

Abstract
In most legume nodules, the N2-fixing rhizobia are present as organelle-like structures inside their host cells. These structures, named symbiosomes, contain one or a few rhizobia surrounded by a plant membrane. Symbiosome formation requires the release of bacteria from cell-wall-bound infection threads. In primitive legumes, rhizobia are hosted in intracellular infection threads that, in contrast to symbiosomes, are bound by a cell wall. The formation of symbiosomes is presumed to represent a major step in the evolution of legume–nodule symbiosis, because symbiosomes facilitate the exchange of metabolites between the two symbionts. Here, we show that the genes, which are essential for initiating nodule formation, are also actively transcribed in mature Medicago truncatula nodules in the region where symbiosome formation occurs. At least one of these genes, encoding the receptor kinase DOES NOT MAKE INFECTIONS 2 (DMI2) is essential for symbiosome formation. The protein locates to the host cell plasma membrane and to the membrane surrounding the infection threads. A partial reduction of DMI2 expression causes a phenotype that resembles the infection structures found in primitive legume nodules, because infected cells are occupied by large intracellular infection threads instead of by organelle-like symbiosomes.
Keywords: infection, Medicago, Rhizobium, Nod factor
Medicago truncatula nodules have meristems at their apices. By division, these meristems continuously add new cells to the various tissues of the root nodule, and, as a consequence, the tissues are of graded age, with the youngest cells adjacent to the meristem. Cell-wall-bound infection threads in these cells grow toward and penetrate cells that are newly added to the central tissue by the meristem. Here, unwalled infection droplets extrude from the infection threads, after which the bacteria are endocytosed into the cytoplasm (1). The rhizobia thus become surrounded by a plant membrane and form organelle-like symbiosomes. Subsequent division of the symbiosomes ultimately results in infected cells that become fully packed with N2-fixing symbiosomes, requiring a major reorganization of the cytoskeletal and endomembrane system of the host cells, with symbiosome membrane biogenesis and demand in infected cells being ≈30 times greater than that required for plasma membrane synthesis (2).
The formation of symbiosomes is presumed to represent a major step in the evolution of legume nodule symbiosis, because symbiosome formation does not occur in nodules formed on legume species that form a symbiosis that is considered to be more primitive (e.g., Andira spp., many species belonging to the Fabaceae subfamily Caesalpinoideae) (3–5), and Parasponia spp., the only nonlegume species that can establish a symbiosis with rhizobia (6). In these species, Rhizobium bacteria are not released into the nodule host cells but remain in infection threads, called fixation threads, which are enclosed by a cell-wall-like structure and in which the rhizobia can fix atmospheric nitrogen. In the more advanced legumes, e.g., M. truncatula, the ability of the rhizobia to fix nitrogen requires the accommodation of the bacteria in the intracellular symbiosomal compartments, where the bacteria find the right conditions for fixing atmospheric nitrogen.
The symbiotic interaction is set in motion by a molecular dialogue between the two partners. The formation of a legume nodule starts with cortical cell divisions, by which a primordium is formed. In addition, the bacteria must enter the plant root. In M. truncatula, entrance into the plant root is established by the formation of infection threads in root hairs that have curled specifically around a few bacteria. These infection threads guide the bacteria to the primordia. There, the bacteria are released from the infection threads into the host cells, upon which the nodule primordium differentiates into a nodule (7). These early steps of the nodulation process, e.g., infection thread and nodule primordium formation, are induced by rhizobial signaling molecules, the so-called Nod factors. These Nod factors are, likely, recognized by a specific LysM domain containing receptor kinases [e.g., LYK3, LYK4 (8), and NFP (ref. 9; C. Gough and R.G., unpublished data)] in M. truncatula that are analogous to NFR1 and NFR5 in Lotus japonicus (10, 11). These LysM receptor kinases trigger a signal-transduction cascade that is essential to induce all early symbiotic events. Besides the LysM receptor kinases, several other components of this Nod-factor-induced signaling cascade have been identified, and, in M. truncatula, these components include the putative cation channel DMI1 (12) [analogous to CASTOR and POLLUX in L. japonicus (13)], the leucine-rich-repeat-containing receptor kinase DMI2 (14) [orthologous to SYMRK in L. japonicus (15)], and the calcium/calmodulin-dependent kinase DMI3 (16, 17).
Loss-of-function mutations in these Nod factor signaling genes cause a block at a very early step of nodule formation. Therefore, such null mutants cannot reveal whether the genes also have a function at later stages of the symbiosis. Here, we show that the Nod factor signaling genes are expressed in the apex of the nodule, in a few cell layers directly adjacent to the meristem, which coincides with the place where rhizobia are released from the infection threads into the host cells. Of these genes, DMI2 showed the highest expression level, and the protein is located in the plasma membrane and infection thread membrane in these cells. By creating mutants in which DMI2 expression is misregulated by both (inducible) RNA interference (RNAi) and expression of DMI2 in the mutant background, by using a promoter with a low activity in the nodule apex, we show that the receptor kinase DMI2, besides being essential for nodule initiation, is a key regulator of symbiosome formation. In this process, DMI2 is shown to be required in the distal part of the infection zone to restrict infection thread growth and to switch to endocytosis of the bacteria.
