


Abstract
To search for restriction endonucleases, we used a novel plant-based cell-free translation procedure that bypasses the toxicity of these enzymes. To identify candidate genes, the related genomes of the hyperthermophilic archaea Pyrococcus abyssi and Pyrococcus horikoshii were compared. In line with the selfish mobile gene hypothesis for restriction–modification systems, apparent genome rearrangement around putative restriction genes served as a selecting criterion. Several candidate restriction genes were identified and then amplified in such a way that they were removed from their own translation signal. During their cloning into a plasmid, the genes became connected with a plant translation signal. After in vitro transcription by T7 RNA polymerase, the mRNAs were separated from the template DNA and translated in a wheat-germ-based cell-free protein synthesis system. The resulting solution could be directly assayed for restriction activity. We identified two deoxyribonucleases. The novel enzyme was denoted as PabI, purified and found to recognize 5′-GTAC and leave a 3′-TA overhang (5′-GTA/C), a novel restriction enzyme-generated terminus. PabI is active up to 90°C and optimally active at a pH of around 6 and in NaCl concentrations ranging from 100 to 200 mM. We predict that it has a novel 3D structure.
INTRODUCTION
Restriction–modification systems consist of at least two enzymes, namely, a restriction endonuclease that recognizes a specific DNA sequence and introduces a double-strand break, and a cognate modification methyltransferase that can methylate the same sequence and thereby render it resistant to the cleavage (1). Restriction endonucleases will cleave foreign DNAs such as viral and plasmid DNA when these DNAs have not been modified by the appropriate modification enzyme. It has been believed that the evolution and maintenance of restriction–modification systems have been driven by the cell's need to protect itself from infection by foreign DNA. However, it has been observed that some restriction–modification gene complexes maintain themselves within their bacterial host by killing cells that have lost them through chromosome cleavage (2). This has led to the proposal that some restriction–modification gene complexes behave as selfish genetic elements, similar to viruses and transposons (3,4).
In agreement with the selfish gene hypothesis, there is increasing evidence for the mobility of restriction–modification gene complexes, particularly from genome analysis. Comparison of two genome sequences within the species Helicobacter pylori has indicated that various types of genome rearrangements are associated with the presence of a restriction–modification gene complex, e.g. the insertion of a restriction–modification gene complex that involved long target duplication and the substitution of a restriction–modification gene homolog adjacent to a large inversion (5,6). Moreover, comparisons of the genomes of two species within the same hyperthermophilic archaeon genus, namely, Pyrococcus abyssi and Pyrococcus horikoshii, revealed that methyltransferase homolog genes were associated with simple substitution, transposition and substitution with adjacent inversion (7). Sequence alignment, analyses of GC contents and codon usage analyses have also revealed potential cases of the horizontal transfer of restriction–modification gene complexes between distantly related bacteria [for review, see (4)]. Further supporting such observations are experiments showing the introduction of large-scale genome rearrangements when restriction–modification gene complexes are threatened under laboratory conditions (8,9).
Restriction endonucleases are indispensable tools for biotechnology and molecular biology, and the discovery of new specificities and new DNA cleavage characters remains desirable. The traditional method used to screen for restriction endonucleases is to culture individual strains and test their extracts for the ability to produce specific fragments from small DNA molecules (10). However, the recent accumulation of genome-wide sequence information now makes it possible to search for novel restriction genes by employing bioinformatics methods (11,12) [for review, see (13)]. While the restriction endonucleases show little, if any, sequence conservation and as such are very hard to identify by bioinformatics approaches, the methyltransferases belong to a conserved protein family. Therefore, the bioinformatics strategy is to first identify methyltransferase genes and then search for their cognate restriction endonuclease genes in the neighboring open reading frames (ORFs). In addition, since restriction–modification gene complexes are often associated with a genome rearrangement, such in silico approaches could employ apparent genome rearrangement in the vicinity of a candidate gene as a selection criterion. Further boosting the validity of this approach is that recently arrived restriction–modification gene complexes (on the evolutionary time scale) tend to retain their enzyme activity (12).
After their identification by bioinformatics, candidate restriction genes are cloned and expressed for use in the biochemical assay. However, the restriction enzymes are cytotoxic when expressed in vivo without appropriate and sufficient methylation on the genome. Various means have been adopted to overcome this problem, not always with satisfactory results. These include cloning and expression of the cognate methyltransferase and the use of tightly repressible expression systems (11). The problem is particularly serious when the restriction enzyme is able to cut the host chromosome at numerous sites (e.g. 4 bp cutters), and when the methyltransferase is of distant descent and cannot be expressed well in the host.
In this study, we present a novel procedure that bypasses this problem. This procedure involves synthesizing the restriction enzyme by using wheat-germ extracts (14) (Figure 1). The procedure has a number of advantages. First, the candidate ORF is separated from its cis-elements for transcription and translation in prokaryotes and is instead connected to a cis-element for translation in plants. This minimizes its expression during the construction and propagation of the recombinant in the host bacterial cells. In addition, the transcripts synthesized in vitro are separated from the template DNA for translation in vitro. This is unlike most other bacteria-based in vitro protein synthesis systems, in which transcription and translation are obligatorily coupled and the template DNA is exposed to the restriction enzymes that are generated. Finally, the solution resulting from the in vitro translation can be directly used to assay restriction activity because the wheat-germ extract contains little, if any, deoxyribonuclease activity (14).
