

Abstract
The risks of cancer and other degenerative diseases caused by reactive oxygen species and electrophiles can be reduced by the up-regulation of detoxifying enzymes. A major mechanism whereby these protective enzymes are induced occurs through activation of the antioxidant response element (ARE) by the oxidative-stress sensor protein Kelch-like ECH-associated protein 1 (Keap1) and the transcription factor NF-E2-related factor 2 (Nrf2). Under basal conditions, Keap1 sequesters Nrf2 in the cytoplasm by binding to its Neh2 domain. Chemical inducers such as sulforaphane are known to react with Keap1 cysteine residues, thereby promoting Nrf2 nuclear accumulation and hence ARE activation. A widely accepted model for Nrf2 nuclear accumulation is that modification of Keap1 cysteines leads directly to dissociation of the Keap1-Nrf2 complex. This model is based on studies with mouse proteins and has served as the experimental basis and hypothesis for numerous investigations. Through a combination of chemical, mass spectrometry, and isothermal titration calorimetry methods, we have tested the direct-dissociation model using a series of ARE inducers: sulforaphane, isoliquiritigenin, 15-deoxy-Δ12,14-prostaglandin-J2, menadione, 1-Cl-2,4-dinitrobenzene, and biotinylated iodoacetamide. Surprisingly, these data suggest that the direct disruption model for Keap1-Nrf2 is incorrect. The relative reactivity of human Keap1 cysteines was determined. In addition to the same five cysteines identified for mouse Keap1, two highly reactive and previously unobserved cysteines were identified. Based on these results, a model is proposed that should aid in the understanding of Keap1-Nrf2 signaling mechanisms.
Keywords: antioxidant response element; sulforaphane; 15-deoxy-Δ12,14-prostaglandin J2; oxidative stress; ubiquitination
The NF-E2-related factor 2 (Nrf2) plays an important role in mediating defenses against electrophilic and oxidative damage to biological macromolecules. Damage to DNA, proteins, and lipids is involved in the aging process as well as the initiation and proliferation of numerous diseases, including cancer and neurodegenerative disorders. The expression of various protective proteins and enzymes is up-regulated by means of Nrf2 binding to the antioxidant response element (ARE), a cis-acting sequence located in the 5′ flanking region of these genes (1). Nrf2-based up-regulation of these enzymes, including NAD(P)H:quinone oxidoreductase 1, glutathione S-transferase, and heme oxygenase-1, protects against cancer, inflammatory diseases, oxidative stress-related diseases (including pulmonary disease), aging, and neurodegenerative disorders (2, 3).
Signaling of Nrf2 induction of the ARE in response to stress agents occurs by induced Nrf2 nuclear accumulation (4). Under basal conditions, Nrf2 is sequestered in the cytoplasm by the Kelch-like ECH-associated protein 1 (Keap1), whose primary sequence is illustrated in Fig. 1. Keap1 is confined to the cytoplasm by means of part of its linker region (5). The Kelch repeat region of Keap1 binds to the Neh2 domain (approximately the first 100 residues) of Nrf2, preventing constitutive Nrf2 nuclear accumulation (6). Keap1-null mice show constitutive Nrf2 activation as well as postnatal lethality, which is counteracted by breeding to Nrf2-deficient mice (7).
Fig. 1.

Primary sequence representation of the mouse and human Keap1 proteins and the most readily modified cysteines. (A) The cysteines in mouse Keap1 most readily modified by dexamethasone 21-mesylate (12) are shown in green. All cysteines shown were modified by BIA in this study, both green- and red-labeled, and those in red were found uniquely in this study. Those labeled with an asterisk were reported to be important in the regulation of Nrf2 ubiquitination (5). (B) The order of reactivity of the cysteines determined in this study is shown, along with the ratio of BIA to Keap1 required to obtain those modifications.
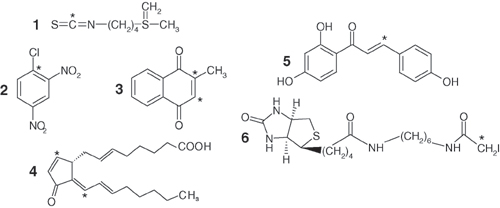
Numerous ARE inducers have been identified in an effort to find cancer preventive agents (8, 9). One well known inducer is sulforaphane (Fig. 7, which is published as supporting information on the PNAS web site), which is obtained through the consumption of cruciferous vegetables. The essential role of Nrf2 in transducing the cancer-preventive effect of sulforaphane is evident, because sulforaphane is significantly less effective in Nrf2 knockout mice (10). The majority of ARE inducers are capable of modifying cysteines, suggesting that Keap1 cysteines are targeted by these compounds in signaling ARE induction (11). Among the 25 cysteines of mouse Keap1, the 5 that are most reactive toward the electrophile dexamethasone 21-mesylate (DexMes) were identified as C257, C273, C288, C297, and C613 (Fig. 1) (12). Of these five cysteines, only C273 and C288 are required for repression of Nrf2 nuclear accumulation in mouse-based ARE-reporter assays (13). The importance of C273 and C288 in human Keap1 repression of ARE activation has been demonstrated by means of mutagenesis studies (5). However, systematic studies to identify the cysteines in human Keap1 most reactive toward chemical inducers have not been reported.
