tokyo). The allele-specific first-round PCR primers used are as follows: 3′ Tg ICR region, LCR-5S1 and ICR-3A6; 3′ endogenous ICR region, ICR-5S5 and the ICR-3A6; 5′ Tg ICR region, ICR-5S1 and BGLB-3A1; 5′ endogenous ICR region, the ICR-5S1 and ICR-3A3; β-globin promoter region, BGLBP-5S1 and BGLBP-3A1. The sequences of second-round PCR primers, common to the Tg and endogenous allele are as follows: 3′ ICR subregion, ICR-5S4 and ICR-3A5; 5′ ICR subregion, ICR-5S2 and ICR-3A1; β-globin promoter subregion, BGLBP-5S2 and BGLBP-3A2. The sequences for oligonucleotides are available on request. The PCR protocol was 94°C for 5 min, followed by 25 (for first-round PCR) or 35 (second round) cycles of 94°C for 1 min, 55°C for 2 min, and 72°C for 2 min. Amplified fragments were cloned into the pGEM-T Easy vector (Promega), and individual clones were sequenced. Two separate sodium bisulfite treatments were performed to verify the results.
Chromatin immunoprecipitation (ChIP) and Semiquantitative PCR. ChIP was performed essentially as described by Sawado et al. (16) with minor modifications. Briefly, spleens were harvested from phenylhydrazine-treated TgM, made into single-cell suspension, and fixed in PBS(-) with 1% formaldehyde for 10 min at 4°C. Immunoprecipitation was performed with anti-CTCF antibody (06-917, Upstate Biotechnology, Lake Placid, NY). A more detailed procedure is available on request.
PCR reactions were performed in the same buffer as that for RT-PCR. The PCR protocol was 94°C for 5 min, followed by 29 (HS5) or 30 (ICR and β-promoter) cycles of 94°C for 30 sec, 58°C (HS5 and ICR) or 51°C (β-promoter) for 30 sec, and 72°C for 1 min. The PCR primers used are as follows: LCR-SEQ-5S2 and ICR-SEQ-3A10 for ectopic H19 ICR, HS5-CTCF-5S and HS5-CTCF-3A for the LCR-HS5, and HBG-CHIP-D5S and HBG-CHIP-3A for the human β-globin promoter amplifications.
Results
Generation of YAC TgM. The human β-globin locus (Fig. 1B) is located on chromosome 11 and expression of transgenic or endogenous β-like globin genes is not subject to imprinting. We have previously shown that human LCR-HS5 conferred enhancer-blocking/insulator activity in the context of a transgenic β-globin YAC (HS5, ref. 14). Hence, we thought it might be feasible to evaluate H19 ICR activity by using the same experimental system. To this end, we introduced a 2.9-kbp ICR fragment, carrying four CTCF binding sites, 3′ to HS1 in the human β-globin YAC (ICR, Fig. 1C). The ICR fragment was floxed to remove it in vivo to regenerate a pseudo wild-type locus as a control for possible transgene site-of-integration effects (loxP, Fig. 1C). Four lines of intact, single-copy YAC TgM were generated (lines 986, 1041, 1048, and 1052), which was verified by copy number (data not shown) and long-range structural analysis (Fig. 1 B and D).
Imprinted β-Globin Gene Expression in TgM. RNA was prepared from the nucleated erythroid cells of adult spleens and β-globin gene expression was analyzed by semiquantitative RT-PCR (Fig. 2A, ICR lines). Although human β-globin gene expression was variable among the lines, it was consistently higher upon paternal inheritance, and expression was independent of the gender of the pups. This parent-of-origin-dependent expression was lost after ICR fragment deletion (Fig. 2 A, loxP lines), demonstrating that the phenotype was due to the inserted ICR fragment.
Fig. 2.