Figure 1.

Procedure used to screen for novel restriction endonucleases. Restriction gene candidates are determined by genome comparison and other bioinformatics methods. PCR is then performed to amplify each candidate ORF from the genome of interest. The primers are designed such that the ORF becomes separated from its promoter and translation signal (SD: Shine–Dalgarno) sequence. The PCR fragments are then inserted into a plasmid so that the ORF becomes connected with the T7 promoter and a plant translation signal. The resulting DNA is subjected to in vitro transcription by T7 RNA polymerase and the mRNA is isolated and subjected to in vitro translation with a wheat-germ extract. The solution can then be used directly to assay for restriction activity.
By employing this in vitro screening method in combination with in silico screening of the P.abyssi and P.horikoshii genomes, we obtained a novel thermoresistant restriction endonuclease named PabI. It cleaves the sequence 5′-GTAC between A and C (GTA/C) to generate a novel type of restricted terminus, namely, a 3′-TA overhang.
MATERIALS AND METHODS
Materials
P.abyssi genomic DNA was provided by Dr Yoshizumi Ishino (University of Kyushu), who prepared it from the cells donated by Dr Patrick Fortre (Institut de Genetique et Microbiologie, France). P.horikoshii genomic DNA was purchased from ATCC. Escherichia coli K12 strain JM109 (recA1, endA1, gyrA96, thi, hsdR17 (), e14− (mcrA−), supE44, relA1, Δ(lac-proAB)/F′ [traD36, proAB+, lac Iq, lacZ ΔM15]) is a gift from Dr Akio Nomoto (University of Tokyo). pEU3-NII containing omega, a plant translation signal, was obtained from Toyobo. The plasmid Litmus38i was purchased from New England Biolabs. Litmus38i has two PabI sites (5′-GTAC): the site located between two NspI sites was removed from Litmus38i by cleavage with NspI and religation (=pKI1). RsaI was purchased from New England Biolabs.
In vitro screening for restriction enzyme activity
The PCR-amplified candidate ORF of the restriction enzyme gene was inserted into the multiple cloning site of pEU3-NII and the resulting plasmid was used to transform E.coli JM109. After overnight culture in 5 ml of Luria–Bertani (LB) medium at 37°C with shaking, the plasmids were purified with a Qiafilter plasmid mini kit (QIAGEN) followed by phenol/chloroform treatment to remove the RNase derived from the kit. The mRNA used for in vitro translation was transcribed with Thermo T7 RNA polymerase (TOYOBO). Thus, 5 μg of template DNA (pEU3-NII inserted with the candidate ORF), 5 μl of the buffer that came with the enzyme, 5 μl of 25 mM NTPs (Amersham), 40 U of RNase inhibitor (TOYOBO), 150 U of Thermo T7 RNA polymerase and de-ionized water used to achieve a total volume of 50 μl were mixed and incubated at 37°C for 4 h. After 2 min of centrifugation (12 000 r.p.m., 13 400 g at 4°C), the transcribed solution was loaded onto a MicroSpin G-25 Column (Amersham) equilibrated with the buffer mix of the PROTEIOS™ (TOYOBO), and centrifuged (3000 r.p.m., 800 g) at 4°C for 2 min. The flow-through fraction containing the mRNA was recovered. Proteins were synthesized from the mRNA preparation by employing the wheat-germ-based cell-free protein synthesis kit PROTEIOS™ (TOYOBO) by the bilayer method (14). The procedure used followed the manufacturer's instructions except that the volume was changed from 300 to 150 μl, and the incubation took 24 h at 26°C. Nuclease activity was tested by incubating at 65°C for 1 h in standard solution (50 mM Tris–HCl, pH 7.5, 100 mM NaCl, 10 mM MgCl2 and 1 mM DTT) 0.3 μg of λDNA with 1 μl of either (i) the protein synthesis buffer after its reaction or (ii) its supernatant after heat treatment (90°C for 10 min) and centrifugation.
Cloning and expression of PabI
For the PabI gene, the following primers were used: 0105-FOR: 5′-ATCCATATGATTCATTTGACTAGTGTAGAAGCGAGTG-3′ and 0105-REV: 5′-CGGGATCCCGTTATGAAGTGCCGATAATACTCCTC-3′. The PCR-amplified PabI ORF was cut with BamHI and inserted into EcoRV- and BamHI-cut pEU3-NII. Large scales were used in transcription and translation (3.6 ml) in vitro for purification of PabI.
Prediction of recognition sequence
The protein synthesis solution containing PabI was heated at 90°C and centrifuged to denature and remove the wheat-germ proteins. The substrate DNA, pUC19 (0.4 μg), was digested to completion at 85°C for 6 h by the resulting PabI-containing supernatant in standard buffer. The product DNA was subjected to 1% agarose gel electrophoresis followed by ethidium bromide staining. The lengths of the PabI digestion products were calculated. Possible isoschizomers of PabI were sought by using REBpredictor, a tool of REBASE (http://rebase.neb.com/rebase/rebase.html).