Materials and Methods
In Situ Hybridizations. In situ hybridizations were conducted on 14-d-old nodules according to procedures described by Van der Wiel et al. in ref. 18. The nodules were fixed in 4% paraformaldehyde, supplemented with 0.25% glutaraldehyde in 10 mM sodium phosphate buffer (pH 7.4) for 3 h. Fixed nodules were dehydrated by passing through a series of graded ethanol baths and embedded into paraffin. Sections (7 μm) were dried overnight on polylysine-coated slides at 37°C, deparaffinized with xylene, and rehydrated by a graded ethanol series. Antisense RNA probes were generated from ≈200-bp subclones of the coding regions of the target genes (DMI1: base pairs 1–203, 205–384, 477–691, 848-1049, 1100–1260, and 1572–1760; DMI2: base pairs 373–617, 784-1039, 1200–1453, 1610–1864, and 1964–2219; DMI3: base pairs 12–229, 221–467, 472–712, and 1198–1388; and LYK3: base pairs 113–342, 483–694, 866-1079, 1258–1486, and 1705–1947). For hybridization, a mixture containing 2 × 106 cpm of each probe was used. After washing, the slides were coated with microautoradiography emulsion LM-1 (Amersham Pharmacia) and exposed for 3 weeks at 4°C. The slides were developed for 5 min in Kodak D-19 developer and fixed in Kodak fixative. Sections were counterstained with toluidine blue and mounted. For imaging, a Nikon Optiphot-2 bright-field microscope was used.
Quantitative RT-PCR (qPCR). qPCR was conducted on RNA isolated from nitrogen-starved, uninoculated roots and nodule apices at 10 d postinoculation with Sinorhizobium meliloti strain 2011 (Sm2011). cDNA was synthesized from 1 μg of total RNA by using the TaqMan gold RT-PCR kit (PerkinElmer Applied Biosystems) in a total volume of 50 μl with the supplied hexamer primers. qPCR reactions were performed in triplicate on 6.5 μl of cDNA by using the SYBR green RT-PCR Master kit (PerkinElmer Applied Biosystems; 40 cycles at 95°C for 10 s and at 60°C for 1 min), and real-time detection was performed on the ABI PRISM 7700 sequence detector (Applied Biosystems) and analyzed by using the program geneamp 5700 sds (PerkinElmer Applied Biosystems). The specificity of the PCR amplification procedures was checked with a heat-dissociation step (from 60°C to 95°C) at the end of the run and by agarose gel electrophoresis. Results were standardized to the MtACTIN2 expression levels. The primers used were MtACTIN2, 5′-TGGCATCACTCAGTACCTTTCAACAG-3′ and 5′-ACCCAAAGCATCAAATAATAAGTCAACC-3′; DMI1, 5′-GTTGCTGCAGATGGAGGGAAGAT-3′ and 5′-GCGCCAGCCACAAAACAGTAT-3′; DMI2, 5′-TGGACCCCTTTTGAATGCCTATG-3′ and 5′-TCCACTCCAACTCTCCAATGCTTC-3′; DMI3, 5′-TCATTGATCCCTTTTGCTTCTCGT-3′ and 5′-GATGCTACTTCCTCTTTGCTGATGC-3′; and LYK3, 5′-TGCTAAGGGTTCAGCTGTTGGTA-3′ and 5′-AAATGCCCTAGAAGTTTGTGGAAG-3′.
Plasmids and Vectors. The full-length DMI2 coding sequence was PCR-amplified from M. truncatula root cDNA by using primers containing NheI and SacI restriction sites 5′-CTAGCTAGCATGATGGAGTTACAAGTTATTAAG-3′ and 5′-TCCGAGCTCTATAGCTCTGTTGAAGTGTC-3′ (restriction sites underlined). The 3,103-bp fragment was subsequently cloned into pGEM-t (Promega). The full-length DMI2 cDNA was cloned by using NheI–SacI into a modified pBluescriptII SK+ vector, containing the CaMV 35S promoter from pMON999 (Monsanto) and a NOS-terminator sequence with an introduced PacI restriction site, and, subsequently, cloned by using HindIII–PacI into the binary vector pRedRoot (19), resulting in 35S::DMI2. To isolate the DMI2 promoter, a 2,200-bp region upstream of the ATG start codon was PCR-amplified on Medicago A17 genomic DNA by using primers containing HindIII and BamHI sites: 5′-AAGCTTCAAATTTGGACCGAACTG-3′ and 5′-GGATCCAACTTGAATCCATGCTAACTAACT-3′. This fragment was used to replace HindIII–BamHI, the 35S promoter, resulting in DMI2p::DMI2. A C-terminal DMI2-GFP fusion was constructed by using the Gateway vector pK7FWG2 (20), modified to contain the red fluorescent marker DsRED1 under the control of the constitutive Arabidopsis Ubiquitin10 promoter (AatI–XbaI). DMI2, in its genomic context, including promoter region and introns, was PCR amplified, cloned into pENTR-D-TOPO (Invitrogen) by using primers 5′-CACCTCTCCTTTTATCTTTTGCTTGTGG-3′ and 5′-TCTCGGTTGAGGGTGTGACAAGG-3′, and recombined into pK7FWG2-Q10::DsRED1 by using LR-Clonase (Invitrogen).