Modification of Keap1 cysteines by ARE inducers is postulated to directly cause dissociation of the Keap1-Neh2 interaction, leading to Nrf2 nuclear accumulation. This model is based primarily on a study by Dinkova-Kostova et al. (12) in which native EMSAs conducted with mouse Keap1 and Neh2 proteins show that addition of sulforaphane disrupts the Keap1-Neh2 complex band. In a subsequent study, it was demonstrated that a C257A-C297A mutant form of mouse Keap1 has approximately half the affinity of wild-type Keap1 for Neh2 (13). These studies have established the widely accepted model that electrophilic modification of Keap1 leads directly to dissociation of the Keap1-Nrf2 complex. Moreover, these studies have significantly influenced work in the fields of oxidative stress response mechanisms and phase 2 enzyme induction (2, 14-20).
Several groups have studied the human Keap1-Nrf2 system under the assumption that the cysteine residues modified by electrophiles in human Keap1 are the same as those modified by electrophiles in mouse Keap1 (5, 15, 21). Because of the importance of the Keap1-Nrf2 system in regulating the ARE and its significance to public health, we thought it important to examine this assumption as well as the direct-disruption model by identifying which cysteines in human Keap1 are most readily modified by chemical inducers, and by quantifying the Kd of Neh2 with unmodified and modified human Keap1. To our surprise, our data suggest that the direct disruption model is invalid for the human system, and that the most reactive cysteines of human Keap1 are not the same as those reported for mouse Keap1. Based on these results, we propose a model to aid in the understanding of Keap1-Nrf2 signaling mechanisms.
Materials and Methods
Protein Expression and Purification. The genes for human Keap1 and the Neh2 domain (amino acids 1-97) of Nrf2 were cloned by using PCR into pET15b vectors. After expression in Escherichia coli, the proteins were purified by using a HiTrap-Ni2+-chelate affinity column followed by a MonoQ anion-exchange column. Detailed procedures for cloning, expression, and purification are presented in Supporting Methods, which is published as supporting information on the PNAS web site.
Keap1-Neh2 Native EMSAs. For binding stoichiometry experiments, reactions containing 15 μM Keap1 and Neh2 from 3.3 to 9.2 μM were incubated on ice for 5 min in reaction buffer [50 mM 4-(2-hydroxyethyl)-1-piperazinepropanesulfonic acid (Hepes), pH 8.0]/0.36 mM Tris(2-carboxyethyl)phosphine hydrochloride (TCEP). Samples were analyzed on Tris·HCl gradient PAGE gels. Similar conditions were followed in experiments in which Keap1 was modified with electrophiles. Incubations of Keap1 with sulforaphane, 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2), isoliquiritigenin, menadione, biotinylated iodoacetamide (BIA), or 1-Cl-2,4-dinitrobenzene (CDNB) were carried out for 70 min at 25°C, and reactions without electrophiles included the same volume of vehicle (dimethyl sulfoxide or acetonitrile) as those in which an electrophile was added. The average number of modifications of Keap1 by each electrophile, except for sulforaphane, was determined by MALDI-TOF mass spectrometry by using a Voyager DEPRO instrument (Applied Biosystems). The extent of Keap1 labeling by sulforaphane in reactions used for EMSA analysis was measured by a two-step ultrafiltration procedure. First, an extinction coefficient of 7 mM-1·cm-1 was determined for the Keap1-sulforaphane conjugate in the absence of TCEP. In the second step, this extinction coefficient was used to quantify the extent of modification of Keap1 by sulforaphane in the presence of TCEP by using ultrafiltration to monitor TCEP-SUL conjugate formation separately from Keap1-sulforaphane conjugate formation. Further details on the ultrafiltration procedure are provided in Supporting Methods. To ensure that an accurate extent of labeling was obtained, an aliquot of the reaction of Keap1 with an electrophile was analyzed for the number of modifications at the same time as Neh2 was added to an aliquot of that reaction. After 5 min of incubation at 25°C, the samples were loaded onto the gel and analyzed as described above.
Isothermal Titration Calorimetry (ITC) Experiments. All ITC experiments were performed on the VP-ITC system (Microcal, Amherst, MA) by using degassed proteins. Keap1, unmodified or modified with CDNB or BIA (see below), was loaded into the sample cell, with Neh2 in the syringe. All experiments were performed at 25°C after a 300-s initial delay with 35 injections at 2 μl per injection spaced by 200 s. The average heat released (kcal/mole of injectant) during the last 11 injections was added to each reading to correct for the heat of dilution. Results were analyzed with MicroCal Systems analysis software (origin 5.0), by using a nonlinear curve fitting and a single-site binding model. Each experiment was performed in at least triplicate. The error for the association constant in each trial ranged from 14 to 24%, which is common for such tight associations determined by ITC (22, 23).
Details of the labeling procedures of Keap1 with BIA (EZ-Link Iodoacetyl-LC-Biotin, Pierce) and CDNB, as well as protein concentration determinations, are given in Supporting Methods. The number of modifications of Keap1 by each electrophile was determined by MALDI-TOF mass spectrometry. The number of modifications of Keap1 by CDNB was estimated after removal of unreacted CDNB by using the extinction coefficient of glutathione (GSH)-conjugated CDNB at 340 nm of 9.6 mM-1·cm-1 (24). Details of the calculation are given in Supporting Methods.