Determination of cleavage sites
To determine the exact cleavage position of PabI in the recognition sequence, the primer extension method was used (15). Thus, single-strand DNA prepared from pUC19 was annealed with either of the following 33P-labeled primers: primer A, 5′-GGCGATTAAGTTGGGTAACGCCAGG-3′, or primer B, 5′-GGAAACAGCTATGACCATGATTACGCC-3′. These primers bind upstream of PabI sites. A primer extension reaction was performed by using the Klenow fragment (Takara). The reaction solution was gel-filtrated by using a MicroSpin G-25 Column (Amersham) followed by phenol/chloroform extraction to remove the Klenow fragment. The recovered DNA was then digested with the heat-denatured wheat-germ products that included active PabI and loaded onto 6% polyacrylamide gel with 8 M urea together with the products of the DNA sequencing reactions from the same primer and template. DNA sequencing was performed by using the BcaBEST sequencing kit (Takara) according to the manufacturer's protocol. dc7GTP in the reaction had to be replaced by dGTP to obtain the exact control with regard to its mobility.
Purification of PabI
The protein synthesis solution containing PabI (3.6 ml) was heated at 90°C for 15 min and the denatured proteins and insoluble materials were removed by centrifugation (8000 r.p.m., 8730 g 4°C, 15 min). The PabI protein in the supernatant was purified by chromatography through Heparin-Sepharose TM CL-6B (Amersham), which was equilibrated with 10 mM Tris–HCl (pH 7.5). Fractions were eluted by a NaCl gradient (0–1.5 M) in 10 mM Tris–HCl buffer. Fractions containing PabI were determined by SDS–PAGE analysis. The fractions containing PabI were dialyzed against 10 mM Tris–HCl (pH 7.5), 200 mM NaCl and 1 mM DTT at 4°C overnight. Glycerol was then added to the dialyzed PabI solution to a final concentration of 5% followed by concentration to 150 μl by Amicon Ultra (Millipore).
Reaction conditions for PabI
The plasmid pKI1 cut with PvuII (TOYOBO) was used as a substrate for PabI. The cleavage by PabI splits this 2559 bp long linear DNA into two DNA fragments of 563 and 1996 bp, respectively. To determine the optimal reaction conditions for PabI, the following experiments were performed:
Temperature. The purified enzyme and substrate DNA were incubated for 1 h in the standard buffer at temperatures ranging from 35 to 95°C.
NaCl concentration. The purified enzyme and the substrate DNA were incubated at 85°C for 1 h in the buffer that only differed from standard buffer in the NaCl concentrations, which ranged from 0 to 400 mM NaCl.
pH. The purified enzyme and substrate DNA were incubated at 85°C for 1 h in buffer that only differed from the standard buffer in the pH determined by 50 mM Tris–HCl pH, which ranged from pH 4.3 to 7.8 at 85°C.
The cleaved DNAs were separated by electrophoresis through a 1% agarose gel and visualized with ultraviolet light after ethidium bromide staining. To determine the optimal conditions, signal intensities of the product bands were compared.
Heat resistance of PabI
Purified PabI (8 ng) was heated at 85°C for varying times ranging from 30 min to 5 h. The enzyme was then used to cleave 200 ng of linearized pKI1 at 85°C. Restriction activity was examined 0.5, 1, 2, 3, 4 and 5 h after starting the incubation. At each time point, 200 ng of the substrate DNA was added to a portion of the solution, which was incubated at 85°C for 1 h.
RESULTS
In silico screening for candidate restriction enzyme genes
We searched for restriction enzyme genes in the P.abyssi and P.horikoshii genomes by using bioinformatics methods. Generally, restriction enzyme genes do not exhibit significant sequence similarity to each other or to any other proteins in the database and it is difficult to find novel restriction enzyme genes by direct homology searching. However, methyltransferase genes do share similarities and thus we first searched for putative modification methyltransferase genes. We selected the following ORFs as being modification enzyme gene candidates: (i) ORFs annotated as putative modification enzymes in REBASE (http://rebase.neb.com/rebase/rebase.html); (ii) ORFs with homology to modification enzyme genes and inferred to be involved in genome rearrangements based on the comparison of the genomes of P.abyssi and P.horikoshii (7) and (iii) ORFs in the database GTOP (http://spock.genes.nig.ac.jp/~genome/gtop.html) hit by key word searching using the words ‘methylase’, ‘methyltransferase’ and ‘modification’. From these candidates, those ORFs annotated as RNA methyltransferase and protein methyltransferase were excluded. Our searches yielded 14 candidate modification gene ORFs (Table 1). Of these, nine were of particular interest since they appeared to be associated with genome rearrangements (7).
Table 1.
Results of in silico and in vitro screening of restriction enzymes in P.abyssi and P.horikoshii
 |
Left part (columns 1–4): Candidate modification enzyme genes were sought by bioinformatics methods in the published genome sequences of P.abyssi and P.horikoshii.
Right part (columns 5–8): ORFs that neighbored one of the modification enzyme gene candidates and were annotated as unknown were selected as candidates for restriction enzyme genes. The restriction enzyme activities of their products were assayed on lambda DNA after their expression in a wheat-germ cell-free protein synthesis system.
The restriction gene candidates in the gray boxes were selected for the first round of screening, while the remainder were used in the second round. Those restriction genes in bold fonts (PAB0105, PH0583) were found to be active.
aListed as a putative modification gene in REBASE.
bHit by keyword searching in GTOP.
cAssociated with a putative genome rearrangement (7).
dProtein expression in the wheat-germ cell-free system as detected by SDS–PAGE and CBB (Coomassie Brilliant blue) staining.
eLambda DNA was cleaved by the putative restriction enzyme.
g‘CGAT’ indicates ‘based on CGAT’ (32).
f‘NT’ indicates not tested.