Plant Material and Rhizobial Strain. Medicago accession Jemalong A17 and dmi2 mutant TR25 containing MtENOD11::GUS (21) were used for transformations. A S. meliloti strain Sm2011 expressing GFP (8) was used to inoculate plants.
Agrobacterium rhizogenes-Mediated Transformation and RNAi. A. rhizogenes-mediated RNAi and root transformation of 5-d-old M. truncatula seedlings was performed according to procedures described by Limpens et al. in ref. 19. Homogenously cotransformed roots were selected, based on the expression of the red fluorescent marker DsRED1. A 555-bp fragment of the DMI2 mRNA sequence (base pairs 1351–1905) was PCR-amplified from nodule cDNA by using primers containing SpeI–AscI and BamHI–SwaI restriction sites (underlined), 5′-ATACTAGTGGCGCGCCACCGTCCTCCTTGCTGATA-3′ and 5′-ATGGATCCATTTAAATCGGGTTCCCTGAGTTGATG-3′, and, subsequently, cloned as an inverted repeat into pRedRoot (as described by Limpens et al. in ref. 19). A Rhizobium-inducible RNAi construct was made by using the Gateway vector pK7GWIWG2(II) (20), modified to contain the red fluorescent marker DsRED1 under the control of the constitutive Arabidopsis Ubiquitin10 promoter (AatI–XbaI). This vector was further modified by replacing SacI–SpeI, the 35S promoter, with the MtENOD12 promoter (22). The MtENOD12 promoter (832 bp) was PCR-amplified from the M. truncatula genomic DNA by using primers containing SacI and SpeI restriction sites (underlined), 5′-GAGCTCGGGTAAAATAGAAAAGAAAAAAGTCAAC-3′ and 5′-ACTAGTTTAAGTAGTAATTTTAATGTTAGTGC-3′. The 555-bp DMI2 fragment was cloned into pENTR-D-TOPO (Invitrogen) and recombined into the modified Gateway pK7GWIWG2(II)-Q10::DsRED binary vector.
Histochemical Analysis and Microscopy. Histochemical β-glucuronidase staining was performed according to the procedure described by Jefferson et al. in ref. 23, with few modifications. Plant material was incubated in 0.05% (wt/vol) X-Gluc (Duchefa, Haarlem, The Netherlands) in 0.1 M sodium phosphate buffer (pH 7) with 3% sucrose, 5 μM potassium ferrocyanide, and 5 μM potassium ferricyanide. The roots were infiltrated for 30 min, under vacuum, and further incubated at 37°C.
For electron microscopy, nodules were fixed for 3.5 h in a mixture of 4% paraformaldehyde and 3% glutaraldehyde in 50 mM potassium phosphate buffer (pH 7.4). The nodules were postfixed for 3 h with1% osmium tetroxide, dehydrated through an ethanol series, and embedded in London resin white. Ultrathin sections (60 nm) were obtained with a Reichert Ultracuts ultratome (Leica), stained with 2% uranyl acetate and Reynolds' lead citrate solution (24), and observed by using an EM208 electron microscope (Philips, Eindhoven, The Netherlands). For light microscopy, semithin sections (0.6–1 μm) were stained with a 1:1 mixture of 1% toluidine blue and 1% methylene blue solution and embedded in Paraplast (Electron Microscopy Sciences, Hatfield, PA). Sections were viewed with a Nikon Optiphot-2 microscope. Images where processed electronically by using the program photoshop 6.0 (Adobe Systems, San Jose, CA). Imaging of DsRED1 or GFP fluorescence was done by using the Leica MZIII fluorescence stereomicroscope and a Nikon Optiphot-2 coupled to a mercury lamp.