Determination of the Identity of Keap1 Cysteines Most Readily Modified by BIA. Keap1 (10 μM) was incubated in the presence of 0.4 mM TCEP with increasing concentrations of BIA from an equimolar amount, relative to Keap1, to a 10-fold excess. The incubation was performed in 25 mM Tris·HCl buffer (pH 8.0), at 25°C for 1 h, followed by quenching with excess DTT. The average molecular weight of modified Keap1 was measured by using MALDI-TOF mass spectrometry. To block remaining free cysteine residues, the reaction was then treated with 1 mM iodoacetamide in excess of the DTT concentration, allowed to react for 30 min, and treated with a final DTT addition. To determine the identity of the modification sites, samples were digested with trypsin, and peptides that were biotinylated by means of reaction of BIA with Keap1 cysteines were purified from the digestion mixture by using an ICAT kit (Applied Biosystems). Affinity-purified peptides were analyzed by using positive ion electrospray liquid chromatography tandem MS (LC-MS/MS) with a Surveyor HPLC system interfaced to an LCQ DecaI ion trap mass spectrometer (Thermo Electron, San Jose, CA). LC-MS/MS data were processed by using bioworks 3.1 software (Thermo Electron). The modification sites were identified based on the tandem mass spectra of the biotinylated peptides following a database search using turbosequest.
Results
Analyzing the Human Keap1-Neh2 Complex by Native EMSA. To determine whether the purified Keap1 and Neh2 proteins were functional, their ability to form a complex was determined by using native EMSA. The Keap1 concentration was held constant as the concentration of Neh2 was increased for each reaction. As shown in Fig. 2, Keap1 is completely bound by Neh2 at the end of the titration. A similar titration of Neh2 with Keap1 protein demonstrated that all of Neh2 was bound by Keap1 (data not shown), indicating that the preparations contain functional proteins. As a further indication that the proteins are properly folded, extensive secondary structure was observed by circular dichroism (data not shown). As observed in Fig. 2, lane 2, Keap1 forms a predominant band on the native gel along with a much less intense lower molecular weight band. Titration of Neh2 results in the formation of two higher molecular bands representing the Keap1-Neh2 complex (lane 5). The formation of two bands suggests that Neh2 binding to Keap1 can induce higher-order oligomeric forms of Keap1 under the conditions used, although this induction could not be confirmed with size exclusion chromatography (data not shown). The titration point at which Keap1 is completely bound by Neh2, shown in lane 8, corresponds to an overall stoichiometry of 2:1 Keap1 to Neh2 molecules. The predominant higher molecular weight form of Keap1 seems to bind Neh2 more readily, because the minor lower molecular form of Keap1 was the last to bind Neh2 (lane 7).
Fig. 2.

Keap1-Neh2 titration showing a binding stoichiometry of 2:1 Keap1:Neh2. Reaction conditions corresponding to lanes 2-10 included 15 μM Keap1 and increasing concentrations of Neh2 from 3.3 to 9.2 μM as shown. Lane 1 corresponds to an incubation containing 9.2 μM Neh2 but with Keap1 omitted. The asterisk in lane 7 indicates the faint band of Keap1 not bound to Neh2 located just above the asterisk.
Modifying Keap1 Cysteines with Sulforaphane Does Not Disrupt Keap1-Neh2 Complex Formation or Binding Stoichiometry. The mouse Keap1-Neh2 complex has been reported to dissociate upon incubation with various electrophiles such as sulforaphane (Fig. 7) (12). Sulforaphane induces cellular accumulation of Nrf2 (15) and stabilizes proteasomal degradation of Neh2 (25). In addition, sulforaphane's cancer preventive effects were significantly diminished in Nrf2 knockout mice (10), making it an excellent choice for our studies.
To date, no method has been developed to determine the number of Keap1 cysteines that are modified by sulforaphane. Because MALDI-TOF mass spectrometry was not suitable for the measurement of sulforaphane modifications, we developed a method to determine an extinction coefficient for the sulforaphane-Keap1 cysteine conjugate using ultrafiltration. The method was developed such that TCEP could be included at the time of reaction, because the inclusion of a reducing agent such as TCEP in the reaction ensures that the most reactive cysteines are in the reduced state at the time of electrophile addition. Keap1 cysteines oxidize with time after removal of TCEP, leading to fewer modifications (data not shown).
Keap1 was modified with increasing concentrations of sulforaphane in the presence of TCEP. As shown in Fig. 3A, lanes 2 and 3, modification of nine Keap1 cysteines by a 15-fold excess of sulforaphane had no apparent effect on the electrophoretic mobility of Keap1. However, a form of Keap1 with very low mobility in the gel was induced by modification of Keap1 with 35- and 80-fold excess of sulforaphane, with 10 and 12 modifications per Keap1.
Fig. 3.

Native EMSA gels for Keap1 modified with sulforaphane (SUL). (A) Keap1 was modified with increasing concentrations of sulforaphane, and its ability to bind to Neh2 was tested. Reaction conditions corresponding to lanes 2-10 include 15 μM Keap1 and 0.4 mM TCEP. Keap1 was modified with sulforaphane at the concentrations in excess of Keap1 as shown before Neh2 addition, except for lane 10, in which Neh2 and Keap1 were prebound before sulforaphane addition. Lanes 1 and 6-10 contain 7.5 μM Neh2. (B) The Keap1-Neh2 binding stoichiometry does not change when Keap1 is preincubated with sulforaphane. Reaction conditions corresponding to lanes 2-9 include 15 μM Keap1 and increasing concentrations of Neh2 from 5 to 7.5 μM as shown. Where indicated, Keap1 was preincubated with 180 μM sulforaphane. Lane 1 contains 7.5 μM Neh2.