We identified candidate restriction enzyme gene ORFs on the basis of the following criteria: (i) ORFs located immediately next to a modification gene candidate or next to its immediate neighbor, and annotated as having an unknown function (ii) ORFs located as described in (i) but annotated as a nuclease, a recombinase, a DNA repair enzyme or a relative of these proteins or (iii) ORFs predicted by bioinformatic methods to show the pattern of secondary structures similar to that of known restriction endonucleases (13). From these ORFs, those shorter than 100 amino acids were excluded. This resulted in 32 candidate restriction enzyme gene ORFs (Table 1, right part). For all these ORFs, secondary structure prediction and protein fold-recognition were carried out by using the GeneSilico meta-server gateway at http://genesilico.pl/meta/ (16) (see Table 1 for results).
Expression by a wheat-germ-based cell-free system and screening for restriction enzyme activity
Each of the candidate restriction enzyme ORFs was amplified from P.abyssi or P.horikoshii genomic DNA by PCR with a unique primer pair. In the process (Figure 1), they were separated from the cis-signals for prokaryotic transcription (promoter) and translation (Shine–Dalgarno sequence) to minimize their in vivo expression in the bacterial host. The PCR fragments were then inserted into a plasmid vector (pEU3-NII) downstream of a plant translation enhancer sequence (omega from Tobacco Mosaic virus) and the T7 promoter. The plasmid construct was recovered and propagated in an E.coli strain lacking T7 RNA polymerase. The template DNA was then transcribed in vitro by T7 RNA polymerase and proteins were synthesized from this mRNA preparation by using the wheat-germ-based cell-free protein synthesis system. This protein synthesis system uncouples transcription and translation, which eliminates the possible reduction of expression due to cleavage of the template DNA by the expressed restriction enzyme. Because the wheat-germ extract shows little, if any, deoxyribonuclease activity (14), the products of this system can be used to directly screen restriction enzyme activity. Indeed the extract, before and after heat treatment, showed no detectable deoxyribonuclease activity on lambda DNA (Figure 2A). The smaller materials at the bottom of gel photographs in Figure 2 are likely RNA molecules present in the wheat-germ extract rather than degradation products from lambda DNA because they are present even in the absence of lambda DNA (Figure 2A) and because they disappeared by treatment with RNase A (data not shown).
Figure 2.

Detection of restriction enzyme activity in the cell-free translation products. (A and B) The mRNA synthesized in vitro was added to wheat-germ-based in vitro translation reaction. In the indicated cases, the solution was heated at 90°C for recovery of supernatant of low-speed centrifugation. Its aliquot was incubated with lambda DNA at 65°C for 1 h. The digests were run through 1% agarose and visualized with ethidium bromide and ultraviolet irradiation. Marker: perfect DNA markers, 0.1–12 kb (Novagen).
The synthesized protein solution from each candidate ORF product was tested for its ability to cleave lambda DNA. In the first round of trials, five of the ORFs identified by bioinformatics, namely, PAB0105, PH0583, PH1305, PH0904 and PH1033 (see Table 1), were tested because they are associated with genome rearrangement (7) and, in some cases, bear some similarity to restriction endonuclease genes. This initial analysis revealed that the products of PAB0105 and PH0583 cleave lambda DNA (Figure 2B, Table 1, lines shaded). In a second round of experiments, the other 27 ORFs were tested (Table 1, lines not shaded). However, no restriction enzyme activity was detected from this screening.
Purification and characterization of PabI
The product of PH0583 has been characterized earlier as PhoI (REBASE) (17). However, the product of PAB0105 has not yet been characterized; we named it PabI because it is, to our knowledge, the first restriction enzyme from P.abyssi (18). To determine the recognition sequence and cleavage site of PabI, the protein synthesis solution containing PabI was heated at 90°C and centrifuged to remove the proteins from the wheat-germ extract (Figure 3). Later, to determine the optimal reaction conditions and heat resistance of PabI, this solution of PabI was further purified by using a Heparin–Sepharose column (Figure 3).
Figure 3.