Genomic imprinting recapitulated in the humanβ-globin locus. (A) Total RNA was prepared from the spleens of anemic TgM (ICR or loxP lines). The relative expression level of the human β-globin gene (hβ), after normalization to that of the endogenous mouse α-globin gene (mα), was determined by RT-PCR. Data were collected from male (odd number) and female (even number) individuals, each inheriting the transgene either maternally (M) or paternally (P). The average and SD, determined by at least three sets of PCR reactions, are graphically depicted. Representative results of RT-PCR for hβ and mα signals are shown below each graph. (B) Total RNA was prepared from the liver (embryonic day 14.5; Left) and the yolk sacs (embryonic day 9.5; Right) of male (lanes 1, 3, 5, 6, 7, 9, 11, and 12) or female (lanes 2, 4, 8, and 10) embryos in two kinds of litters; one from the intercross of male TgM and female wild-type animals (P for paternal) and the other from the opposite combination (M for maternal). Representative results of RT-PCR analysis of the human ε (hε),γ (hγ), hβ, and mα expression in TgM lines ICR and WT are shown below each graph. The average and SD, after normalization to the endogenous mα gene signal, was determined as described in A. (C) Reprogramming of imprinting in the transgenic locus. Total RNA from the spleens of ICR-TgM was analyzed as described in A. Data were collected from male and female individuals, each inheriting the transgene either maternally (gray bars) or paternally (filled bars).
Although CTCF binding sites in the H19 ICR have several CpG motifs, that of human LCR-HS5 has no CpG sequences (17). It is therefore predicted that HS5 insulator activity is not regulated by methylation-dependent CTCF binding. We analyzed β-globin gene expression in adult spleens of HS5 TgM and found no parent-of-origin-dependent difference (data not shown).
β-like globin gene expression was next examined in the fetal liver and yolk sac erythroid cells of ICR TgM. In the fetal liver (Fig. 2B Left), both γ- and β-globin gene expression was significantly attenuated in comparison with wild-type TgM (31-WT). In addition, the magnitude of attenuation was far more evident in the maternally inherited allele, confirming imprinted gene expression. Again, this virtually monoallelic gene expression pattern was not observed in the HS5 TgM (data not shown).
In primitive erythroid cells of the yolk sac, γ-globin gene expression was not significantly affected in the HS5 TgM, whereas ε expression was severely attenuated (14), regardless of their parent of origin (data not shown). Because this expression pattern was also observed in the HS5/dCTCF TgM, in which the CTCF binding site was deleted from the HS5, we considered the possibility that the CTCF-dependent insulator is not active in primitive erythroid cells (14). In the paternally inherited allele in the ICR TgM (Fig. 2B, lanes 7 and 8), γ-globin expression was not severely attenuated in comparison with the wild-type control (Fig. 2B Lower, lanes 11 and 12), similar to the HS5 TgM. Surprisingly, however, it was severely attenuated when the locus was maternally inherited (lanes 9 and 10). This observation lead to two conclusions: first, the ICR exhibits insulator activity in primitive erythroid cells, and second, the paternally inherited ICR conferred modest insulator activity, which is suggestive of a high degree of CpG methylation. Furthermore, if the latter implication is correct, severely attenuated ε-globin gene expression, even in the paternal allele, must not solely be due to ICR insulator activity (Fig. 2B Upper). As we proposed in ref. 14, insertional disruption of a postulated ε gene-specific activator sequence around HS1 and/or a distance effect between the LCR and the ε promoter might explain this phenotype. We conclude that the imprinted gene expression pattern in the transgenic β-globin locus is not a gene- or developmental stage-specific phenomenon.
Reprogrammable β-Globin Gene Expression. We next asked whether the imprinted β-globin expression program is reset between generations. As shown in Fig. 2C, adult spleens were recovered from TgM from different generations and were subjected to RNA expression analysis. The maternally inherited (silent) β-globin gene in males (lane 4, for example) became active in the next generation (lanes 5 and 6). In contrast, paternally inherited (active) β-globin genes in females (lane 13, for example) were silenced in the next generation (lane 14). These results clearly demonstrated that imprinted β-globin gene expression in TgM was reprogrammable in a manner consistent with imprinting. We focused on line 1048 in the subsequent analyses, because the parent-of-origin-dependent phenotype was most prominent in this line.