Neh2 was then added to the modified Keap1 mixtures, followed by native EMSA analysis. Surprisingly, there was no apparent difference in the mobility of the Keap1-Neh2 complex upon modification of nine Keap1 residues by sulforaphane (Fig. 3A, lanes 6 and 7). Increasing concentrations of sulforaphane (lanes 8 and 9) led to some free Neh2 protein, but this result was likely due to the inability of Neh2 to bind to the low-mobility form of Keap1 because no unbound Keap1 was observed. Finally, the effect of sulforaphane on a prebound Keap1-Neh2 complex was tested, by using an 80-fold excess of sulforaphane. In contrast to results obtained for the mouse proteins, no human Keap1-Neh2 complex disruption was observed. Interestingly, Neh2 seemed to protect Keap1 from over-modification, because Keap1 was not found in the bottom of the wells (comparing lanes 9 and 10). Correspondingly, the number of cysteine modifications was reduced from 12 to 10 when modification occurred in the presence of Neh2 compared with in its absence.
To determine whether any change in the binding stoichiometry of the Keap1-Neh2 complex might occur when Keap1 is modified with sulforaphane, Neh2 titrations of modified and unmodified Keap1 were evaluated by native EMSA analysis. Keap1 was modified with a 12-fold excess of sulforaphane in the presence of TCEP, resulting in eight modifications per Keap1 molecule on average. As shown in Fig. 3B, both modified and unmodified Keap1 show the same binding stoichiometry.
Modification of Keap1 by Michael Reaction Acceptors Does Not Disrupt Keap1-Neh2 Complex Formation. Sulforaphane reacts with Keap1 by means of an isothiocyanate group that is fairly unique among ARE electrophile inducers. The most commonly found electrophilic moiety among ARE inducers is the α,β-unsaturated carbonyl group, also known as a Michael reaction acceptor (8, 9, 26). Three Michael reaction acceptors that are known ARE inducers were selected to test for their effect on Keap1-Neh2 complex formation. These compounds (Fig. 7) include menadione, 15d-PGJ2, and isoliquiritigenin, a chalcone found in a variety of edible plant sources including shallots, licorice, and tonka bean seeds (8). Isoliquiritigenin is a potent inducer of quinone reductase and shows a significant response in the mouse mammary organ culture assay (8). The molecule 15d-PGJ2, a lipid oxidation product that accumulates during acute inflammation, has been shown to induce nuclear accumulation of Nrf2 (15). The nuclear accumulation seemed to be accompanied by elimination of Nrf2 from the cytoplasm, indicating possible dissociation of the Keap1-Nrf2 complex. In addition, biotinylated 15d-PGJ2 has been shown to bind to native Keap1 in vivo by using rat hepatocyte cells (14). Menadione has been shown to induce Nrf2 cellular accumulation (15).
Keap1 was incubated with each of the Michael reaction acceptors in a 32-fold excess over the Keap1 concentration in the presence of TCEP. Identical reactions were also performed in which Neh2 was preincubated with Keap1 before the addition of electrophile. MALDI-TOF mass spectrometry was used to determine the average number of modifications per Keap1 molecule, and simultaneously, aliquots of the reactions without Neh2 were combined with Neh2. All reactions were then evaluated immediately by native EMSA analysis. As shown in Fig. 4, Keap1 modified with each of the three electrophiles was still capable of being completely bound by Neh2 (lanes 2, 5, 8, and 11). The extent of Keap1 modification by each of the electrophiles is indicated. Again, as observed with sulforaphane, addition of the electrophiles did not disrupt the Keap1-Neh2 complex (lanes 3, 6, 9, and 12). The presence of Neh2 partially protected Keap1 from modification by 15d-PGJ2, resulting in four modifications versus five. As shown in lanes 1 and 4, modification of Keap1 by five molecules of 15d-PGJ2 on average noticeably increased the mobility of Keap1. The increase in mobility toward the cathode is probably due to the introduction of the negatively charged carboxylic acid moiety on the electrophile into the protein. In comparison, modification of Keap1 by an average of eight molecules of menadione, a largely nonionizable compound, leads to only a slight increase in the mobility of Keap1 (lane 10).
Fig. 4.

Native EMSA of Keap1 and Neh2 with Keap1 incubated with various Michael reaction acceptor electrophiles alone or in the presence of Neh2. Reaction conditions corresponding to all lanes include 15 μM Keap1, and, in lanes 2, 5, 8, and 11, Keap1 was preincubated with the electrophiles indicated before addition of 7.5 μM Neh2. In the reactions corresponding to lanes 3, 6, 9, and 12, Neh2 was prebound to Keap1 before addition of the electrophile. The average number of modifications per Keap1 molecule as determined by MALDI-TOF is indicated. nd, Not determined; isoliquirit, isoliquiritigenin.
Modification of Keap1 by Electrophiles Does Not Affect the Keap1-Neh2 Dissociation Constant. Modification of Keap1 in vitro by various ARE inducers does not seem to lead to disruption of the Keap1-Neh2 complex or interfere with complex formation as determined by native EMSA. However, it is possible that there is a change in the Kd of Keap1 and Neh2 upon modification of Keap1 with electrophiles that is of a magnitude that cannot be detected by using the EMSA technique. Therefore, ITC was used to quantify any change in the Kd of Keap1 and Neh2 after modification of Keap1 by the irreversible-binding electrophiles CDNB and BIA. BIA was chosen because it was useful in identifying the most readily modified cysteines (see following section). CDNB is a potent inducer of quinone reductase (27) and was shown to induce HO-1 exclusively through the Nrf2 pathway in Nrf2 knockout mice (28). After Keap1 modification by CDNB or BIA, MALDI-TOF mass spectrometric analysis was used to detect an average of 5 ± 1 Keap1 residues modified by BIA and 6 ± 2 modified by CDNB. The higher error in the CDNB-modified Keap1 signal is due to a higher concentration of Hepes buffer in that sample, which partially suppresses ionization during MALDI-TOF mass spectrometry. Because the unreacted CDNB was removed, the number of cysteines modified by CDNB was also estimated based on the extinction coefficient of CDNB conjugated with glutathione (GSH) at 340 nm (24). On average, 4.3 ± 0.3 residues were modified by CDNB, in agreement within error with that obtained by using mass spectrometry.