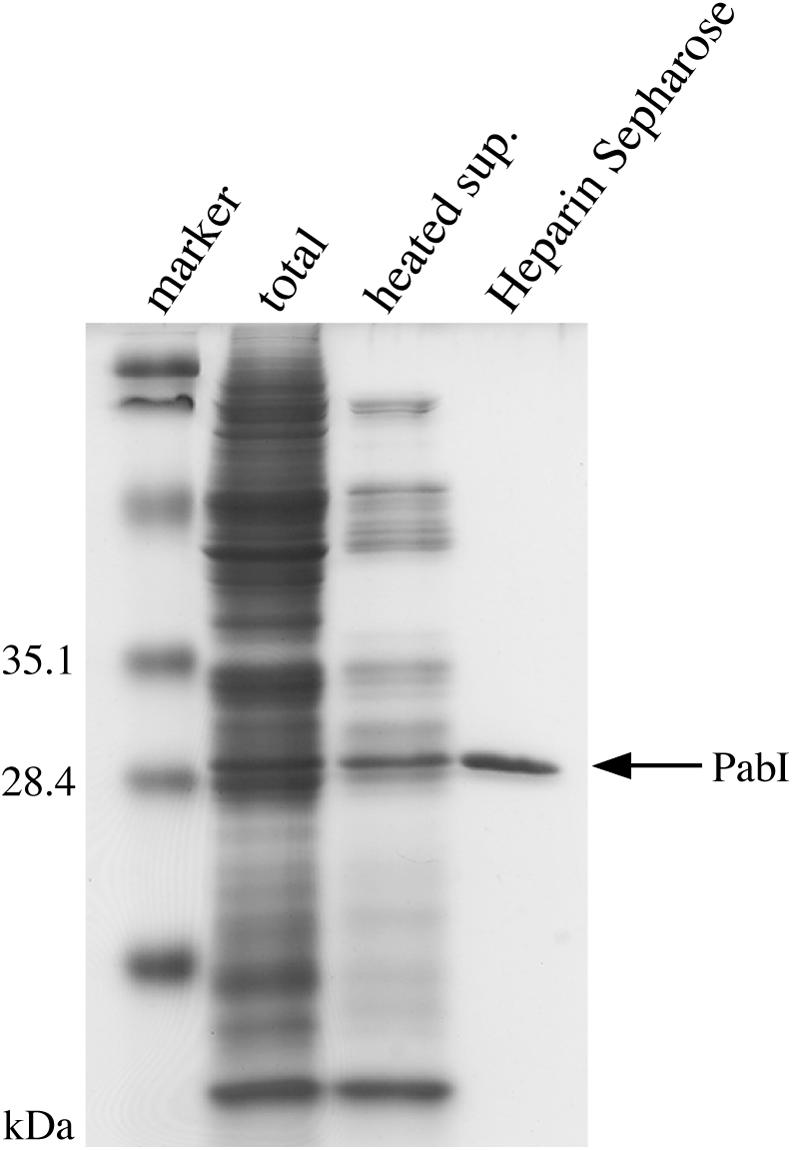
Purification of PabI. The purification steps were monitored by SDS–PAGE electrophoresis. Marker, the protein size markers [Prestained SDS–PAGE Standards Low Range (BIO-RAD)]; total, wheat-germ-based cell-free protein synthesis solution containing expressed PabI; heated sup., the protein synthesis solution after its treatment at 90°C and low-speed centrifugation; purified PabI, the heated supernatant after Heparin–Sepharose column chromatography.
In order to determine the recognition sequence of PabI, pUC19 was digested with PabI. Its recognition sequence was predicted as the same as that of RsaI, namely, 5′-GTAC (19). Indeed, the DNA digestion patterns of pUC19, pBR322 and phiX174 with PabI and RsaI turned out to be indistinguishable (Figure 4). PabI cleavage site was also determined to be between the A and C of the recognition sequence (GTA/C) (Figure 5A and B). The cleavage site of PabI differs from that of RsaI. Thus, PabI represents a neoschizomer of RsaI (Figure 5C). According to REBASE, PabI is the first restriction enzyme that produces a 3′ overhang of TA.
Figure 4.

PabI and RsaI both cut at 5′-GTAC and show identical restriction patterns. The PabI- or RsaI-cleaved DNA substrates pUC19, pBR322 and phiX174 were separated by electrophoresis through a 1% agarose gel and visualized under UV light after ethidium bromide staining. Marker: perfect DNA markers, 0.1–12 kb (Novagen).
Figure 5.

Cleavage sites of PabI. (A and B) Gel electrophoresis of the primer extension reaction followed by RsaI or PabI digestion. The cleavage positions were determined by comparing the product bands with sequence ladders run in parallel. (C) The recognition sequence and the cleavage positions of PabI and RsaI.
Heparin-purified PabI was employed in a series of reactions to determine its optimal reaction conditions. This revealed the optimum pH to be about 6 and the optimum NaCl concentrations to range between 100 and 200 mM at 85°C. It was active from 60 to 90°C with apparent maximum at 85°C (Figure 6A). It retained approximately half of its activity after 1 h of incubation at 85°C (Figure 6B).
Figure 6.

Thermoresistance of PabI. (A) DNA cleavage activity of PabI at different temperatures. Linearized pKI1 was cleaved with PabI for 1 h at the specified temperature. RsaI: DNA cleaved with RsaI. PabI is active at temperatures ranging from 60 to 90°C. Although its activity increased at temperatures ranging from 60 to 85°C, the substrates and product DNAs seem to be altered at higher temperatures (90 and 95°C). (B) Heat resistance of PabI at 85°C. PabI was pre-heated at 85°C for the indicated duration and then used to cleave linearized pKI1 at 85°C for 1 h. The DNA products were separated through a 1% agarose gel and visualized under UV light after ethidium bromide staining. RsaI: DNA cleaved with RsaI.
DISCUSSION
Use of wheat-germ-based cell-free protein synthesis for restriction enzyme screening
Our results demonstrated how powerful the wheat-germ-based cell-free protein synthesis system is in the search of a novel restriction enzyme. This is likely to be due to three features of this system. First, this method can be used to express proteins that are cytotoxic to the bacteria. Initially, we sought to express PabI in E.coli with pLT7K, which has been constructed to tightly repress the expression of cloned genes until their induction with isopropyl-β-d-thiogalactopyranoside (11). However, E.coli transformants were not obtained with pLT7K ligated with the PabI gene. This is presumably because of the residual activity of this 4 bp cutter at 37°C on E.coli chromosome. The PabI gene was successfully cloned into the pEU3-NII expression vector in E.coli. This suggests that expression of PabI is repressed more tightly on pEU3-NII than on pLT7K in E.coli at 37°C. This is probably because the cis-signal for its translation is replaced by a plant translation enhancer, which shows only low activity in E.coli (20). This suggests that our strategy is worth trying for restriction enzymes active at 37°C. Furthermore, if the template restriction enzyme gene is prepared by PCR in vitro rather than by cloning in vivo, the cytotoxicity, if any, would not represent any obstacle at all in its expression, identification or characterization. This completely in vitro approach reported for other proteins (21) should be applicable to screening of any sort of restriction endonucleases, either thermophilic, mesophilic or cryophilic and other deoxyribonucleases.