Confocal imaging of DMI2-GFP was done by using a Zeiss LSM 510 confocal laser scanning microscope [excitation 488 (GFP), 534 nm (propidium iodide)]. GFP emission was detected by using a 505- to 530-nm band-pass filter; propidium iodide emission was detected in another channel with a 560- to 615-nm band pass. Nodules were hand-sectioned, incubated for 10 min in 0.2 μg/ml propidium iodide, and washed with water.
Results
Expression of the Nod Factor Signaling Genes in the Nodule. To determine the expression pattern of the Nod factor signaling genes in the nodule, in situ hybridizations were performed by using LYK3, DMI1, DMI2, and DMI3 as probes. These in situ hybridizations show that LYK3, DMI1, DMI2, and DMI3 are expressed in the apices of nitrogen-fixing Medicago nodules (Fig. 1), consistent with the possibility that the Nod factor signaling genes, besides being essential for initiating nodule formation, also have a function at later stages of nodule development. In all cases, the highest level of expression occurs in only a few cell layers directly adjacent to the meristem, except for DMI1, which is expressed in a slightly broader region. DMI2, especially, was very abundant in those cell layers directly adjacent to the meristem, which form the most distal part of the so-called infection zone and correspond to the region where bacteria are released from infection threads into the host cells and symbiosomes subsequently divide (1). Real-time qPCR analysis on RNA isolated from the nodule apex confirmed the relatively strong expression of DMI2 in this region (Fig. 2). Real-time qPCR analysis further showed that all three DMI genes are expressed at markedly higher levels in the nodule apex, as compared with roots, whereas expression of LYK3 is reduced.
Fig. 1.

In situ localization of DMI2, DMI1, DMI3, and LYK3 mRNA in longitudinal sections of 14-d-old Medicago nodules. (A, C, E, and G) Bright-field images of nodule sections hybridized with 35S-UTP labeled antisense DMI2 (A), DMI1 (C), DMI3 (E), and LYK3 (G) probes. The signal appears as silver grains (black). (B, D, F, and H) Epipolarization images of A, C, E, and G, respectively. M, meristem; ZII, infection zone; ZIII, fixation zone. (Scale bars: 200 μm.)
Fig. 2.

Quantification of mRNA levels of the Nod factor signaling genes LYK3, DMI1, DMI2, and DMI3 in roots (white) and the nodule apex (gray). Relative transcript levels were determined by real-time qPCR and normalized by using MtACTIN2 as reference.
DMI2 Is Essential for Symbiosome Formation. Because DMI2 transcripts especially are highly abundant in the distal part of the infection zone of the nodule, we reasoned that a partial reduction of DMI2 expression could specifically affect its function in the nodule, while still allowing the first steps of Nod factor signaling that lead to the formation of a nodule. Therefore, we made mutants with reduced DMI2 expression by using RNAi, and we expressed DMI2 in the mutant background, under the control of a promoter with a lower activity in the nodule apex than the DMI2 promoter.
RNAi generally results in a range of knockdown levels of a targeted gene and can, thus, be used as a tool to identify weak phenotypes. We exploited this characteristic to knock down DMI2 expression by using Agrobacterium rhizogenes-mediated root transformations and looked for phenotypes in the nodule. A. rhizogenes-mediated RNAi, with the (constitutive) CaMV 35S promoter, caused 40–90% knockdown of DMI2 RNA levels (Fig. 3), and, on only ≈15% of the transformed RNAi roots (9 of 57), some nodules formed upon inoculation with Sm2011. Histological analysis of 12 of these nodules revealed, in 6 of them, the presence of numerous wide infection threads in the central tissue of the nodule but no symbiosomes (Fig. 4A). The other 6 nodules showed infected cells filled with symbiosomes, similar to control nodules (Fig. 4 C and F). This finding suggests that partial knockdown of DMI2 expression causes extensive infection thread growth in the nodule and blocks the release of rhizobia from these infection threads.
Fig. 3.

Quantification of mRNA levels in five independent DMI2 knockdown (RNAi) roots determined by real-time qPCR using MtACTIN2 as reference. The average of three independent control roots transformed with an empty binary vector is shown.
Fig. 4.

Histology of nodules with reduced DMI2 expression levels and wild-type nodules. (A) Longitudinal sections (5 μm) of 2-week-old nodules formed on (partial) DMI2 knockdown (RNAi) roots. Most cells in the central zone are filled with infection threads. (B) Longitudinal section (1 μm) of a 35S::DMI2-transformed TR25 nodule, showing wide infection threads occupying most cells of the central tissue. (C) Longitudinal section (1 μm) of a control nodule (transformed with an empty binary vector). A typical zonation of indeterminate nodules can be seen. M, meristem; zII, infection zone; zIII, fixation zone. The arrow indicates extension of the fixation zone in the direction of the nodule base. (D) Transmission electron microscopy of a 35S::DMI2-transformed TR25 nodule section, showing an infection thread inside a cell without releasing bacteria. The infection thread is surrounded by a fibrillar wall (arrowhead) and a membrane. (E) Closeup of boxed area in B, showing infection threads in the distal part of the 35S::DMI2-transformed TR25 nodule occupying major parts of the cells but with no release of bacteria. (F) Closeup of boxed area in C, showing part of the infection zone of a control nodule. Bacteria are released from infection threads, which are stained dark blue. it, infection thread. (Scale bars: A–C, 100 μm; D, 1 μm; and E and F, 20 μm.)