The Kd for the unmodified Keap1-Neh2 interaction was determined in triplicate, and a typical thermograph and binding isotherm are shown in Fig. 5. A very tight association was found to exist between the two proteins (Kd of 9 nM). Formation of the Keap1-Neh2 complex is almost entirely enthalpy driven, with an average ΔH value for three trials of -23.0 ± 1.6 kcal/mol. The average value for TΔS calculated from these experiments was -12.0 ± 1.7 kcal/mol, with a ΔS value of -0.040 kcal/(K·mol).
Fig. 5.

ITC profiles of Neh2 with unmodified or modified Keap1. Representative profiles are shown for 39.5 μM Neh2 titrated into 1.5 μM Keap1, unmodified or modified with BIA or CDNB as labeled. (Upper) ITC thermographs. (Lower) The fitted binding isotherms.
Typical thermographs and binding isotherms for experiments with CDNB- or BIA-modified Keap1 are shown in Fig. 5. The binding isotherms closely resemble those obtained for nonmodified Keap1. The average Kd values for Neh2 binding to BIA- and CDNB-modified Keap1 were 12 nM and 9 nM. The average ΔH values measured were -23.2 ± 0.9 kcal/mol and -28.4 ± 1.9 kcal/mol, similar to that of nonmodified Keap1. The modification of Keap1 by both BIA and CDNB was also analyzed by native EMSA (data not shown). The EMSA results are equivalent to those obtained with the other electrophiles by EMSA analysis, demonstrating that Keap1 modified with BIA and CDNB readily binds to Neh2 with the same stoichiometry as does unmodified Keap1.
Identification of the Human Keap1 Cysteines Most Readily Modified by BIA. The cysteines of human Keap1 that are most readily modified by an electrophile were determined by using a method that utilizes a biotin tag attached to the ARE inducer iodoacetamide (BIA, Fig. 7). BIA itself is an ARE activator and induces Nrf2 nuclear accumulation (D. Liebler, personal communication). After reaction of Keap1 with BIA, biotinylated peptides obtained by tryptic digestion were purified by means of their biotin tag. At a reaction ratio of 10 BIA molecules per Keap1 protein, the mass of Keap1 was observed to increase by 2,132 Da, or 6 ± 1 additions of BIA to Keap1 on average (Fig. 8, which is published as supporting information on the PNAS web site). The width of the mass spectrum peak was observed to widen after modification (Fig. 8), indicating a population of modified Keap1 proteins. Correspondingly, seven cysteine-containing biotinylated peptides were detected by using LC-MS/MS. The identities of the peptides were confirmed based on a database search of their tandem mass spectra (Fig. 9, which is published as supporting information on the PNAS web site). To assess the reactivity order of these seven cysteines, the identities of cysteines modified at reaction ratios of 1, 2, and 10 BIA molecules per Keap1 molecule were determined (Fig. 1). The three cysteines of human Keap1 most readily modified were identified as C151, C288, and C297. The next most reactive cysteine was found to be C319, followed by C257, C273, and C613. It should be noted that previous reports indicated that cysteines 151 and 319 were not reactive in mouse Keap1 (12, 13).
Discussion
Contrary to the widely accepted model that electrophilic modification of Keap1 leads to direct dissociation of the Keap1-Nrf2 complex, we find that addition of electrophilic ARE inducers to a preformed Keap1-Neh2 complex, or to Keap1 before complex formation, does not lead to complex disruption. We also find that the human Keap1 and Neh2 proteins bind with very high affinity, with a Kd of 9 nM. Modification of human Keap1 by a variety of ARE inducers does not alter the affinity of Keap1 for the Neh2 domain protein of Nrf2, nor does it alter the binding stoichiometry.
We determined the average number of modifications of Keap1 by each electrophile in these experiments, typically four to eight cysteines, using chemical and mass spectrometry-based techniques. In the case of sulforaphane, up to 12 modifications were detected. In every case, we were unable to detect any disruption of the binding of Keap1 and Neh2. In the case of the ARE inducer BIA, the seven most reactive cysteines in human Keap1 were identified, and their order of reactivity was established (Fig. 1). These seven cysteine residues include five of the cysteines (C257, C273, C288, C297, and C613) in mouse Keap1 that were found to be the most readily modified by dexamethasone 21-mesylate (12), and most interestingly, two additional cysteines, (C151 and C319) that have not been identified previously as being highly reactive to electrophiles. All seven of these residues are conserved among mammals, and C151, C288, C297, and C613 are conserved from humans to zebrafish (Fig. 10, which is published as supporting information on the PNAS web site).