The second advantage of the wheat-germ-based cell-free protein synthesis is that it is especially suited to expressing deoxyribonucleases because it is uncoupled from the transcription system and can take place in the absence of the template DNA. In bacterial systems, where translation is coupled to transcription, the restriction gene product could attack its gene, thereby repressing its transcription and translation. For example, in an E.coli transcription–translation coupled system, linearized template DNA reduces β-gal synthesis by 35%, even when the recB-deletion strain was used to prepare the cell extract (22).
The third advantage of the system we used is that since the wheat-germ extract contains little deoxyribonuclease activity, if any (14), it is possible to directly use the product solution, without a long purification step, for assaying restriction enzyme activity and even for determining the recognition sequence. In the case of the thermo-resistant restriction enzyme PabI, we were also able to determine the cleavage site by using the supernatant of the product solution after it was heated.
Sequence analysis and structure prediction of PabI
In our initial in silico screening, we focused on ORFs that were annotated as unknown in that they lacked detectable sequence similarity with any known genes. A reasonable expectation from this strategy is that we might encounter a protein of novel structure.
Sequence searches of databases at NCBI (non-redundant, unfinished genomes and environmental sequencing) and REBASE revealed only one intact homolog of PAB0105 (PabI), that is jhp0455 from H.pylori J99 (expectation value 2 × 10−14, 28% amino acid sequence identity to PAB0105) (Figure 7). Its close homolog (90% sequence identity) was found disrupted (HP0504 and HP0505) in H.pylori 26695. Another incomplete sequence of a PabI homolog (hypothetical protein CUP0001 truncated at the N-terminus) was identified in the genome of Campylobacter upsaliensis RM3195 (Figure 7).
Figure 7.

Sequence alignment of PabI with its homologs. Amino acid residues are colored according to their physico-chemical properties (green, aliphatic and aromatic; magenta, polar; blue, positive; red, negative; yellow, glycine and proline). The conserved residues are shaded. SS indicates the consensus secondary structure prediction [E: extended (β), H: helical (α) conformation].
The pattern of predicted secondary structures (Figure 7) deviated strongly from that observed in PD-(D/E)XK-superfamily nucleases (23) as well as in other known nuclease folds, including HNH, GIY-YIG, or PLD/Nuc [review (24)]. Analysis of the predicted structures and sequence conservation between PabI and jhp0455 suggests that the following residues of PAB0105 may be important for catalysis and/or DNA binding: K30, R32, K34, E63, Q65, E77, R117, K152, Q155, Q161, E199, K202, H211 and D214.
We sought to predict the 3D fold of PabI by using the fold-recognition approach [review (13)]. However, it failed to reveal any reliable matching to any known protein structures. Moreover, preliminary de novo folding simulations (data not shown) revealed a two-domain structure that did not resemble any of the known restriction endonuclease structures. Thus, PabI is an interesting candidate for experimental structure determination, as it may exhibit a novel tertiary fold or a very unusual variation of one of the known folds.
Search for restriction enzyme genes by systematic genome sequence comparison of related bacterial species within the same genus
Restriction genes are specific to particular strains of the same species of bacteria. Sequencing revealed that their chromosomal loci are not occupied by them in the other strains (25–28). Systematic comparison of many restriction–modification systems between two strains of H.pylori with known genome sequences revealed that strain-specific restriction genes tend to be active (12). Genome-wide comparison of these two genomes revealed their linkage with genome rearrangements (6).
Our results in this paper further demonstrate that the screening for restriction enzymes can be greatly facilitated by systematic comparison of genome sequences of less related bacteria—between different species within a genus.
The success of the genome comparison method we used is probably due to the nature of the restriction–modification gene complexes as mobile genetic elements. Since early genome comparisons revealed restriction–modification gene complexes are associated with large-scale genome polymorphisms (3,5,6,12), we used large-scale genomic rearrangements in the vicinity of a putative restriction–modification system as a screening criterion.
Horizontal transfer of PabI restriction–modification system
Indeed, our results further support the concept that restriction–modification gene complexes act as mobile genetic elements in a broad sense (3) because comparison with P.horikoshii suggested the region containing the PabI restriction gene and its putative cognate modification gene, PAB2246, appears to be inserted into the P.abyssi genome (7). The GC content of the inserted region is lower than that of the rest of the P.abyssi genome (7).
The Helicobacter homologs of PabI (jhp0455 in J99 and HP0505/HP0504 in 26695) are linked with a homolog of this putative M.PabI (jhp0454 and HP0502) (29). These gene complexes are probably related to PabI/PAB2246 restriction–modification system in evolution through vertical and horizontal transfer (3,4).
