

Abstract
Background
These studies investigate the role of mitoKATP channels, protein kinase C (PKC) and Mitogen activated protein kinase (p38MAPK) on the cardioprotection of ischemic (IP) and pharmacological preconditioning (PP) of the human myocardium and their sequence of activation.
Results
Right atrial appendages from patients undergoing elective cardiac surgery were equilibrated for 30 min and then subjected to 90 min of simulated ischemia followed by 120 min reoxygenation. At the end of each protocol creatinine kinase leakage (CK U/g wet wt) and the reduction of MTT to formazan dye (mM/g wet wt) were measured. Similar protection was obtained with α1 agonist phenylephrine, adenosine and IP and their combination did not afford additional cardioprotection. Blockade of mitoKATP channels with 5-hydroxydecanoate, PKC with chelerythrine, or p38MAPK with SB203580 abolished the protection of IP and of PP. In additional studies, the stimulation of mitoKATP channels with diazoxide or activation of PKC with PMA or p38MAPK with anisomycin induced identical protection to that of IP and PP. The protection induced by diazoxide was abolished by blockade of PKC and by blockade of p38MAPK. Furthermore, the protection induced by PMA was abolished by SB203580 but not by 5-hydroxydecanoate, whereas the protection induced by anisomycin was unaffected by either 5-hydroxydecanoate or chelerythrine.
Conclusions
Opening of mitoKATP channels and activation of PKC and p38MAPK are obligatory steps in the signal transduction cascade of IP and PP of the human myocardium with PKC activation being downstream of the opening of mitoKATP channels and upstream of p38MAPK activation.
Background
Ischemic preconditioning (IP) is a powerful protective endogenous adaptive response of the heart against a prolonged ischemic insult. [1,2] However, the application of IP requires a physical cut of the blood supply which can be difficult or impractical in many clinical situations. A way to circumvent these potential problems associated with the clinical application of IP may be preconditioning by pharmacological (PP) means or manipulation of the signalling pathway involved in the protection. A number of membrane receptors are involved in the phenomenon of IP including α1[3-5] and β-adrenoceptors, [6] opioid [7] and adenosine A1 and A3 receptors. [8,9] Other factors such as heat shock proteins, [10] bradykinin, [11] calcium [12] and nitric oxide synthase activity [13] have also been shown to participate in the protection of IP, however, whether the various forms of PP share the same molecular mechanism with IP is not fully elucidated.
The intracellular sequence of events that translate the binding of the various agonists to their membrane receptors into the protection of preconditioning remains under intense investigation. It has been reported that α1-adrenoceptors are coupled with protein kinase C (PKC) through phospholipase activity, [14] that in turn activate p38 Mitogen Activated Protein Kinase (p38MAPK) in some cardiac preparations. [15] ATP sensitive potassium channels have also been implicated in the signal transduction mechanism of IP, [16,17] and recent evidence from several investigators [18-20] including ourselves [21] has shown that the mitochondrial and not the sarcolemmal KATP channels are involved. The order of involvement of the above mediators remains controversial although recently it has been suggested that mitochondrial KATP channels are the triggers in the signal transduction mechanism rather than the end effectors. [18]
The aims of the present series of studies were to investigate the efficacy of pharmacological preconditioning of the human myocardium with α1-adrenoceptor and adenosine receptor agonists as compared to ischemic preconditioning and to elucidate the contribution and sequence of activation of PKC, p38MAPK and mitoKATP channels.
Results
All samples entering the studies completed the applied protocol and were included in the analysis.
Ischemic versus pharmacological preconditioning (Study 1)
Figures 4A and 4B demonstrate that SI/R alone resulted in a significant increase in CK leakage and decrease in MTT reduction when compared to the aerobic controls. They also show that PP with phenylephrine or adenosine administered prior to SI/R is as protective as IP and that their use in combination does not result in additive protection.
Figure 4.
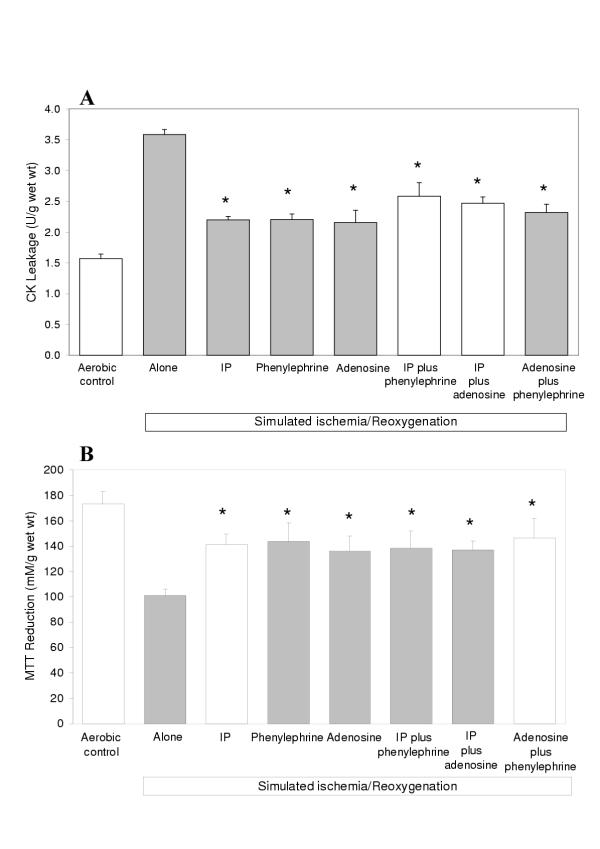
Creatine Kinase (CK) leakage into the media (A) during the 120 min reoxygenation period and MTT reduction by the slices (B) at the end of the reoxygenation period in human atrial myocardium subjected to various protocols (see Figure 1) to investigate the efficacy of preconditioning via α1-adrenoreceptors or adenosine receptors alone and in combination with IP (Study 1). Data are expressed as mean ± SEM of six experiments. *p < 0.05 vs SI/R alone group.