Similar reactivities of the cysteines in human and mouse Keap1 were expected based on the high degree (94%) of sequence identity between these proteins. However, the identification of two additional cysteines as being highly reactive in human Keap1 was unexpected. The first cysteine, C319, was identified as being the fourth most reactive cysteine in human Keap1. Mutagenesis studies on C319 have not yet been undertaken, and it is only conserved in mammalian Keap1 proteins. Therefore, insight into its potential role in electrophile signaling by means of Keap1 is limited. The other cysteine, C151, was found to be one of the three most reactive cysteines toward BIA and is 100% conserved between species.
Cysteine 151 seems to play a critical role in electrophile signaling. Under basal conditions, the Neh2 domain of Nrf2 is ubiquitinated by means of the Cullin3-based E3-ligase ubiquitination complex, with Keap1 acting as a bridge between Cul3 and Neh2 (19, 21, 29). Addition of sulforaphane to cells down-regulates ubiquitination of the Neh2 domain and thereby limits turnover of Nrf2. This mechanism has been proposed to induce Nrf2 accumulation by electrophiles (19, 21, 29). Interestingly, in a C151S mutant of human Keap1, this down-regulation in response to sulforaphane does not occur (21). A simple explanation for these results is that modification of C151 by an electrophile is required for signaling to occur by means of down-regulation of Nrf2 ubiquitination. This mechanism is supported by our identification of human Keap1 C151 as being highly reactive to electrophiles such as BIA.
Based on our observations that C151 is highly reactive toward an ARE inducer, and that the Keap1-Neh2 interaction is maintained in the presence of electrophiles, we propose a model for Nrf2 nuclear accumulation that encompasses a large amount of experimental data concerning the Keap1-Nrf2 system. This model is described in detail below and is illustrated in Fig. 6. Although modification of reactive cysteines other than C151 likely contributes to the signaling mechanism, our mechanistic model solely encompasses modification of human Keap1 C151 by BIA. Indeed, C151 is crucial for the signaling mechanism, based on the nonresponsiveness of the Keap1 C151S mutant to sulforaphane treatment (5).
Fig. 6.

A model depicting how modification of Keap1 C151 by ARE inducers could lead to Nrf2 nuclear accumulation. (A) Under basal conditions, Keap1 forms a bridge between Cul3 and Nrf2, leading to ubiquitination of the Neh2 domain of Nrf2. Upon introduction of electrophiles, modification of Keap1 C151 leads to a change in the conformation of the BTB domain by means of perturbing the homodimerization site, disrupting Neh2 ubiquitination, and causing ubiquitination of Keap1. Modification of Keap1 cysteines by electrophiles does not lead to disruption of the Keap1-Nrf2 complex. Rather, the switch of ubiquitination from Nrf2 to Keap1 leads to Nrf2 nuclear accumulation. (B) Structural representation of the putative Keap1 BTB domain homodimerization interface and BTB-Cul3 interaction interface. The PLZF BTB domain was aligned structurally with Skp1 (Fig. 11, which is published as supporting information on the PNAS web site), with which it shares significant structural similarity, to map the Cul3-BTB interaction site. The sequences of the PLZF and Keap1 BTB domains were aligned (Fig. 11) to obtain the locations of the Keap1 residues illustrated. The location of Keap1 C151 (PLZF D98) is mapped as a cysteine. The locations of the Keap1 residues 125-127 and 162-164, in the putative Cul3 binding region, are shown colored in yellow on the PLZF backbone.
Cysteine 151 is located in the Broad complex, Tramtrack, Bric-a-Brac (BTB) domain of Keap1 (Fig. 1), which is predicted to interact with Cul3 (29-31). However, mutations in the BTB domain of human Keap1 at the predicted interface between BTB and Cul3 did not disrupt the Keap1-Cul3 interaction, but led to down-regulation of Neh2 ubiquitination as well as up-regulation of Keap1 ubiquitination (21). Therefore, modification of C151 by an electrophile might also induce a switch from ubiquitination of the Neh2 domain of Nrf2 to ubiquitination of Keap1 (Fig. 6A) by altering the BTB-Cul3 interaction. Alteration of the Keap1-Cul3 interaction by modification of C151 is suggested by the results of Zhang et al. (21) who demonstrated that, whereas the Keap1-Cul3 interaction was reduced in cell lysates by the presence of sulforaphane, mutation of Keap1 C151 to serine partly restored the interaction. As a final contribution to our model, contacts outside the BTB domain and within the linker region seem to contribute significantly to Keap1-Cul3 binding (19), indicating a means by which disruption of the BTB-Cul3 interaction might lead to ubiquitination of Keap1 (Fig. 6A). This model would explain both our results and the recent results from Furukawa and Xiong (31), who demonstrated that silencing of Cul3 expression by means of a short hairpin RNA leads to Nrf2 accumulation primarily in the cytoplasm, as opposed to the nucleus.
We have constructed a structural model (Fig. 6B) of the complex of the BTB domain of human Keap1 with Cul3 based on the crystal structures of the BTB domain of promyelocytic leukemia zinc finger (PLZF) and the Skp1-Cul1 complex (32, 33). In this model, C151 lies directly next to an obligate homodimerization region of the BTB domain. This positioning suggests that C151 modification by electrophiles could alter the conformation of the Keap1 BTB domain by altering the homodimerization interface, and in so doing disrupt the BTB-Cul3 interaction.
The greatest difference between our results for the human Keap1 and Neh2 proteins reported here and those reported previously for the mouse proteins is in the effect of electrophiles on the Keap1-Neh2 complex. Electrophilic modification of human Keap1 by sulforaphane did not disrupt the Keap1-Neh2 complex, as determined by native EMSA. However, Dinkova-Kostova et al. (12) showed by the same method that addition of sulforaphane caused the Keap1-Neh2 complex band to disappear. It is possible that mouse Keap1 responds differently to electrophiles, which could be reflected in the much higher Kd of 580 nM for the mouse Keap-Neh2 complex determined by surface plasmon resonance (4).