Methylation Imprinting Activity of the H19 ICR in Erythroid Cells. We next examined the methylation status of the ICR by sodium bisulfite conversion of the DNA, followed by PCR amplification and sequencing. Genomic DNA was prepared from adult spleens from four categories of individuals: male or female mice, each inheriting the transgenes either paternally or maternally. To distinguish transgenic from endogenous ICRs, we conducted nested PCR with locus-specific first-round and common second-round primer sets (Fig. 3A). Because of the difficulty in amplifying the whole ICR fragment by locus-specific first-round PCR, we focused on sequences surrounding three of the four CTCF binding sites. The 5′ segment of the ICR (5′ICR, 751 bp) covers CTCF binding sites 1 and 2, whereas the 3′ segment (3′ICR, 383 bp) covers site 4 (Fig. 3A). Although individual transgenic ICR DNA molecules showed a wide range of methylation (Fig. 3B Left), paternally inherited sequences (P, Left) were more heavily methylated, irrespective of the gender of the pup. In addition, we examined the methylation status of the transgenic ICR in all four TgM lines by using combined bisulfite and restriction analysis (COBRA) and found that the paternal ICR was always more heavily methylated, consistent with the expression results (data not shown).
Fig. 3.

Methylation analysis of erythroid cell-derived DNA by bisulfite sequencing. (A) Schematic representation of endogenous (Left) and transgenic (Right) allele-specific ICR DNA amplification. The position of the primers used for nested PCR is shown by arrows: a, ICR-5S5; b, ICR-3A6; c, ICR-5S1; d, ICR-3A3; e, ICR-5S4; f, ICR-3A5; g, ICR-5S2; h, ICR-3A1; i, LCR-5S1; j, BGLB-3A1. (B and C) Genomic DNA from nucleated erythroid cells (line 1048) was prepared from four categories of individuals: male (Upper) or female (Lower) mice, each inheriting the transgenes either paternally (P) or maternally (M). Genomic DNA was digested with XbaI, treated with sodium bisulfite, and amplified by nested PCR. PCR products were subcloned and DNA sequences were determined. Each horizontal row represents a single DNA template molecule. Methylated (filled circles) and unmethylated (open circles) CpG motifs are shown. Gray bar indicates the locations of the CTCF binding sites. The β-globin promoter was also amplified by nested PCR, and its sequences were determined in ICR (B) as well as loxP (C) TgM, in which the ICR fragment was removed by cre-loxP recombination. The positions of CpG motifs within the β-globin promoter are shown relative to the transcription initiation site (open circles). Primers used: k, BGLBP-5S1; l, BGLBP-5S2; m, BGLBP-3A1; n, BGLBP-3A2.
Considering the multiple roles of the ICR in the endogenous Igf2/H19 locus, the paternally methylated ICR in the transgenic β-globin locus could control preferential β-globin gene expression by either of two means: i.e., promoter DNA methylation, which could interfere with repressor protein binding and/or by enhancer blocking through methylation-sensitive CTCF binding. We therefore examined the methylation status of the β-globin promoter. As shown in Fig. 3 B and C, four CpG motifs in the β-globin promoter, before and after ICR fragment deletion, respectively, were similarly methylated regardless of their parent of origin. Thus, the imprinted β-globin gene expression was not due to differential promoter methylation but rather through the enhancer blocking function of the ICR.