They might also recognize (or have recognized) 5′-GTAC as PabI. This sequence is much less abundant in H.pylori genomes than expected (REBASE, http://tools.neb.com/~vincze/cutfreq/GTAC.html). Such restriction avoidance in these genomes might reflect selection by attack by these restriction–modification systems as proposed for other cases (30,31).
SUMMARY
The restriction endonucleases in the genomes of P.abyssi and P.horikoshii were screened by a combination of genome comparison and the use of a plant-based uncoupled in vitro protein synthesis system. One of the restriction endonucleases that were identified was PabI, a novel thermophilic restriction enzyme that cleaves 5′-GTAC and leaves 3′-TA overhangs. Since restriction enzymes that leave such an end have not yet been reported, this enzyme may be useful in biotechnology. This novel screening procedure would be useful in the search for additional novel restriction enzymes, deoxyribonucleases and other such cytotoxic proteins in general.
Acknowledgments
We thank Dr Yoshizumi Ishino for providing the P.abyssi genomic DNA, Dr Keiko Kita and Dr Yoshizumi Ishino for advice in biochemical experiments, and Dr Naofumi Handa and Dr Marat R. Sadykov for comments on a version of the manuscript. We are also grateful to Dr Asao Ichige for suggestion about the restriction avoidance in H.pylori. This work was supported by Japanese national project PROTEIN 3000 and a grant-in-aid by MEXT (Genome Homeostasis, Kiban). The work of J.M.B. was supported by the Polish Ministry of Scientific Research and Information Technology and by the Young Investigator award from EMBO and HHMI. Funding to pay the Open Access publication charges for this article was provided by a grant from MEXT.
Conflict of interest statement. None declared.
REFERENCES
- 1.Roberts R.J. Restriction enzymes and their isoschizomers. Nucleic Acids Res. 1990;18:2331–2365. doi: 10.1093/nar/18.suppl.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Naito T., Kusano K., Kobayashi I. Selfish behavior of restriction–modification systems. Science. 1995;267:897–899. doi: 10.1126/science.7846533. [DOI] [PubMed] [Google Scholar]
- 3.Kobayashi I. Behavior of restriction–modification systems as selfish mobile elements and their impact on genome evolution. Nucleic Acids Res. 2001;29:3742–3756. doi: 10.1093/nar/29.18.3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kobayashi I. Restriction–modification systems as minimal forms of life. In: Pingoud A., editor. Restriction Endonucleases. Berlin: Springer-Verlag; 2004. pp. 19–62. [Google Scholar]
- 5.Nobusato A., Uchiyama I., Ohashi S., Kobayashi I. Insertion with long target duplication: a mechanism for gene mobility suggested from comparison of two related bacterial genomes. Gene. 2000;259:99–108. doi: 10.1016/s0378-1119(00)00456-x. [DOI] [PubMed] [Google Scholar]
- 6.Alm R.A., Ling L.S., Moir D.T., King B.L., Brown E.D., Doig P.C., Smith D.R., Noonan B., Guild B.C., de Jonge B.L., et al. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature. 1999;397:176–180. doi: 10.1038/16495. [DOI] [PubMed] [Google Scholar]
- 7.Chinen A., Uchiyama I., Kobayashi I. Comparison between Pyrococcus horikoshii and Pyrococcus abyssi genome sequences reveals linkage of restriction–modification genes with large genome polymorphisms. Gene. 2000;259:109–121. doi: 10.1016/s0378-1119(00)00459-5. [DOI] [PubMed] [Google Scholar]
- 8.Handa N., Nakayama Y., Sadykov M., Kobayashi I. Experimental genome evolution: large-scale genome rearrangements associated with resistance to replacement of a chromosomal restriction–modification gene complex. Mol. Microbiol. 2001;40:932–940. doi: 10.1046/j.1365-2958.2001.02436.x. [DOI] [PubMed] [Google Scholar]
- 9.Sadykov M., Asami Y., Niki H., Handa N., Itaya M., Tanokura M., Kobayashi I. Multiplication of a restriction–modification gene complex. Mol. Microbiol. 2003;48:417–427. doi: 10.1046/j.1365-2958.2003.03464.x. [DOI] [PubMed] [Google Scholar]
- 10.Schildkraut I.S. Screening for and characterizing restriction endonucleases. In: Setlow J.K., Hollaender A., editors. Genetic Engineering, Principles and Methods. Vol. 6. NY: Plenum Press; 1984. pp. 117–140. [Google Scholar]
- 11.Kong H., Lin L.F., Porter N., Stickel S., Byrd D., Posfai J., Roberts R.J. Functional analysis of putative restriction–modification system genes in the Helicobacter pylori J99 genome. Nucleic Acids Res. 2000;28:3216–3223. doi: 10.1093/nar/28.17.3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin L.F., Posfai J., Roberts R.J., Kong H. Comparative genomics of the restriction–modification systems in Helicobacter pylori. Proc. Natl Acad. Sci. USA. 2001;98:2740–2745. doi: 10.1073/pnas.051612298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bujnicki J.M. Crystallographic and bioinformatic studies on restriction endonucleases: inference of evolutionary relationships in the ‘midnight zone’ of homology. Curr. Protein Pept. Sci. 2003;4:327–337. doi: 10.2174/1389203033487072. [DOI] [PubMed] [Google Scholar]