Role of PKC, p38MAPK and mitoKATP channels in IP and PP (Study 2)
Figures 5A and 5B show that both IP and PP with phenylephrine or adenosine are equally abolished by the PKC antagonist chelerythrine, the p38MAPK inhibitor SB203580 or the mitoKATP blocker 5-hydroxydecanoate suggesting that these steps are necessary in the signal transduction cascade of preconditioning.
Figure 5.
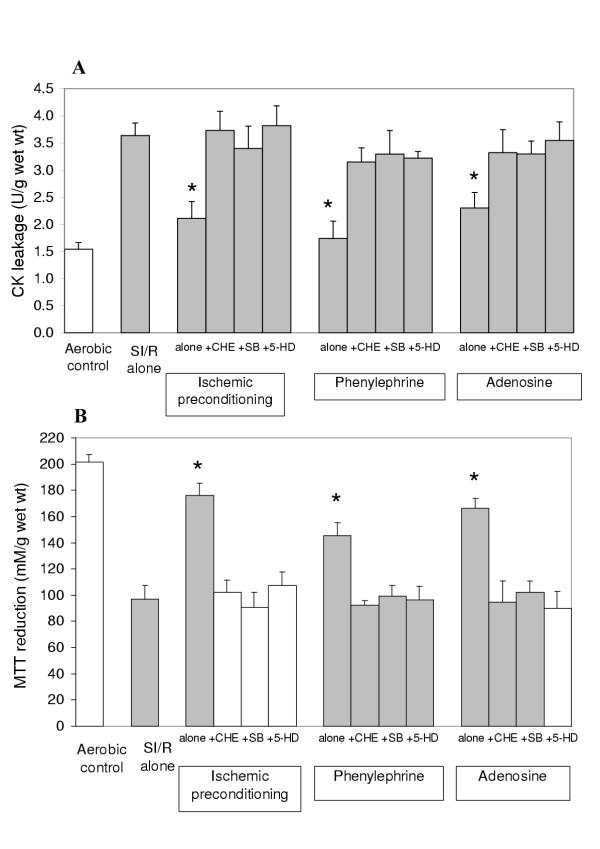
Creatine Kinase (CK) leakage into the media (A) during the 120 min reoxygenation period and MTT reduction by the slices (B) at the end of the reoxygenation period in human atrial myocardium subjected to various protocols (see Figure 2) to investigate the role of PKC, p38MAPK and mitoKATP channels in ischemic and pharmacological preconditioning with phenylephrine (P) or adenosine (Study 2). Data are expressed as mean ± SEM of six experiments. *p < 0.05 vs. SI/R alone group. (5-HD: 5-hydroxydecanoate, SB: SB203580).
Sequence of activation of PKC, p38MAPK and mitoKATP channels (Study 3)
The results on CK leakage and MTT reduction shown in Figures 6A and 6B demonstrate that identical protection is obtained with the mitoKATP channel opener diazoxide, the PKC activator PMA and the p38MAPK activator anisomycin. Importantly, they also show that whilst the protection of diazoxide is abolished by the PKC antagonist chelerythrine and the p38MAPK antagonist SB203580, the protective effect of PMA is abolished by SB203580 but not by the mitoKATP channel blocker 5-hydroxydecanoate, and the protective action of anisomycin is unaffected by chelerythrine and 5-hydroxydecanoate.
Figure 6.
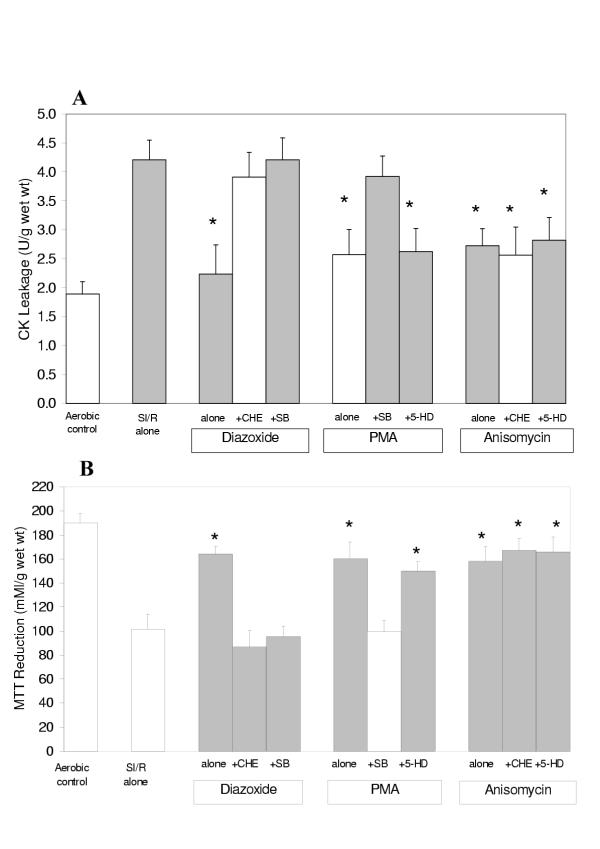
Creatine Kinase (CK) leakage into the media (A) during the 120 min reoxygenation period and MTT reduction by the slices (B) at the end of the reoxygenation period in human atrial myocardium subjected to various protocols (see Figure 3) to investigate the sequence of activation of the mediators of pharmacological and ischemic preconditioning (Study 3). Data are expressed as mean ± SEM of six experiments. *p < 0.05 vs SI/R alone group. (CHE: chelerythrine, 5-HD: 5-hydroxydecanoate, SB: SB203580, PMA: phorbol-12-myristate-13-acetate).