Preferential CTCF Recruitment to the Maternally Inherited Transgenic ICR. To further demonstrate a role of CTCF-dependent insulator activity in imprinted β-globin gene expression, we performed a ChIP analysis by using nucleated erythroid cells from spleens of the ICR TgM. Three regions within the locus, LCR-HS5 (HS5-CTCF, Fig. 4A), ectopic murine ICR (ICR-CTCF), and β-globin (β-promoter), were analyzed by allele-specific PCR. Because the HS5 TgM exhibited the same low levels of β-globin gene expression irrespective of their parental inheritance (data not shown), we predicted that HS5-CTCF sites on both alleles would be equally occupied by CTCF. ChIP analyses of two sets of TgM pairs (lines 1048, upper three panels; 1052 lower three panels, Fig. 4B) revealed that CTCF was recruited to HS5-CTCF sites on both alleles. In striking contrast, CTCF was preferentially recruited to the CTCF site 4 of the maternally inherited transgenic ICR (ICR-CTCF, Fig. 4B). The difference in enrichment between maternal and paternal alleles was 6- to 8-fold by using CTCF binding to the HS5-CTCF site as an internal control (Fig. 4C). No enrichment of CTCF was observed in the β-globin promoter, which has no reported CTCF binding sites, confirming the specificity of immunoprecipitation by the CTCF antibody (β-promoter, Fig. 4B). These results demonstrate that H19 ICR is able to imprint a heterologous β-globin gene locus by acting as a parent-of-origin-dependent, methylation-sensitive insulator.
Fig. 4.
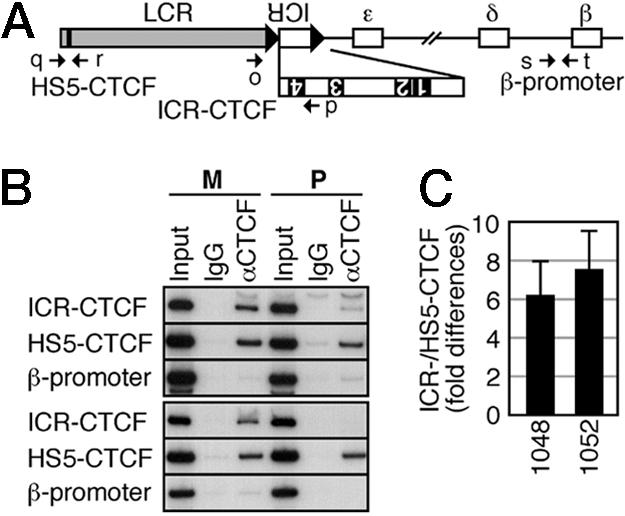
Allele-specific ChIP analysis. (A) Structure of the ICR transgene locus. The positions of the CTCF binding sites 1-4 are shown as filled rectangles in the enlarged ICR map. PCR primers are indicated by arrows. Primers used are as follows: o, LCR-SEQ-5S2; p, ICR-SEQ-3A10; q, HS5-CTCF-5S; r, HS5-CTCF-3A; s, HBG-CHIP-D5S; t, HBG-CHIP-3A. (B) ChIP analysis of occupancy of CTCF at HS5-CTCF, ICR-CTCF, and β-promoter regions of maternally (M) or paternally (P) inherited transgene loci. Chromatin was immunoprecipitated with control IgG or anti-CTCF antibodies. The upper and lower half represent results obtained from lines 1048- and 1052-ICR TgM, respectively. (C) The ICR-/HS5-CTCF signal ratio in the maternal Tg allele was divided by that in the paternal Tg allele to determine the fold differences in the CTCF recruitment to the site 4 of the ectopic ICR fragments. Semiquantitative PCR was repeated three times, and mean ± SD values were calculated for TgM lines 1048 and 1052.