However, it seems likely that the mouse Keap1-Neh2 complex behaves in the same manner as does the human system. The EMSA data of Dinkova-Kostova et al. (12) show that, upon addition of sulforaphane, a form of Keap1 appears that does not migrate appreciably into the gel, rather than showing that the mouse Keap1-Neh2 complex dissociates stoichiometrically into free Keap1 and Neh2. We observed the formation of a very similar band upon addition of sulforaphane to human Keap1 (Fig. 3A). This form of Keap1, which we observe at 35- to 80-fold excess sulforaphane in the absence of Neh2, is likely a highly aggregated form that is incapable of binding Neh2 tightly (Fig. 3A, lane 9). Importantly, at 80-fold excess sulforaphane, nonaggregated Keap1 still binds to Neh2, because no free nonaggregated Keap1 is visible in lane 9. Although the presence of Neh2 helps to protect Keap1 from the formation of an aggregate at 80-fold excess sulforaphane (lane 10), the 2,000-fold excess of sulforaphane added to a preformed Keap1-Neh2 complex in the experiments of Dinkova-Kostova et al. likely was high enough to induce the aggregated form of Keap1 and therefore cause a disappearance of the Keap1-Neh2 complex band. Therefore, at least for the human proteins, and likely the mouse proteins as well, nonaggregated Keap1 binds to Neh2 equally well whether unmodified or modified by electrophiles.
Although electrophilic modification of Keap1 does not alter its affinity for Neh2, other factors might result in a decreased affinity of Keap1 for Nrf2. For example, Neh2 phosphorylation at serine 40 was shown to decrease the affinity of Keap1 for Neh2 (34). However, the Keap1-Nrf2 interaction in the cell does not seem to be abolished in response to electrophiles, because a significant amount of Nrf2 is precipitated with Keap1 from total cell extracts after treatment with sulforaphane (5, 21). As an alternative to Keap1-Nrf2 complex disruption, Nrf2 nuclear accumulation might occur by means of a switch from Cul3-mediated ubiquitination of Nrf2 to Keap1 ubiquitination upon electrophilic modification of Keap1 cysteines, e.g., C151. Although further research is required to elucidate the mechanistic details of the electrophilic signal transduction mechanism of Nrf2 nuclear accumulation, our results support a model whereby electrophilic modification of Keap1 alone does not disrupt the Keap1-Nrf2 complex.
Supplementary Material
Acknowledgments
We thank Dr. Klavs Dolmer for help with the ITC experiments [National Institutes of Health (NIH) Grant 1510RR15958]. This work was supported by NIH Grants 5 P01 CA48112 and S10RR014686.
Author contributions: A.L.E., G.L., and A.D.M. designed research; A.L.E. and G.L. performed research; A.L.E. contributed new reagents/analytic tools; A.L.E., G.L., J.M.P., R.B.v.B., and A.D.M. analyzed data; and A.L.E., G.L., J.M.P., R.B.v.B., and A.D.M. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: ARE, antioxidant response element; CDNB, 1-Cl-2,4-dinitrobenzene; BIA, biotinylated iodoacetamide; TCEP, tris(2-carboxyethyl)phosphine hydrochloride; 15d-PGJ2, 15-deoxy-Δ12,14-prostaglandin J2; Nrf2, NF-E2-related factor 2; Keap1, Kelch-like ECH-associated protein 1; ITC, isothermal titration calorimetry; BTB, Broad complex, Tramtrack, Bric-a-Brac; PLZF, promyelocytic leukemia zinc finger.
References
- 1.Ishii, T., Itoh, K. & Yamamoto, M. (2002) Methods Enzymol. 348, 182. [DOI] [PubMed] [Google Scholar]
- 2.Kobayashi, M. & Yamamoto, M. (2005) Antioxid. Redox Signal. 7, 385. [DOI] [PubMed] [Google Scholar]
- 3.Talalay, P., Dinkova-Kostova, A. T. & Holtzclaw, W. D. (2003) Adv. Enzyme Regul. 43, 121. [DOI] [PubMed] [Google Scholar]
- 4.Itoh, K., Wakabayashi, N., Katoh, Y., Ishii, T., Igarashi, K., Engel, J. D. & Yamamoto, M. (1999) Genes Dev. 13, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang, D. D. & Hannink, M. (2003) Mol. Cell. Biol. 23, 8137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kobayashi, M., Itoh, K., Suzuki, T., Osanai, H., Nishikawa, K., Katoh, Y., Takagi, Y. & Yamamoto, M. (2002) Genes Cells 7, 807-820. [DOI] [PubMed] [Google Scholar]
- 7.Wakabayashi, N., Itoh, K., Wakabayashi, J., Motohashi, H., Noda, S., Takahashi, S., Imakado, S., Kotsuji, T., Otsuka, F., Roop, D. R., et al. (2003) Nat. Genet. 35, 238. [DOI] [PubMed] [Google Scholar]
- 8.Jang, D. S., Park, E. J., Hawthorne, M. E., Vigo, J. S., Graham, J. G., Cabieses, F., Santarsiero, B. D., Mesecar, A. D., Fong, H. H., Mehta, R. G., et al. (2003) J. Nat. Prod. 66, 583. [DOI] [PubMed] [Google Scholar]