Discussion
The present studies have demonstrated that protection with PP by activation of α1-adrenoreceptors or adenosine receptors is identical to that of IP in the human myocardium. In addition, they have shown that mitoKATP channels, PKC and p38MAPK are an integral part of the cellular signal transduction involved in this cardioprotection in which mitoKATP channels are placed upstream and p38MAPK placed downstream of PKC. These studies provide novel information to understand the underlying mechanism of protection by preconditioning of the human myocardium and the results have obvious clinical importance that warrants further discussion.
Pharmacological versus ischemic preconditioning
The demonstration that activation of α1-adrenoreceptors with phenylephrine is as protective as IP of the human myocardium in these studies is supported by other results in the rat heart [3,5] but contrast with the results reported by Cleveland et al [4] in the human myocardium. The differing results may be due to the use of 50 μM phenylephrine by Cleveland et al, [4] a concentration 500 times the one used in our studies. Using an identical preparation, we have recently shown [22] that phenylephrine exerts maximal protection at 0.1 μM and that protection is lost at concentrations ≥ 10 μM. It should also be pointed out that the human trabeculae preparation used by Cleveland et al [4] differed from our preparation in that muscles were electrically stimulated and subjected to only 45 min of simulated ischemia which may be another potential explanation.
The protection induced by adenosine was also identical to that of IP and the use of the two in combination did not result in additional benefit up and above that seen with each intervention alone. These results in the human myocardium are supported by studies in other animal species [23] but contrast with the results reported by Leesar et al [24] in the vivo human heart and by McCully et al [25] in the isolated rabbit heart. In the study by Leesar et al [24] patients undergoing percutaneous transluminal coronary angioplasty were subjected to three 2-minute balloon inflations 5 minutes apart. Under these conditions the administration of adenosine was more effective in limiting ST-segment shift than the balloon inflation protocol. Although we have previously shown [2] that 4 to 5 min of ischemia are sufficient to precondition the human myocardium, independent of whether this time is attained with one or two cycles of ischemia, it is conceivable that in their study [24] the balloon inflation protocol may not have been sufficient to induce enough ischemia to trigger preconditioning. This is a strong possibility under clinical conditions where collateral flow may lessen the severity of ischemia. Furthermore, in that study [24] changes in ST-segment, that are modulated by sarcolemmal KATP channels, [26] were used as the main end-point and it is now recognized that the protection of preconditioning may be mediated by mitochondrial rather than by sarcolemmal KATP channels. [18-21] The results reported by McCully et al [25] that adenosine is more potent than and extends the cardioprotection of IP in the rabbit heart are difficult to explain because they used a protocol of 5 min ischemia/5 min reperfusion for preconditioning which is identical to the one used in our studies and shown to afford optimal protection in other preparations. [5,9] However, since adenosine and IP use identical cellular signal transduction mechanism to induce protection, the most likely explanation of their results may be the presence of some unknown factor that may have influenced the severity of the IP insult and therefore its protective efficacy.
Mechanism of preconditioning
The elucidation of the factors involved in the signal transduction pathway of preconditioning has been the subject of intense investigation and although the participation of factors such as PKC [17,19,27] and mitoKATP channels [16-21] is well established, their relevance and the sequence of activation remains controversial. The present studies are the first to demonstrate that PP and IP of the human myocardium share identical signal transduction cascade that involves mitoKATP channels, PKC and p38MAPK in that order. Wang et al [28] showed that PKC inhibition with chelerythrine or calphostin C completely abolished the beneficial effects of diazoxide in the isolated rat heart, thus providing further support that mitoKATP channels are upstream of PKC. However, Pain et al [18] and Miura et al [29] using chelerythrine and calphostin C to block PKC were unable to confirm this observation in the rabbit, suggesting that these differences could arise as a result of different animal species. In spite of this, it is interesting to note that Pain et al [18] reported that genistein, a tyrosine kinase antagonist, blocked the protection induced by diazoxide which indicates that activation of kinases also lies downstream of mitoKATP channels in the rabbit heart.
Our demonstration that activation of any one of the components of the transduction cascade investigated in our studies (mitoKATP channels, PKC, p38MAPK) can provide identical protection and that blockade of any of them individually completely abolishes protection indicates that in the human myocardium there is only one pathway of protection by preconditioning. The failure to obtain additional protection when more than one agent was used to induce PP or when these agents were used in combination with IP further support this thesis. But again, the mechanism of preconditioning the human myocardium may not be applicable to all species as suggested by the need to combine the inhibition of PKC and tyrosine kinase to abort the protection of preconditioning in pigs. [30]
The molecular interactions between the various components of the preconditioning pathway are not well understood. There is evidence that mitoKATP channel opening increases radical oxygen species production which in turn may activate PKC, [18] but it is well known that mitoKATP channels are also modulated by PKC [19]. The above and the realization of selective translocation of PKC with preconditioning, [27,28,31] suggest that PKC may play a dual role upstream and downstream of mitoKATP channels. Our observation that blockade of PKC abolishes the protection by diazoxide does not eliminate that possibility.
Our results have shown that activation of p38MAPK is a crucial step in the transduction pathway of preconditioning in the human myocardium. Activation of p38MAPK has also been connected to preconditioning in the rabbit heart, [32] however the relationship could not be established in rat [33] and pig [34] hearts. Therefore, it seems that once more the components of the signal transduction pathway of preconditioning are species-dependent.