Methylation Status of the ICR in Testis Germ Cells. It is generally believed that sex-specific epigenetic differences are established during gametogenesis. The H19 ICR, for example, is methylated in sperm, but not in oocytes, and this imprinted pattern is maintained during the development of all somatic tissues (18). We therefore examined the methylation status of the transgenic and endogenous H19 ICRs in germ cells (Fig. 5). DNA was extracted from the testes, each inheriting the transgene either paternally or maternally, and was subjected to sodium bisulfite treatment. Bisulfite sequencing revealed that the transgenic ICR fragment in germ cells was almost completely devoid of methylation (Fig. 5A), whereas the endogenous ICR was heavily methylated (Fig. 5B). Results from paternally or maternally inherited transgenic DNA were not significantly different, meaning that any contamination by somatic DNA was negligible. These results demonstrated that imprinted gene expression does not require establishment of methylation imprinting of the transgenic ICR in germ cells, and that the imprinting mark can be established after fertilization.
Fig. 5.
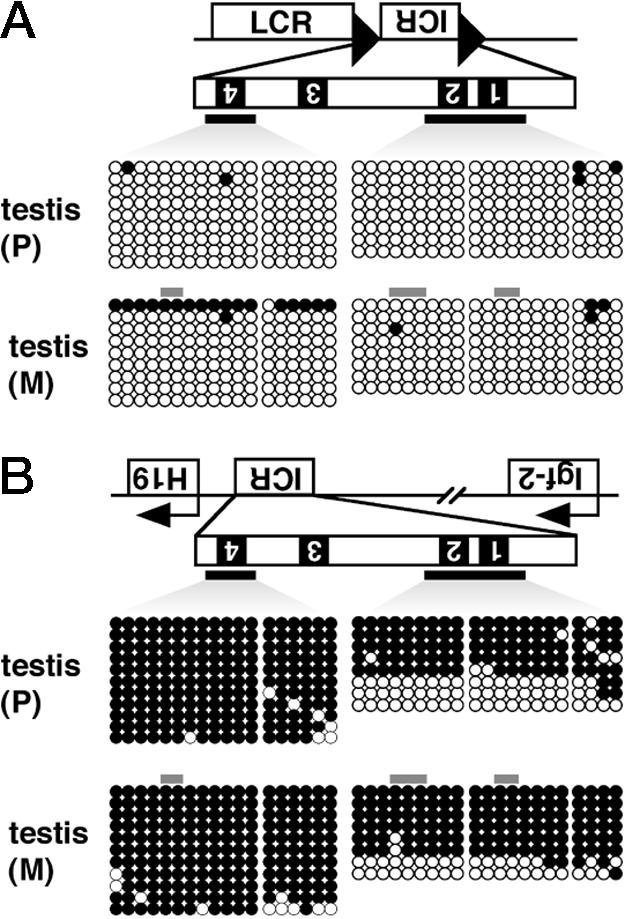
Methylation of the ICR in testis DNA. Testis DNA was extracted from 1- to 2-month-old male mice, in which the transgene was paternally (P) or maternally (M) inherited. After XbaI digestion and bisulfite treatment, two distinct regions of the ICR were amplified by allele-specific nested PCR, as described in Fig. 3 legend, and products were cloned and sequenced. The methylation status of the transgenic (A) and endogenous (B) ICR fragments is shown.
Discussion
Imprinted genes are characterized by regions of parent-of-origin-specific DNA methylation (differentially methylated region, DMR). However, neither the elements that are responsible for establishment of the methylation mark, nor its developmental timing, including the important question of whether marking occurs in the germ line or postzygotically, is fully understood. Some DMRs, such as DMR 1 and 2 in the Igf2 locus and the H19 ICR, are heavily methylated in sperm and unmethylated in oocytes. After fertilization, however, paternal methylation in the Igf2 DMRs is erased by the morula and blastocyst stages (19), whereas the H19 ICR is protected against demethylation and methylation is maintained through all developmental stages.