Because the vast majority of the DMI2 knockdown lines did not develop nodules, we tried to circumvent this early block of nodulation by using a Rhizobium-inducible RNAi system specifically to knock down DMI2 expression in the nodule. To this end, we used the Medicago ENOD12 promoter to drive the DMI2 RNAi hairpin construct (ENOD12::DMI2i). The ENOD12 gene is specifically and strongly up-regulated upon rhizobial infection and highly expressed in the distal part of the infection zone (18). All transgenic ENOD12::DMI2i roots formed nodules (7 nodules per root, n = 14) upon inoculation with Sm2011. Sectioning these nodules revealed, in ≈30% of the transgenic nodules (7 of 23), an infection phenotype similar to that observed in the CaMV 35S partial knockdown lines, namely, extensive infection thread growth in the central tissue of the nodule but no symbiosome formation.
In addition to partial knockdown of DMI2 expression by RNAi, we expressed DMI2 in the mutant background under the control of a constitutive promoter with low activity in the nodule apex. Based on β-glucuronidase-expression data, we knew that a CaMV 35S-derived promoter has a markedly lower expression level than does the DMI2 promoter in the apex of the nodule (data not shown). Introduction of 35S::DMI2 into the dmi2 knockout mutant TR25, by using A. rhizogenes transformation, partially restored nodulation ability, and, on average, two nodules formed on a 35S::DMI2-transformed TR25 root (n = 43). Sectioning these nodules (n = 30) revealed, in all cases, a phenotype similar to that in DMI2 knockdown nodules, extensive infection thread growth in the central tissue of the nodule, and no symbiosomes (Fig. 4 B and E). Because the used 35S promoter is expressed in almost the entire nodule, we tested whether this ectopic expression has an effect on bacterial release. Wild-type roots expressing 35S::DMI2 were efficiently nodulated (8 nodules per root, n = 8) and the cytology of these nodules was indistinguishable from that of wild-type nodules, showing that ectopic expression of DMI2 does not affect infection thread growth or symbiosome formation. Furthermore, the introduction of DMI2, under the control of its endogenous promoter, fully complemented the TR25 mutant (8 nodules per root, n = 12), with nodules showing a wild-type histology (data not shown). These data indicate that the phenotype observed in the 35S::DMI2 TR25 nodules is due to misregulation of DMI2 expression in the nodule apex.
In a section of wild-type nodules, infection threads are observed in only 10–20% of the infected cells (Fig. 4 C and F). In contrast, the vast majority of the cells of the central tissue in 1-μm-thick sections of the 35S::DMI2 TR25 nodules contain numerous large infection threads, indicating that these infection threads fill a major part of these cells (Fig. 4 B and E). Transmission electron microscopy showed that the mutant infection threads contained numerous bacteria embedded in a matrix with a low electron density and are bound by a cell wall and a membrane (Fig. 4D). In comparison with wild-type infection threads, these threads showed an increase in diameter and extensive branching. Furthermore, these analyses confirmed the absence of symbiosomes. Therefore, DMI2 in the distal part of the infection zone of the nodule is required to restrict infection thread growth and to switch to endocytosis, by means of which, bacteria are released and N2-fixing symbiosomes are formed.
Both the DMI2 knockdown and the 35S::DMI2 TR25 mutant nodules are white and so, most likely, lack leghemoglobin, a protein required to facilitate oxygen transport to the symbiosomes, essential for nitrogen fixation. In wild-type M. truncatula nodules, the rhizobial nif (nitrogen fixation) genes are first induced in the fixation zone, when symbiosomes have fully filled the infected cells (R. Mirabella, personal communication). The induction of the rhizobial nif genes at such a relatively late stage in the development of wild-type nodules seems consistent with a lack of nitrogen fixation in the DMI2 mutant nodules. Furthermore, the 35S::DMI2 TR25 mutants were unable to grow under nitrogen-limiting conditions. These data indicate that the mutant nodules were unable to fix nitrogen.