It is still unknown whether the activation of p38MAPK is the last step of the transduction cascade that phosphorylates the end-effector and whether there is a simple or multiple effectors and their location. p38MAPK can phosphorylate a wide range of proteins, some of which may be potential candidates for end-effectors of preconditioning. Thus, for example, the low molecular weight heat shock protein HSP27 may be phosphorylated by p38MAPK via the intermediate MAPKAPK2 [35] and this may lead to polymerization of actin [36] and to increase tolerance of the cytoskeleton to stress. [37] Translocation of PKC isoforms to mitochondrial sites, intercalated discs and nucleus may suggest that p38MAPK activation in these places may activate enzymes involved with energy production, intercellular communication through cell junctions or gene transcriptions.
Our results suggest that mitoKATP channels are not the end-effectors of cardioprotection by preconditioning of the human myocardium. The concept that mitoKATP channels may be the end-effectors of preconditioning has been based on the efficacy of diazoxide, a highly selective mitoKATP channel opener, to mimic cardioprotection by preconditioning [20,21] and the blockade of this protection by 5-hydroxydecanoate, [20,21] a specific mitoKATP channel blocker; however, it was never clear whether the actions of these channels were limited to the mitochondria or were part of a more complex signal transduction cascade with effects on other cellular structures. The effect of opening the mitoKATP channels remains controversial and whereas some investigators have reported large decreases in mitochondrial membrane potential affecting respiration and resulting in a reduction in Ca2+ uptake into mitochondria, [38] others have observed little effect on membrane potential, bioenergetics or Ca2+ uptake but important changes on matrix and inter-membrane space volumes. [39] The reason given for these discrepancies was the use of high doses of mitoKATP channel openers in the former studies [39] but this does not clarify how changes in mitochondrial volume are connected to PKC activation, that is downstream in the signal transduction cascade.
The diagram in Figure 7 describes our proposal of the signal transduction cascade of preconditioning in the human myocardium. It is suggested that upon activation of sarcolemmal receptors (e.g. adenosine receptors, α1-adrenoreceptors) mitoKATP channels are opened via Gi proteins and PKC, possibly PKC-δ [28]. The opening of mitoKATP channels will activate PKC possibly via the production of radical oxygen species [18]. PKC may then translocate to various cellular sites including the mitochondria, sarcolemma and intercalated discs, and nucleus, a phenomena that may involve specific PKC isoforms, where p38MAPK will be activated. In turn, p38MAPK may activate a simple or multiple end-effectors directly or via MAPK intermediates. It is clear that more studies are required to fully elucidate the signal transduction pathway of preconditioning and the coupling between its components.
Figure 7.
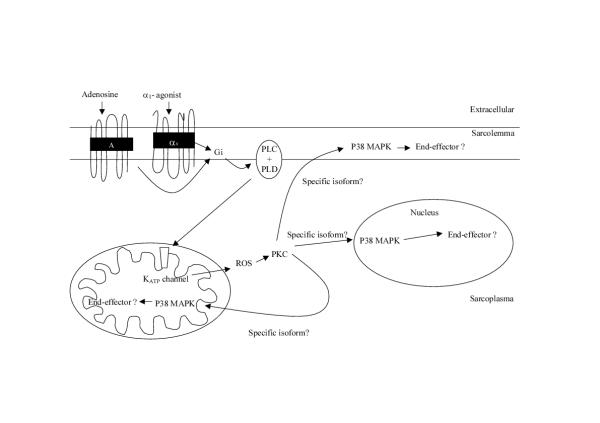
Proposed schematic representation of the signal transduction mechanism leading to cardioprotection by pharmacological and ischemic preconditioning of the human myocardium. Upon activation of sarcolemmal receptors mitoKATP channels are opened via G proteins and possibly PKC-δ. The opening of mitoKATP channels will activate PKC possibly via the production of radical oxygen species (ROS). PKC may then translocate to various cellular sites including the mitochondria, sarcolemma and intercalated discs, and nucleus, a phenomenon that may involve specific PKC isoforms, where p38MAPK will be activated. In turn, p38MAPK may activate a single or multiple end-effectors directly or via MAPK intermediates. PLC: phospholipase C, PLD: phospholipase D.
Clinical implications
The findings that the cardioprotection achieved by activation of α1-adrenoreceptors or adenosine receptors is as potent as the one obtained with IP and that their use in combination does not result in additional benefit have obvious major clinical implications because maximal protection can be attained without the need to occlude the coronary arteries to induce ischemia. The procurement of cardioprotection with stimuli of receptors as diverse as α1-adrenoreceptors and adenosine receptors can be clinically advantageous because some of these agents may be contraindicated in certain conditions (e.g. α1-adrenoreceptor agonists in hypertension, adenosine in the presence of alterations of the cardiac conduction system). These interventions can be useful to combat ischemic injury in different clinical conditions such as coronary angioplasty, cardiac surgery and heart transplantation; however, it is necessary to mention that our studies were performed in an in-vitro preparation and therefore any extrapolation to the clinical setting should be made with caution.
The realization that cardioprotection by PP and IP of the human myocardium is mediated by one obligatory signal transduction pathway also opens the therapeutical window for direct manipulation of its components. Recently, we have demonstrated that although the myocardium from patients with poor left ventricular function (ejection fraction < 30%) or with diabetes cannot be protected with IP, the mitoKATP channel opener diazoxide elicited protection in the former but not in the latter [40]. This suggests that if part of the signal transduction cascade is affected by disease states, cardioprotection can still be obtained by bypassing the defective components. It is also of clinical relevance that blockade at any stage of the signal transduction involved in preconditioning does not seem to exacerbate injury suggesting that this pathway is solely used for cardioprotection and not in tissue injury.