To address the mechanism underlying the “establishment and maintenance” of methylation, the contribution of the H19 ICR CTCF binding sites to imprinting has been tested by mutating the CTCF sites in mice. A mutant ICR bearing deleted CTCF sites was hypermethylated and hypomethylated in male and female germ cells, respectively, demonstrating that CTCF binding is not necessary to establish differential methylation of the ICR (20-23). In further support of this observation, methylation imprinting in the mutant ICR, in which nine CpG motifs within the CTCF binding sites were mutated (H19DMD-9CG), was unaffected in germ cells (24). Because this mutant ICR contains constitutively accessible, methylation-insensitive CTCF binding sites, this result indicates that binding of CTCF is not inhibitory for the establishment of methylation patterns in the ICR during spermatogenesis. According to these results, although CTCF is present in male and female germ cells, it is not required for establishing the epigenetic state of the ICR during gametogenesis. One exceptional observation to this notion is that depletion of CTCF in oocytes by RNA interference lead to increased methylation of the H19 ICR (25), demonstrating an inhibitory role of the CTCF in establishing a methylation-free status in maternal ICR. Thus, the role of CTCF in the establishment of methylation status in germ cells is yet to be determined.
Although it was shown that a DNA methyltransferase, Dnmt1a, is essential for the establishment of H19 ICR methylation in sperm (26), attempts to identify proteins that regulate allelic methylation establishment in germ cells have not been successful (27, 28). The results presented here, however, demonstrate that establishing methylation imprinting in the germ line is not mandatory for successfully executing genomic imprinting during later stages of development.
It has been postulated that CTCF is required for postzygotic maintenance of the hypomethylated maternal, and hypermethylated paternal, H19 ICR. The mutant maternal ICR with deleted CTCF sites became methylated after fertilization, indicating that CTCF binding is required for maintaining the hypomethylated state of the ICR in somatic cells by preventing recruitment of methyltransferases to the region (20-23). In contrast, paternal ICR methylation was partially lost in the H19DMD-9CG allele, demonstrating that continuous CTCF binding interferes with the maintenance of methylation (24). Thus, the transacting factor involved in maintaining the methylation status of the H19 ICR after fertilization is likely to be CTCF. If this hypothesis is the case, the methylated status of the H19 ICR, which is established in sperm, is a prerequisite for maintaining methylation imprinting by preventing CTCF binding to the ICR, which antagonizes a putative de novo methyltransferase (29). The observation that the transgenic paternal ICR was almost completely devoid of methylation in the testis clearly demonstrated that “establishment of differential methylation” is not an autonomous function of the ICR in germ cells. Surprisingly, however, allele-specific ICR methylation was acquired after fertilization. This observation is not compatible with the notion that methylation imprinting in sperm is required to maintain the methylated status of the genome.
Even after implantation and early postimplantation development, there is a small but considerable amount of de novo methylation activity, which is probably mediated by Dnmt3a and 3b (30). Park et al. (11) reported that the H19 ICR knocked-in at the afp locus was de novo methylated around the gastrulation stage. Because the enzyme should theoretically act on both parental alleles, the maternal H19 ICR must be protected from being methylated. Although allele-specific processes that establish ICR methylation after fertilization are undefined, several hypothetical mechanisms seem possible. For example, “methylation imprinting centers (MIC)” that are differentially methylated in germ cells may be scattered throughout the genome. After fertilization, methyltransferases could recognize the methylation state of the regulatory MIC by some currently undefined mechanism and introduce methyl groups into the transgenic ICR in cis. This scenario seems unlikely, however, as the transgenic ICR appeared more heavily methylated on the paternal allele (Figs. 2 and 3 and data not shown), whereas most of the endogenous DMR is more frequently methylated on the maternal allele (19, 31). Alternatively, parental genomes may be differentially marked with modifications other than DNA methylation in germ cells, which in turn either cause or prevent postfertilization DNA methyltransferase recruitment to the ICR in an allele-specific manner. This hypothesis implies the existence of specific sequences in the ICR that are capable of marking the parental chromosomes in germ cells. In vivo dissection of the ICR fragment in this present experimental system might disclose the identity of such sequences.