DMI2 Is Located in the Plasma Membrane and Infection Thread Membrane. Because DMI2 is required to restrict infection thread growth and to switch to the release of rhizobia in the distal part of the infection zone of the nodule, we investigated whether DMI2 would be located on the infection thread membrane in this region. Therefore, a C-terminal GFP fusion was constructed by using DMI2 in its genomic context; including introns and the 1.7-kb promoter region. The corresponding binary construct was introduced into the dmi2–1 mutant TR25 (21) via A. rhizogenes-mediated root transformation, and transgenic roots were selected by using the red-fluorescent-selectable marker DsRED1 (9). Upon inoculation with Sm2011, N2-fixing nodules were formed, showing that this construct was able to fully complement the dmi2 mutant TR25. To determine the localization of the DMI2 protein, transgenic nodules were sectioned and analyzed by confocal laser scanning microscopy. Strong fluorescent labeling could be observed in the plasma membrane as well as infection thread membranes in cells in the distal part of the infection zone (Fig. 5). In contrast, no GFP fluorescence was observed in symbiosome membranes surrounding bacteria released into the host cytosol.
Fig. 5.
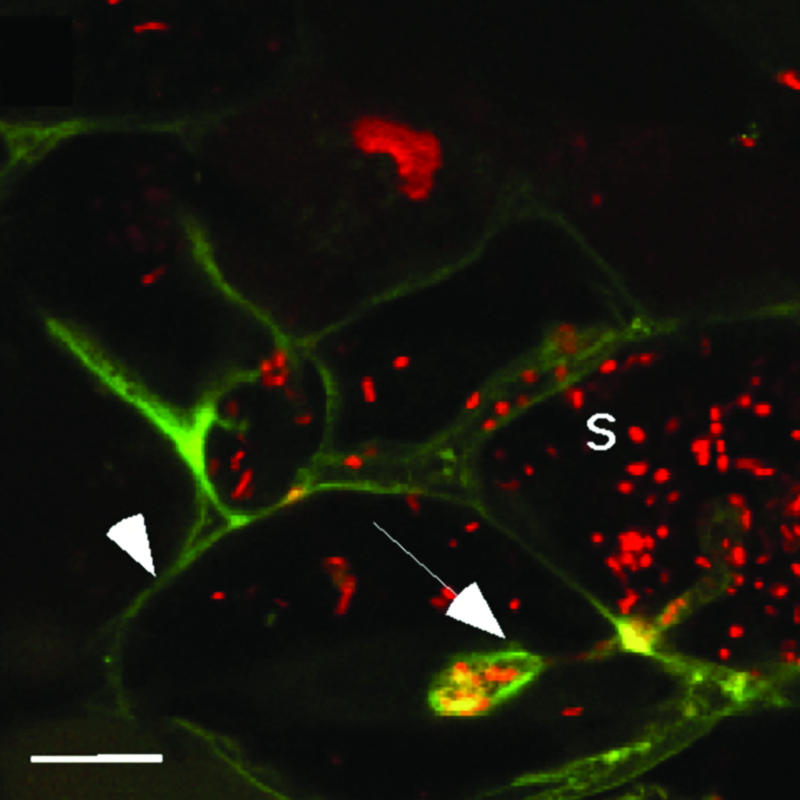
Subcellular localization of DMI2 in the distal part of the infection zone. DMI2-GFP localizes to the plasma membrane (arrowhead) and infection thread membrane (arrow). A confocal cross-section through an infection thread is shown. No GFP labeling is observed around symbiosomes (S). Rhizobia are stained with propidium iodide. (Scale bar: 20 μm.)
Discussion
The receptor kinase DMI2 is an essential component of the Nod factor signaling cascade, inducing the early steps of nodulation, such as infection thread and nodule primordium formation (14, 21). Here, we show that DMI2 is highly expressed in the apex of the nodule directly adjacent to the meristem, where the receptor kinase is present in the host cell plasma membrane as well as in the membrane surrounding infection threads. There, DMI2 is a key regulator of organelle-like symbiosome formation.
In situ hybridizations revealed high DMI2 expression levels in two to three cell layers of the infection zone adjacent to the meristem. Other components of the Nod factor signaling pathway, e.g., DMI1, DMI3, and LYK3, were also expressed in these cell layers, which correspond to the nodule cells where release of bacteria from the infection thread takes place and symbiosomes subsequently divide. It has been shown that the rhizobial nod genes that are involved in Nod factor synthesis are still active in these cell layers (25, 26), making it likely that Nod factor signaling occurs in these cells. However, it cannot be excluded that DMI2 controls symbiosome formation in a Nod-factor-independent manner (27).