Possible limitations of the study
A potential limitation of our studies was the use of atrial tissue as opposed to ventricular myocardium and the fact that this was a necrosis in vitro model, and therefore any extrapolation must be conducted with caution. Another limitation might be that right atrial appendages were obtained from patients subjected to various medical treatments such as nitrates and beta blockers, which may have influenced the simulated ischemia/reoxygenation injury and the protection induced by preconditioning. However this effect is unlikely to be significant as all the specimens responded similarly to ischemic injury and preconditioning.
Conclusions
The current studies demonstrate that protection with pharmacological preconditioning by activation of α1-adrenoreceptors or adenosine receptors is identical to that of ischemic preconditioning in the human myocardium. In addition, they show that mitoKATP channels, PKC and p38MAPK are an integral part of the cellular signal transduction involved in this cardioprotection in which mitoKATP channels are placed upstream and p38MAPK placed downstream of PKC.
Materials and methods
Experimental preparation
Experiments were performed on trabeculae obtained from the right atrial appendage of patients undergoing elective coronary artery bypass graft surgery or aortic valve replacement in a cell necrosis model that was developed and characterised in our laboratory and that has been described previously [41]. The investigation conforms with the principles outlined in the Decleration of Helsinki. Donor patients were excluded if they had enlarged atriums, atrial arrhythmias, poor left ventricular function (ejection fraction <30%), right ventricular failure or were taking oral hypoglycaemic agents, opioid analgesia, KATP channel openers or catecholamines. Local ethical committee approval was obtained for the harvesting technique. Temperature was maintained at 37°C throughout the experiments and simulated ischemia was induced by bubbling the medium with 95% N2/5% CO2 (pH 6.8–7.0) instead of 95% O2/5% CO2 (pH 7.36 and 7.45) and replacing D-glucose with 2-deoxy-D-glucose.
Solutions and chemicals
The incubation medium was prepared daily with de-ionized distilled water and contained (in mM): NaCl2 (118), KCl (4.8), NaHCO3 (27.2), MgCl2 (1.2), KH2PO4 (1.0) CaCl2 (1.25), HEPES (20) and D-glucose (10) or 2-deoxy-D-glucose (10). All the chemicals were purchased from Sigma Chemicals.
Experimental time course
All the muscles were equilibrated for a 30 min period before being randomly assigned to serve as time-matched aerobic controls or subjected to a 90 min period of simulated ischemia (SI). The muscles were then reoxygenated (R) for another 120 min by incubation in 10 ml of oxygenated medium with added glucose. At the end of the experimental protocols, samples from the incubation media used during the reoxygenation period were collected for the assessment of creative kinase (CK) leakage and the tissue was taken for the assessment of viability (reduction of 3- [4,5-dimethylthiazol-2-yl]-2,5-diphenyltetiazolium bromide (MTT). All agents tested were added for 5, 10 or 20 min at the end of the equilibration period and before the induction of SI. The doses of the agent used in the present studies (phenylephrine at 0.1 μM, adenosine at 100 μM, chelerythrine at10 μM, 5-hydroxydecanoate at 1 mM, SB203580 at 10 μM, PMA at 1 μM diazoxide at 100 μM anisomycin at 1 nM) were selected following preliminary dose-response studies for each of the drugs.
Study groups
In Study 1, the efficacy of preconditioning via α1-adrenoceptors or adenosine receptors alone and in combination with IP was investigated following the protocol described in Figure 1.
Figure 1.

Protocol for Study 1 to investigate the efficacy of preconditioning via α1-adrenoceptors or adenosine (A) receptors alone and in combination with IP (n = 6 specimens/group): (i) time-matched aerobic control, (ii) SI/R alone, (iii) IP induced with 5 min of SI/ 5 min R before SI, (iv) phenylephrine (P) for 5 min and 5 min washout (W) before SI, (v) adenosine (A) for 5 min and 5 min washout (W) before SI, (vi) phenylephrine (P) for 5 min and 5 min washout (W) before IP, (vii) adenosine (A) for 5 min and 5 min washout (W) before IP and (viii) adenosine (A) for 5 min and 5 min washout (W) followed by phenylephrine (P) for 5 min and 5 min washout (W) prior to SI.
Study 2 investigated the role of PKC, p38MAPK and mitoKATP channels in the cardioprotective effect of IP and of PP with phenylephrine and adenosine following the protocol described in Figure 2.
Figure 2.

Protocol for Study 2 to investigate the role of PKC, p38MAPK and mitoKATP channels in the cardioprotective effect of IP and PP with phenylephrine (P) or adenosine (A) (n = 6 specimens/group): (i) time-matched aerobic control, (ii) SI/R alone, (iii) IP alone prior to SI, (iv) chelerythrine (CHE) for 10 min prior to IP, (v) SB203580 (SB) for 10 min prior to IP, (vi) 5-hydroxydecanoate (5-HD) for 10 min prior to IP, (vii) phenylephrine (P) for 5 min and 5 min washout (W) before SI, (viii) chelerythrine (CHE) for 10 min prior to phenylephrine (P) for 5 min and 5 min washout (W), (ix) SB203580 (SB) for 10 min prior to phenylephrine (P) for 5 min and 5 min washout (W), (x) 5-hydroxydecanoate (5-HD) for 10 min prior to phenylephrine (P) for 5 min and 5 min washout (W), (xi) adenosine (A) for 5 min and 5 min washout (W) before SI, (xii) chelerythrine (CHE) for 10 min prior to adenosine (A) for 5 min and 5 min washout (W), (xiii) SB203580 (SB) for 10 min prior to adenosine (A) for 5 min and 5 min washout (W) and (xiv) 5-hydroxydecanoate (5-HD) for 10 min prior to adenosine (A) for 5 min and 5 min washout (W).