- 9.Kinghorn, A. D., Su, B. N., Jang, D. S., Chang, L. C., Lee, D., Gu, J. Q., Carcache-Blanco, E. J., Pawlus, A. D., Lee, S. K., Park, E. J., et al. (2004) Planta Med. 70, 691. [DOI] [PubMed] [Google Scholar]
- 10.Ramos-Gomez, M., Kwak, M. K., Dolan, P. M., Itoh, K., Yamamoto, M., Talalay, P. & Kensler, T. W. (2001) Proc. Natl. Acad. Sci. USA 98, 3410-3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dinkova-Kostova, A. T., Massiah, M. A., Bozak, R. E., Hicks, R. J. & Talalay, P. (2001) Proc. Natl. Acad. Sci. USA 98, 3404-3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dinkova-Kostova, A. T., Holtzclaw, W. D., Cole, R. N., Itoh, K., Wakabayashi, N., Katoh, Y., Yamamoto, M. & Talalay, P. (2002) Proc. Natl. Acad. Sci. USA 99, 11908-11913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wakabayashi, N., Dinkova-Kostova, A. T., Holtzclaw, W. D., Kang, M. I., Kobayashi, A., Yamamoto, M., Kensler, T. W. & Talalay, P. (2004) Proc. Natl. Acad. Sci. USA 101, 2040-2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Itoh, K., Mochizuki, M., Ishii, Y., Ishii, T., Shibata, T., Kawamoto, Y., Kelly, V., Sekizawa, K., Uchida, K. & Yamamoto, M. (2004) Mol. Cell. Biol. 24, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levonen, A. L., Landar, A., Ramachandran, A., Ceaser, E. K., Dickinson, D. A., Zanoni, G., Morrow, J. D. & Darley-Usmar, V. M. (2004) Biochem. J. 378, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strachan, G. D., Morgan, K. L., Otis, L. L., Caltagarone, J., Gittis, A., Bowser, R. & Jordan-Sciutto, K. L. (2004) Biochemistry 43, 12113-12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zipper, L. M. & Mulcahy, R. T. (2002) J. Biol. Chem. 277, 36544. [DOI] [PubMed] [Google Scholar]
- 18.Papaiahgari, S., Kleeberger, S. R., Cho, H.-Y., Kalvakolanu, D. V. & Reddy, S. P. (2004) J. Biol. Chem. 279, 42302. [DOI] [PubMed] [Google Scholar]
- 19.Kobayashi, A., Kang, M. I., Okawa, H., Ohtsuji, M., Zenke, Y., Chiba, T., Igarashi, K. & Yamamoto, M. (2004) Mol. Cell. Biol. 24, 7130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karapetian, R. N., Evstafieva, A. G., Abaeva, I. S., Chichkova, N. V., Filonov, G. S., Rubtsov, Y. P., Sukhacheva, E. A., Melnikov, S. V., Schneider, U., Wanker, E. E. & Vartapetian, A. B. (2005) Mol. Cell. Biol. 25, 1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang, D. D., Lo, S.-C., Cross, J. V., Templeton, D. J. & Hannink, M. (2004) Mol. Cell. Biol. 24, 10941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chesnokova, L. S., Slepenkov, S. V., Protasevich, I. I., Sehorn, M. G., Brouillette, C. G. & Witt, S. N. (2003) Biochemistry 42, 9028-9040. [DOI] [PubMed] [Google Scholar]
- 23.Stuart, J. K., Myszka, D. G., Joss, L., Mitchell, R. S., McDonald, S. M., Xie, Z., Takayama, S., Reed, J. C. & Ely, K. R. (1998) J. Biol. Chem. 273, 22506. [DOI] [PubMed] [Google Scholar]
- 24.Habig, W. H., Pabst, M. J. & Jakoby, W. B. (1974) J. Biol. Chem. 249, 7130. [PubMed] [Google Scholar]
- 25.McMahon, M., Itoh, K., Yamamoto, M. & Hayes, J. D. (2003) J. Biol. Chem. 278, 21592. [DOI] [PubMed] [Google Scholar]
- 26.Talalay, P., De Long, M. J. & Prochaska, H. J. (1988) Proc. Natl. Acad. Sci. USA 85, 8261-8265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spencer, S. R., Xue, L. A., Klenz, E. M. & Talalay, P. (1991) Biochem. J. 273, 711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishii, T., Itoh, K., Takahashi, S., Sato, H., Yanagawa, T., Katoh, Y., Bannai, S. & Yamamoto, M. (2000) J. Biol. Chem. 275, 16023. [DOI] [PubMed] [Google Scholar]
- 29.Cullinan, S. B., Gordan, J. D., Jin, J., Harper, J. W. & Diehl, J. A. (2004) Mol. Cell. Biol. 24, 8477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pintard, L., Willems, A. & Peter, M. (2004) EMBO J. 23, 1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Furukawa, M. & Xiong, Y. (2005) Mol. Cell. Biol. 25, 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ahmad, K. F., Engel, C. K. & Prive, G. G. (1998) Proc. Natl. Acad. Sci. USA 95, 12123-12128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zheng, N., Schulman, B. A., Song, L., Miller, J. J., Jeffrey, P. D., Wang, P., Chu, C., Koepp, D. M., Elledge, S. J., Pagano, M., et al. (2002) Nature 416, 703-709. [DOI] [PubMed] [Google Scholar]
- 34.Bloom, D. A. & Jaiswal, A. K. (2003) J. Biol. Chem. 278, 44675. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}