Because loss-of-function mutations in the Nod factor signaling genes cause a block at a very early step of nodule formation, we decided to reduce the expression of DMI2 to affect DMI2 functions in the nodule, while still allowing a nodule to be formed. Knockdown of DMI2 expression via (inducible) RNAi, as well as expression of DMI2 in the mutant background by means of a 35S-derived promoter (with relatively low activity in the nodule apex when compared with the DMI2 promoter), resulted in extensive growth of infection threads and blocked the subsequent release of bacteria from these infection threads. In wild-type nodules, a switch from infection thread growth to release of bacteria in the distal part of the infection zone is required to facilitate symbiosome formation. Because this switch is not occurring in the mutant nodules, it could be that the sustained infection thread growth and suppressed release of bacteria are interlinked processes. These data suggest that a threshold level of DMI2 expression in the distal part of the infection zone needs to be reached to induce the switch from infection thread growth to release of bacteria. Because the expression level of DMI2 is only partially reduced, it is possible that, in addition to symbiosome formation, several other processes in the nodule are controlled by DMI2, because these processes might not be affected by this partial reduction. The subcellular localization of DMI2 to the plasma membrane, as well as to the membrane surrounding the infection threads, suggests a direct function for DMI2 in the internalization of the rhizobia.
A similar block of symbiosome formation was observed in strong DMI2 knockdown lines in Sesbania rostrata, as described in the accompanying paper by Capoen et al. (28). In Sesbania, the epidermal responses required for nodulation can be circumvented through intercellular colonization of the cortex at lateral root bases, resulting in the formation of infection pockets from which infection threads penetrate the cortex and nodules are formed. It was shown that even 90% DMI2 knockdown still allowed the formation of such infection pockets at lateral root bases and subsequent nodule organ formation but blocked the release of the bacteria from infection threads in the nodule (28). In M. truncatula, the epidermal steps required for nodulation cannot be circumvented, and similar knockdown levels cause an epidermal block of nodulation (8).
The infection phenotype in mutant nodules with reduced DMI2 expression shows some striking similarities to that of the recently identified M. truncatula mutant numerous infections polyphenolics (nip) (29). This mutant, similarly, contains numerous bulbous infection threads in the central tissue of the nodule, without the release of bacteria. However, in contrast to dmi2 mutants, nip shows additional defects in lateral root development, indicating that NIP has a more general function during plant development. dmi2 mutants do not show any defects in lateral root development, suggesting that DMI2 does not play a role in lateral root development. Given the fact that DMI2 encodes a receptor kinase and already functions early in the Nod factor signal-transduction cascade, either DMI2 regulates NIP during nodule formation to facilitate symbiosome formation or DMI2 and NIP control symbiosome formation via independent pathways.
Although the rhizobia present in the dmi2 mutant nodules do not fix nitrogen, the infection phenotype in these nodules is reminiscent of that of nodules formed on primitive legumes and Parasponia (1, 3–6). In those nodules, bacteria are not released, but, instead, N2-fixing rhizobia are hosted within intracellular infection threads. The reason for this evolutionary step from, in principle, “extracellular” accommodation of the rhizobia to the more intimate intracellular accommodation in organelle-like symbiosomes could be to improve the exchange of metabolites between the two partners, because of the absence of a cell wall and a maximized interface. It is intriguing that this evolutionary step of forming organelle-like symbiosomes has come under the control of at least one of the genes essential for Nod factor signaling in the epidermis.
Acknowledgments
We thank D. G. Barker for providing the TR25-ENOD11::GUS line and S. de Nooijer, who participated in this work as an undergraduate student. This work was supported by Horizon Breakthrough Grant 050-71-010 from the Dutch Genomics Initiative.
Author contributions: H.F., T.B., and R.G. designed research; E.L., R.M., E.F., C.F., and R.G. performed research; R.M., E.F., and C.F. contributed new reagents/analytic tools; E.L., R.M., E.F., H.F., and R.G. analyzed data; and E.L., T.B., and R.G. wrote the paper.
Abbreviation: qPCR, qualitative PCR.
References
- 1.Brewin, N. J. (2004) Crit. Rev. Plant Sci. 23, 293-316. [Google Scholar]
- 2.Verma, D. P. S. & Hong, Z. (1996) Trends Microbiol. 4, 364-368. [DOI] [PubMed] [Google Scholar]
- 3.De Faria, S. M., Sutherland, J. M. & Sprent, J. I. (1986) Plant Sci. 45, 143-147. [Google Scholar]
- 4.De Faria, S. M., McInroy, S. G. & Sprent, J. I. (1987) Can. J. Bot. 65, 553-558. [Google Scholar]
- 5.Sprent, J. I. (2001) Nodulation in Legumes (R. Bot. Gard., Kew, U.K.), 146 p.