Study 3 was designed as described in Figure 3 to elucidate the sequence of the participation of involvement of mitoKATP channels, PKC and p38MAPK in the signal transduction cascade of cardioprotection.
Figure 3.
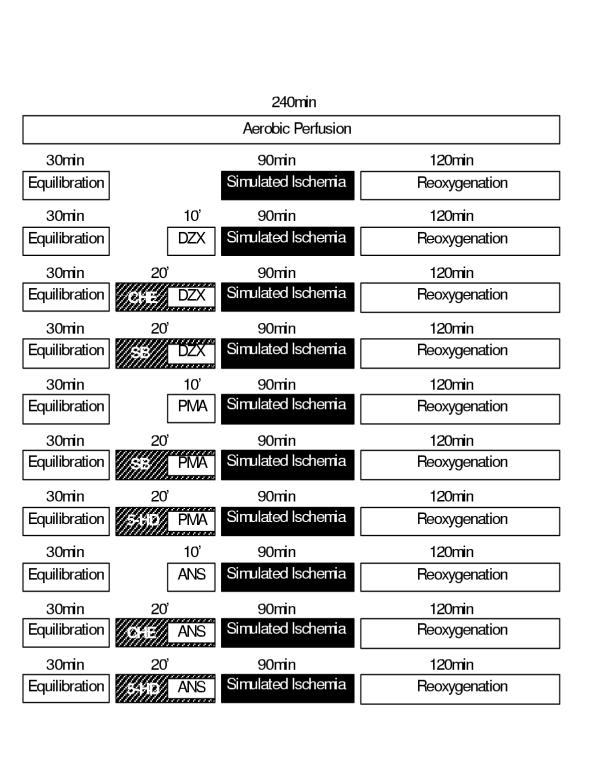
Study 3 protocol designed to elucidate the sequence of the participation of involvement of mitoKATP channels, PKC and p38MAPK in the signal transduction cascade of cardioprotection. For this purpose, in addition to the (i) aerobic time-matched control and (ii) SI/R alone the following groups were studied (n = 6 specimens/group): (iii) diazoxide alone for 10 min prior to SI, (iv) chelerythrine (CHE) for 20 min with diazoxide added for the last 10 min prior to SI, (v) SB203580 (SB) for 20 min with diazoxide added for the last 10 min prior to SI, (vi) PMA alone for 10 min prior to SI, (vii) SB203580 (SB) for 20 min with PMA added for the last 10 min before SI, (viii) 5-hydroxydecanoate (5-HD) for 20 min with PMA added in the last 10 min before SI, (ix) anisomycin (ANS) alone for 10 min prior to SI, (x) chelerythrine (CHE) for 20 min with anisomycin (ANS) added for the last 10 min prior to SI and (xi) 5-hydroxydecanoate (5-HD) for 20 min with anisomycin (ANS) added for the last 10 min prior to SI.
In the three studies, n = 6 specimens from different subjects/group were used.
Assessment of tissue injury and viability
Tissue injury was determined by measuring the leakage of CK into the incubation medium during the 120 min reoxygenation period. This was assayed by a kinetic ultraviolet method based on the formation of NAD (Sigma Catalogue No. 1340-K) and the results expressed as U/g wet wt.
Tissue viability was assessed by the reduction of MTT to a blue formazan product at the end of the experimental time. The absorbance of the blue formazan formed was measured on a plate reader (Benchmark, Bio-Rad Laboratories, California, USA) at 550 nm and the results expressed as mM/g wet wt [41].
Statistical analysis
All data are presented as mean ± SEM. All values were compared by ANOVA with application of a post hoc Tukey's test. Statistical significance was taken as p < 0.05.
Authors' Contribution
ML participated in the design of the study, carried out all the studies, collated and analyzed the data and drafted the manuscript. MG participated in the design of the study, analysis and presentation of the data and revision of the manuscript. Both authors read and approved the final manuscript.
Acknowledgments
Acknowledgments
This work was supported in part by a grant from Mason Medical Research Foundation and by individual contribution from Professor M Galiñanes.
Contributor Information
Mahmoud Loubani, Email: mahmoud.loubani@ntlworld.com.
Manuel Galiñanes, Email: mg50@le.ac.uk.