In addition to the role of CTCF in interpreting the methylation status of binding sites within the H19 ICR to control imprinted gene expression and to protect differential methylation states at and around its target sites during development, CTCF might also be required to postzygotically establish differential methylation patterns in other loci, as exemplified by this transgenic β-globin locus bearing the H19 ICR. Based on the results obtained from an analysis of the methylation status of the endogenous H19 ICR with deleted CTCF sites (20-23), it was assumed that incomplete methylation of the paternal (mutant) ICR is due to a loss of methylation after fertilization, because it was initially fully methylated in the sperm. Apparently, postzygotic methylation acquisition and involvement of CTCF in this “initiation” process has not been considered previously. This issue can be examined by using the zygotically unmethylated ICR as the starting point in the β-globin TgM assays.
In summary, H19 ICR sequences in the heterologous transgenic human β-globin locus were subject to methylation imprinting. As a consequence, genomic imprinting was recapitulated in the locus, both in primitive and definitive erythroid cells. Finally, methylation imprinting in the β-globin locus was not acquired during gametogenesis, revealing the existence of a mechanism for establishing methylation imprinting during the postfertilization period.
Acknowledgments
This work was supported by research grants from the National Institutes of Health (HL24415 and HL73465 to J.D.E.), the Inamori Foundation (to K.T.), the Naito Foundation (to K.T.), the 21st Century Center of Excellence Program (to A.F.) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (MEXT), and Grants-in-Aid for Scientific Research (MEXT, to A.F.), Scientific Research on Priority Areas (MEXT, to K.T.) and Young Scientists (MEXT, to K.T.).
Author contributions: K.T., J.B., J.D.E., and A.F. designed research; K.T., M.S., H.M., and A.O. performed research; K.T. and M.S. analyzed data; and K.T., J.B., and J.D.E. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: ChIP, chromatin immunoprecipitation; HS, hypersensitive site; ICR, imprinting control region; LCR, locus control region; TgM, transgenic mouse; YAC, yeast artificial chromosome.
References
- 1.Bartolomei, M. S. & Tilghman, S. M. (1997) Annu. Rev. Genet. 31, 493-525. [DOI] [PubMed] [Google Scholar]
- 2.Bell, A. C. & Felsenfeld, G. (2000) Nature 405, 482-485. [DOI] [PubMed] [Google Scholar]
- 3.Hark, A. T., Schoenherr, C. J., Katz, D. J., Ingram, R. S., Levorse, J. M. & Tilghman, S. M. (2000) Nature 405, 486-489. [DOI] [PubMed] [Google Scholar]
- 4.Kanduri, C., Holmgren, C., Pilartz, M., Franklin, G., Kanduri, M., Liu, L., Ginjala, V., Ulleras, E., Mattsson, R. & Ohlsson, R. (2000) Curr. Biol. 10, 449-457. [DOI] [PubMed] [Google Scholar]
- 5.Kanduri, C., Pant, V., Loukinov, D., Pugacheva, E., Qi, C. F., Wolffe, A., Ohlsson, R. & Lobanenkov, V. V. (2000) Curr. Biol. 10, 853-856. [DOI] [PubMed] [Google Scholar]
- 6.Holmgren, C., Kanduri, C., Dell, G., Ward, A., Mukhopadhya, R., Kanduri, M., Lobanenkov, V. & Ohlsson, R. (2001) Curr. Biol. 11, 1128-1130. [DOI] [PubMed] [Google Scholar]
- 7.Leighton, P. A., Saam, J. R., Ingram, R. S., Stewart, C. L. & Tilghman, S. M. (1995) Genes Dev. 9, 2079-2089. [DOI] [PubMed] [Google Scholar]
- 8.Bartolomei, M. S., Webber, A. L., Brunkow, M. E. & Tilghman, S. M. (1993) Genes Dev. 7, 1663-1673. [DOI] [PubMed] [Google Scholar]
- 9.Szabo, P. E., Tang, S. H., Reed, M. R., Silva, F. J., Tsark, W. M. & Mann, J. R. (2002) Development (Cambridge, U.K.) 129, 897-904. [DOI] [PubMed] [Google Scholar]
- 10.Reinhart, B., Eljanne, M. & Chaillet, J. R. (2002) Mol. Cell. Biol. 22, 2089-2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park, K. Y., Sellars, E. A., Grinberg, A., Huang, S. P. & Pfeifer, K. (2004) Mol. Cell. Biol. 24, 3588-3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stamatoyannopoulos, G. & Neinhuis, A. W. (1994) in Hemoglobin Switching, eds. Stamatoyannopoulos, G., Nienhuis, A. W., Majerus, P. & Varmus, H. (Saunders, Philadelphia), Chapter 4, pp. 107-155.