- 14.Sawasaki T., Hasegawa Y., Tsuchimochi M., Kamura N., Ogasawara T., Kuroita T., Endo Y. A bilayer cell-free protein synthesis system for high-throughput screening of gene products. FEBS Lett. 2002;514:102–105. doi: 10.1016/s0014-5793(02)02329-3. [DOI] [PubMed] [Google Scholar]
- 15.Komori K., Fujita N., Ichiyanagi K., Shinagawa H., Morikawa K., Ishino Y. PI-PfuI and PI-PfuII, intein-coded homing endonucleases from Pyrococcus furiosus. I. Purification and identification of the homing-type endonuclease activities. Nucleic Acids Res. 1999;27:4167–4174. doi: 10.1093/nar/27.21.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kurowski M.A., Bujnicki J.M. GeneSilico protein structure prediction meta-server. Nucleic Acids Res. 2003;31:3305–3307. doi: 10.1093/nar/gkg557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei A., Yuan A., Fawcett G., Butler A., Davis T., Xu S.Y., Salkoff L. Efficient isolation of targeted Caenorhabditis elegans deletion strains using highly thermostable restriction endonucleases and PCR. Nucleic Acids Res. 2002;30:e110. doi: 10.1093/nar/gnf109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roberts R.J., Belfort M., Bestor T., Bhagwat A.S., Bickle T.A., Bitinaite J., Blumenthal R.M., Degtyarev S., Dryden D.T., Dybvig K., et al. A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res. 2003;31:1805–1812. doi: 10.1093/nar/gkg274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lynn S.P., Cohen L.K., Kaplan S., Gardner J.F. RsaI: a new sequence-specific endonuclease activity from Rhodopseudomonas sphaeroides. J. Bacteriol. 1980;142:380–383. doi: 10.1128/jb.142.2.380-383.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gallie D.R., Sleat D.E., Watts J.W., Turner P.C., Wilson T.M. A comparison of eukaryotic viral 5′-leader sequences as enhancers of mRNA expression in vivo. Nucleic Acids Res. 1987;15:8693–8711. doi: 10.1093/nar/15.21.8693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sawasaki T., Ogasawara T., Morishita R., Endo Y. A cell-free protein synthesis system for high-throughput proteomics. Proc. Natl Acad. Sci. USA. 2002;99:14652–14657. doi: 10.1073/pnas.232580399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen H.Z., Zubay G. Prokaryotic coupled transcription-translation. Methods Enzymol. 1983;101:674–690. doi: 10.1016/0076-6879(83)01047-2. [DOI] [PubMed] [Google Scholar]
- 23.Bujnicki J.M., Rychlewski L. Grouping together highly diverged PD-(D/E)XK nucleases and identification of novel superfamily members using structure-guided alignment of sequence profiles. J. Mol. Microbiol. Biotechnol. 2001;3:69–72. [PubMed] [Google Scholar]
- 24.Bujnicki J.M. Understanding the evolution of restriction–modification systems: clues from sequence and structure comparisons. Acta Biochim. Pol. 2001;48:935–967. [PubMed] [Google Scholar]
- 25.Lin T.L., Shun C.T., Chang K.C., Wang J.T. Isolation and characterization of a HpyC1I restriction–modification system in Helicobacter pylori. J. Biol. Chem. 2004;279:11156–11162. doi: 10.1074/jbc.M311639200. [DOI] [PubMed] [Google Scholar]
- 26.Xu Q., Stickel S., Roberts R.J., Blaser M.J., Morgan R.D. Purification of the novel endonuclease, Hpy188I, and cloning of its restriction–modification genes reveal evidence of its horizontal transfer to the Helicobacter pylori genome. J. Biol. Chem. 2000;275:17086–17093. doi: 10.1074/jbc.M910303199. [DOI] [PubMed] [Google Scholar]
- 27.Xu Q., Morgan R.D., Roberts R.J., Blaser M.J. Identification of type II restriction and modification systems in Helicobacter pylori reveals their substantial diversity among strains. Proc. Natl Acad. Sci. USA. 2000;97:9671–9676. doi: 10.1073/pnas.97.17.9671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sekizaki T., Otani Y., Osaki M., Takamatsu D., Shimoji Y. Evidence for horizontal transfer of SsuDAT1I restriction–modification genes to the Streptococcus suis genome. J. Bacteriol. 2001;183:500–511. doi: 10.1128/JB.183.2.500-511.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nobusato A., Uchiyama I., Kobayashi I. Diversity of restriction–modification gene homologues in Helicobacter pylori. Gene. 2000;259:89–98. doi: 10.1016/s0378-1119(00)00455-8. [DOI] [PubMed] [Google Scholar]
- 30.Gelfand M.S., Koonin E.V. Avoidance of palindromic words in bacterial and archaeal genomes: a close connection with restriction enzymes. Nucleic Acids Res. 1997;25:2430–2439. doi: 10.1093/nar/25.12.2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rocha E.P., Danchin A., Viari A. Evolutionary role of restriction/modification systems as revealed by comparative genome analysis. Genome Res. 2001;11:946–958. doi: 10.1101/gr.gr-1531rr. [DOI] [PubMed] [Google Scholar]
- 32.Uchiyama I., Higuchi T., Kobayashi I. CGAT: Comparative genome analysis tool for closely related microbial genomes. In: Dunker A., Konagaya S., Miyano T. Takagi, editors. Genome Informatics 2000. Tokyo, Japan: Universal Academy Press; 2000. pp. 341–343. [Google Scholar]