References
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Standen NB, Galiñanes M. Preconditioning the human myocardium by simulated ischemia: Studies on the early and delayed protection. Cardiovasc Res. 2000;45:339–350. doi: 10.1016/s0008-6363(99)00353-3. [DOI] [PubMed] [Google Scholar]
- Banerjee A, Locke-Winter C, Rogers KB, et al. Preconditioning against myocardial dysfunction after ischemia and reperfusion by an alpha-1 adrenergic mechanism. Circ Res. 1993;73:656–670. doi: 10.1161/01.res.73.4.656. [DOI] [PubMed] [Google Scholar]
- Cleveland JC, Meldrum DR, Rowland RT, et al. Ischemic preconditioning of human myocardium: protein kinase C mediates a permissive role for α1-adrenoceptors. Am J Physiol. 1997;273:H902–H908. doi: 10.1152/ajpheart.1997.273.2.H902. [DOI] [PubMed] [Google Scholar]
- Meng X, Shamer BD, Pulido EJ, et al. Adrenergic induction of bimodal myocardial protection: signal transduction and cardiac gene reprogramming. Am J Physiol. 1999;276:R1525–R1533. doi: 10.1152/ajpregu.1999.276.5.R1525. [DOI] [PubMed] [Google Scholar]
- Lochner A, Genade S, Tromp E, Podzuweit T, Moolman JA. Ischemic preconditioning and the β-adrenergic signal transduction pathway. Circulation. 1999;100:958–966. doi: 10.1161/01.cir.100.9.958. [DOI] [PubMed] [Google Scholar]
- Schultz JJ, Hsu AK, Gross GJ. Ischemic preconditioning is mediated by a peripheral opioid receptor mechanism in the intact rat heart. J Mol Cell Cardiol. 1997;29:1355–1362. doi: 10.1006/jmcc.1996.0369. [DOI] [PubMed] [Google Scholar]
- Downey JM, Liu GS, Thornton JD. Adenosine and the anti-infarct effects of preconditioning. Cardiovasc Res. 1993;27:3–8. doi: 10.1093/cvr/27.1.3. [DOI] [PubMed] [Google Scholar]
- Carr CS, Hill RJ, Masamune H, et al. Evidence for a role for both A1 and A3 receptors in protection of isolated human atrial muscle against simulated ischemia. Cardiovasc Res. 1997;36:52–59. doi: 10.1016/s0008-6363(97)00160-0. [DOI] [PubMed] [Google Scholar]
- Joyeux M, Godin-Ribuot D, Ribuot C. Resistance to myocardial infarction induced by heat stress and the effect of ATP-sensitive potassium channel blockade in the rat isolated heart. Br J Pharmacol. 1998;123:1085–1088. doi: 10.1038/sj.bjp.0701710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brew EB, Mitchell MB, Rehring TF, et al. The role of bradykinin in cardiac functional protection after global ischemia-reperfusion in the rat heart. Am J Physiol. 1995;269:H1370–H1378. doi: 10.1152/ajpheart.1995.269.4.H1370. [DOI] [PubMed] [Google Scholar]
- Meldrum DR, Cleveland JC, Sheridan BC, Rowland RT, Banerjee A, Harken AH. Cardiac preconditioning with calcium: clinically accessible myocardial protection. J Thorac Cardiovasc Surg. 1996;112:778–786. doi: 10.1016/S0022-5223(96)70065-X. [DOI] [PubMed] [Google Scholar]
- Jones WK, Flaherty MP, Tang XL, et al. Ischemic preconditioning increases iNOS transcript levels in conscious rabbits via a nitric oxide-dependent mechanism. J Mol Cell Cardiol. 1999;31:1469–1481. doi: 10.1006/jmcc.1999.0983. [DOI] [PubMed] [Google Scholar]
- Strasser RH, Braun-Dullaeus RB, Walendzik H, Marquetant R. α1-receptor-dependent activation of protein kinase C in acute myocardial ischemia: mechanisms of sensitization of the adenylyl cyclase system. Circ Res. 1992;70:1304–1312. doi: 10.1161/01.res.70.6.1304. [DOI] [PubMed] [Google Scholar]
- Lazou A, Sugden PH, Clerk A. Activation of mitogen-activated protein kinases (p38-MAPKs, SAPKs/JNKs and ERKs) by the G-protein coupled receptor agonist phenylephrine in the perfused rat heart. Biochem J. 1998;332:459–465. doi: 10.1042/bj3320459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross GJ, Auchampach JA. Blockade of ATP-dependent potassium channels prevents myocardial preconditioning. Circ Res. 1992;70:223–233. doi: 10.1161/01.res.70.2.223. [DOI] [PubMed] [Google Scholar]
- Speechly-Dick ME, Grover GJ, Yellon DM. Does ischemic preconditioning in the human involve protein kinase C and the ATP-dependent K channel? Studies in an atrial in vitro model. Circ Res. 1995;77:1030–1035. doi: 10.1161/01.res.77.5.1030. [DOI] [PubMed] [Google Scholar]
- Pain T, Yang X, Critz SD, et al. Opening of mitochondrial KATP channels triggers the preconditioned state by generating free radicals. Circ Res. 2000;87:460–466. doi: 10.1161/01.res.87.6.460. [DOI] [PubMed] [Google Scholar]
- Sato T, O'Rourke B, Marban E. Modulation of mitochondrial ATP-dependent K+ channels by protein kinase C. Circ Res. 1998;83:110–114. doi: 10.1161/01.res.83.1.110. [DOI] [PubMed] [Google Scholar]
- Garlid KD, Paucek P, Yarov-Yarovoy V, et al. Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels: Possible mechanism of cardioprotection. Circ Res. 1997;81:1072–1082. doi: 10.1161/01.res.81.6.1072. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Standen NB, Galiñanes M. Evidence for mitochondrial KATP channels as effectors of human myocardial preconditioning. Cardiovasc Res. 2000;45:934–940. doi: 10.1016/s0008-6363(99)00407-1. [DOI] [PubMed] [Google Scholar]