- 13.Tanimoto, K., Liu, Q., Grosveld, F., Bungert, J. & Engel, J. D. (2000) Genes Dev. 14, 2778-2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanimoto, K., Sugiura, A., Omori, A., Felsenfeld, G., Engel, J. D. & Fukamizu, A. (2003) Mol. Cell. Biol. 23, 8946-8952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu, R. N., Ito, M., Saunders, T. L., Camper, S. A. & Jameson, J. L. (1998) Nat. Genet. 20, 353-357. [DOI] [PubMed] [Google Scholar]
- 16.Sawado, T., Halow, J., Bender, M. A. & Groudine, M. (2003) Genes Dev. 17, 1009-1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Farrell, C. M., West, A. G. & Felsenfeld, G. (2002) Mol. Cell. Biol. 22, 3820-3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tremblay, K. D., Duran, K. L. & Bartolomei, M. S. (1997) Mol. Cell. Biol. 17, 4322-4329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reik, W. & Walter, J. (2001) Nat. Genet. 27, 255-256. [DOI] [PubMed] [Google Scholar]
- 20.Schoenherr, C. J., Levorse, J. M. & Tilghman, S. M. (2003) Nat. Genet. 33, 66-69. [DOI] [PubMed] [Google Scholar]
- 21.Pant, V., Mariano, P., Kanduri, C., Mattsson, A., Lobanenkov, V., Heuchel, R. & Ohlsson, R. (2003) Genes Dev. 17, 586-590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pant, V., Kurukuti, S., Pugacheva, E., Shamsuddin, S., Mariano, P., Renkawitz, R., Klenova, E., Lobanenkov, V. & Ohlsson, R. (2004) Mol. Cell. Biol. 24, 3497-3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Szabo, P. E., Tang, S. H., Silva, F. J., Tsark, W. M. & Mann, J. R. (2004) Mol. Cell. Biol. 24, 4791-4800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Engel, N., West, A. G., Felsenfeld, G. & Bartolomei, M. S. (2004) Nat. Genet. 36, 883-888. [DOI] [PubMed] [Google Scholar]
- 25.Fedoriw, A. M., Stein, P., Svoboda, P., Schultz, R. M. & Bartolomei, M. S. (2004) Science 303, 238-240. [DOI] [PubMed] [Google Scholar]
- 26.Kaneda, M., Okano, M., Hata, K., Sado, T., Tsujimoto, N., Li, E. & Sasaki, H. (2004) Nature 429, 900-903. [DOI] [PubMed] [Google Scholar]
- 27.Bowman, A. B., Levorse, J. M., Ingram, R. S. & Tilghman, S. M. (2003) Mol. Cell. Biol. 23, 8345-8351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szabo, P. E., Pfeifer, G. P. & Mann, J. R. (2004) Mol. Cell. Biol. 24, 4858-4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Szabo, P. E. & Mann, J. R. (1995) Genes Dev. 9, 3097-3108. [DOI] [PubMed] [Google Scholar]
- 30.Okano, M., Bell, D. W., Haber, D. A. & Li, E. (1999) Cell 99, 247-257. [DOI] [PubMed] [Google Scholar]
- 31.Delaval, K. & Feil, R. (2004) Curr. Opin Genet. Dev. 14, 188-195. [DOI] [PubMed] [Google Scholar]