- 6.Trinick, M. J. (1979) Can. J. Microbiol. 25, 565-578. [DOI] [PubMed] [Google Scholar]
- 7.Oldroyd, G. D. & Downie, J. A. (2004) Nat. Rev. Mol. Cell Biol. 5, 566-576. [DOI] [PubMed] [Google Scholar]
- 8.Limpens, E., Franken, C., Smit, P., Willemse, J., Bisseling, T. & Geurts, R. (2003) Science 302, 630-633. [DOI] [PubMed] [Google Scholar]
- 9.Amor, B. B., Shaw, S. L., Oldroyd, G. E. D., Maillet, F., Penmetsa, R. V., Cook, D., Long, S. R., Dénarié, J. & Gough, C. (2003) Plant J. 34, 495-506. [DOI] [PubMed] [Google Scholar]
- 10.Madsen, E. B., Madsen, L. H., Radutoiu, S., Olbryt, M., Rakwalska, M., Szczyglowski, K., Sato, S., Kaneko, T., Tabata, S., Sandal, N. & Stougaard, J. (2003) Nature 425, 637-640. [DOI] [PubMed] [Google Scholar]
- 11.Radutoiu, S., Madsen, L. H., Madsen, E. B., Felle, H. H., Umehara, Y., Gronlund, M., Sato, S., Nakamura, Y., Tabata, S., Sandal, N. & Stougaard, J. (2003) Nature 425, 585-592. [DOI] [PubMed] [Google Scholar]
- 12.Ané, J. M., Kiss, G. B., Riely, B. K., Penmetsa, R. V., Oldroyd, G. E., Ayax, C., Levy, J., Debellé, F., Baek, J. M., Kaló, P., et al. (2004) Science 303, 1364-1367. [DOI] [PubMed] [Google Scholar]
- 13.Imaizumi-Anraku, H., Takeda, N., Charpentier, M., Perry, J., Miwa, H., Umehara, Y., Kouchi, H., Murakami, Y., Mulder, L., Vickers, K., et al. (2005) Nature 433, 527-531. [DOI] [PubMed] [Google Scholar]
- 14.Endre, G., Kereszt, A., Kevei, Z., Mihacea, S., Kaló, P. & Kiss, G. B. (2002) Nature 417, 962-966. [DOI] [PubMed] [Google Scholar]
- 15.Stracke, S., Kistner, C., Yoshida, S., Mulder, L., Sato, S., Kaneko, T., Tabata, S., Sandal, N., Stougaard, J., Szczyglowski, K. & Parniske, M. (2002) Nature 417, 959-962. [DOI] [PubMed] [Google Scholar]
- 16.Levy, J., Bres, C., Geurts, R., Chalhoub, B., Kulikova, O., Duc, G., Journet, E. P., Ané, J. M., Lauber, E., Bisseling, T., et al. (2004) Science 303, 1361-1364. [DOI] [PubMed] [Google Scholar]
- 17.Mitra, R. M., Gleason, C. A., Edwards, A., Hadfield, J., Downie, J. A., Oldroyd, G. E. & Long, S. R. (2004) Proc. Natl. Acad. Sci. USA 101, 4701-4705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van De Wiel, C., Norris, J. H., Bochenek, B., Dickstein, R. & Bisseling, T. (1990) Plant Cell 2, 1009-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Limpens, E., Ramos, J., Franken, C., Raz, V., Compaan, B., Franssen, H., Bisseling, T. & Geurts, R. (2004) J. Exp. Bot. 55, 983-992. [DOI] [PubMed] [Google Scholar]
- 20.Karimi, M., Inze, D. & Depicker, A. (2002) Trends Plant Sci. 7, 193-195. [DOI] [PubMed] [Google Scholar]
- 21.Catoira, R., Galera, C., De Billy, F., Penmetsa, R. V., Journet, E. P., Maillet, F., Rosenberg, C., Cook, D., Gough, C. & Dénarié, J. (2000) Plant Cell 12, 1647-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pichon, M., Journet, E. P., Dedieu, A., de Billy, F., Truchet, G. & Barker, D. G. (1992) Plant Cell 4, 1199-1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jefferson, A. R., Kavanagh, T. A. & Bevan, M. W. (1987) EMBO J. 6, 3901-3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reynolds, E. S. (1963) J. Cell Biol. 17, 208-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharma, S. B. & Signer, E. R. (1990) Genes Dev. 4, 344-356. [DOI] [PubMed] [Google Scholar]
- 26.Schlaman, H. R., Horvath, B., Vijgenboom, E., Okker, R. J. & Lugtenberg, B. J. (1991) J. Bacteriol. 173, 4277-4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.D'Haeze, W., Gao, M., De Rycke, R., Van Montagu, M., Engler, G. & Holsters, M. (1998) Mol. Plant–Microbe Interact. 11, 999-1008. [Google Scholar]
- 28.Capoen, W., Goormachtig, S., De Rycke, R., Schroeyers, K. & Holsters, M. (2005) Proc. Natl. Acad. Sci. USA 102, 10369-10374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Veereshlingam, H., Haynes, J. G., Penmetsa, R. V., Cook, D. R., Sherrier, D. J. & Dickstein, R. (2004) Plant Physiol. 136, 3692-3702. [DOI] [PMC free article] [PubMed] [Google Scholar]