- Loubani M, Galiñanes M. Alpha 1 adrenoceptors during simulated ischemia and reoxygenation of the human myocardium: effect of the dose and time of administration. J Thorac Cardiovasc Surg. 2001;122:103–112. doi: 10.1067/mtc.2001.114778. [DOI] [PubMed] [Google Scholar]
- Liu GS, Thornton J, Van Winkle DM, Stanley AW, Olsson RA, Downey JM. Protection against infarction afforded by preconditioning is mediated by A1 adenosine receptors in rabbit heart. Circulation. 1991;84:350–356. doi: 10.1161/01.cir.84.1.350. [DOI] [PubMed] [Google Scholar]
- Leesar MA, Stoddard M, Ahmed M, Broadbent J, Bolli R. Preconditioning of human myocardium with adenosine during coronary angioplasty. Circulation. 1997;95:2500–2507. doi: 10.1161/01.cir.95.11.2500. [DOI] [PubMed] [Google Scholar]
- McCully JD, Toyoda Y, Uematsu M, Stewart RD, Levitsky S. Adenosine-enhanced ischemic preconditioning adenosine receptor involvement during ischemia and reperfusion. Am J Physiol. 2001;280:H591–H602. doi: 10.1152/ajpheart.2001.280.2.H591. [DOI] [PubMed] [Google Scholar]
- Li RA, Leppo M, Miki T, Seino S, Marban E. Molecular basis of electrocardiographic ST-segment elevation. Circ Res. 2000;87:837–839. doi: 10.1161/01.res.87.10.837. [DOI] [PubMed] [Google Scholar]
- Mitchell MB, Meng X, Ao L, Brown JM, Harken AH, Banerjee A. Preconditioning of isolated rat heart is mediated by protein kinase C. Circ Res. 1995;76:73–81. doi: 10.1161/01.res.76.1.73. [DOI] [PubMed] [Google Scholar]
- Wang Y, Hirai K, Ashraf M. Activation of mitochondrial ATP-sensitive K(+) channel for cardiac protection against ischemic injury is dependent on protein kinase C activity. Circ Res. 1999;85:731–741. doi: 10.1161/01.res.85.8.731. [DOI] [PubMed] [Google Scholar]
- Miura T, Liu Y, Kita H, Ogawa T, Shimamoto K. Roles of mitochondrial ATP-sensitive K channels and PKC in anti-infarct tolerance afforded by adenosine A1 receptor activation. J Am Coll Cardiol. 2000;35:238–245. doi: 10.1016/s0735-1097(99)00493-3. [DOI] [PubMed] [Google Scholar]
- Vahlhaus C, Schulz R, Post H, Rose J, Heusch G. Prevention of ischemic preconditioning only by combination of protein kinase C and protein tyrosine kinase in pigs. J Mol Cell Cardiol. 1998;38:197–209. doi: 10.1006/jmcc.1997.0609. [DOI] [PubMed] [Google Scholar]
- Ping P, Zhang J, Qiu Y, et al. Ischemic preconditioning induces selective translocation of protein kinase C isoforms epsilon and eta in the heart of conscious rabbits without subcellular redistribution of total protein kinase C activity. Circ Res. 1997;81:404–414. doi: 10.1161/01.res.81.3.404. [DOI] [PubMed] [Google Scholar]
- Nakano A, Baines CP, Kim SO, et al. Ischemic preconditioning activates MAPKAPK2 in the isolated rabbit heart: evidence for involvement of p38 MAPK. Cir Res. 2000;84:144–151. doi: 10.1161/01.res.86.2.144. [DOI] [PubMed] [Google Scholar]
- Schneider S, Chen W, Hou J, Steenbergen C, Murphy E. Inhibition of p38 MAPK α/β reduces ischemic injury and does not block protective effects of preconditioning. Am J Physiol. 2000;280:H449–H508. doi: 10.1152/ajpheart.2001.280.2.H499. [DOI] [PubMed] [Google Scholar]
- Behrends M, Schulz R, Post H, et al. Inconsistent relation of MAPK activation to infarct size reduction by ischemic preconditioning in pigs. Am J Physiol Heart Circ Physiol. 2000;279:H1111–H1119. doi: 10.1152/ajpheart.2000.279.3.H1111. [DOI] [PubMed] [Google Scholar]
- Stokoe D, Engel K, Campbell DG, Cohen P, Gaestel M. Identification of MAPKAP kinase 2 as a major enzyme responsible for the phosphorylation of the small mammalian heat shock proteins. FEBS Lett. 1992;313:307–313. doi: 10.1016/0014-5793(92)81216-9. [DOI] [PubMed] [Google Scholar]
- Guay J, Lambert H, Gingras-Breton G, Lavoie JN, Huot J, Landry J. Regulation of actin filament dynamics by p38 map kinase-mediated phosphorylation of heat shock protein 27. J Cell Sci. 1997;110:357–368. doi: 10.1242/jcs.110.3.357. [DOI] [PubMed] [Google Scholar]
- Huot J, Houle F, Spitz DR, Landry J. HSP 27 phosphorylation-mediated resistance against actin fragmentation and cell death induced by oxidative stress. Cancer Res. 1996;56:273–279. [PubMed] [Google Scholar]
- Liu Y, Sato T, Seharaseyon J, Szewczky A, O'Rourke B, Marban E. Mitochondrial ATP-dependent potassium channels. Viable candidate effectors of ischemic preconditioning. Ann N Y Acad Sci. 1999;874:27–37. doi: 10.1111/j.1749-6632.1999.tb09222.x. [DOI] [PubMed] [Google Scholar]
- Kowaltowski AJ, Seetharaman S, Paucek P, Garlid KD. Bioenergetic consequences of opening the ATP-sensitive K(+) channel of heart mitochondria. Am J Physiol Heart Circ Physiol. 2001;280:H649–H657. doi: 10.1152/ajpheart.2001.280.2.H649. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Standen NB, Galiñanes M. Failure to precondition pathological human myocardium. J Am Coll Cardiol. 2001;37:711–718. doi: 10.1016/s0735-1097(00)01161-x. [DOI] [PubMed] [Google Scholar]
- Zhang JG, Ghosh S, Ockelford C, Galiñanes M. Characterization of an in vitro model for the study of the short and prolonged effects of myocardial ischaemia and reperfusion in man. Clin Sci. 2000;99:443–453. [PubMed] [Google Scholar]