

Abstract
DYSMORPHOLOGY IS THE BRANCH OF CLINICAL GENETICS in which clinicians and researchers study and attempt to interpret the patterns of human growth and structural defects. Reaching an accurate diagnosis for children with dysmorphic signs is important to their families, because it makes available all the accumulated knowledge about the relevant condition and may provide the family with the opportunity for interaction with patient or parent support groups. I show in this review that reaching a diagnosis in dysmorphology involves an approach that is not fundamentally different from that of other medical disciplines. Cytogenetic and molecular techniques continue to improve our ability to make precise syndrome diagnoses; however, these tests are expensive and should be used selectively.
Case 1
This boy was born at 38 weeks' gestation weighing 2750 g (> 10th percentile). Initial feeding problems improved by 7 months, and he became quite a voracious eater by the age of 6 years. His motor and language development were moderately delayed. At the age of 10 years, a cranial CT scan showed mild bilateral frontal “atrophy.” A G-banded (Giemsa-stained) chromosome study showed a lengthened short (i.e., “p”) arm of chromosome 14 (14p+), which was interpreted as being the common normal variant that is due to variable amounts of repetitive DNA in that region. Neither parent had a 14p+ “variant” and the developmental delay was unexplained. At 12 years, this boy was referred to a genetics clinic. He was at the 90th percentile for height and had a head circumference and weight that were above the 97th percentile. Bifrontal narrowing, a slight upslant to the palpebrae (eyelids), which were of normal length, and some Brushfield spots (small white spots on the periphery of the iris) were noted. His mouth was small, and his hands were at the 60th percentile for length.
Case 2
This girl was born at term weighing 2280 g (3rd–10th percentile). The family history was noncontributory. There were early feeding difficulties and marked growth and developmental delay. Chromosome studies were done twice, because this girl's appearance was reminiscent of that typical of Down's syndrome. At 4 years, she was diagnosed as having Williams1 syndrome, but at the age of 13 years this diagnosis was ruled out using a fluorescence in situ hybridization (FISH) probe specific for the elastin gene on chromosome 7, which showed that the gene was not missing from one of the chromosomes as is seen to occur in Williams syndrome. The family moved and requested an assessment at the local genetics clinic. At the age of 13 years, this girl's history featured the recent onset of seizures, long-standing behaviour problems including stubbornness, impulsiveness, violent temper outbursts and self-abuse, and a disturbed sleep pattern. She also had a history of putting objects in orifices. At 13 years, her height was that of an 8 year old, and her head circumference was much lower than the third percentile. Physical signs included mild brachycephaly, malar hypoplasia, slightly upslanting palpebral fissures and scooped nasal shape, with the philtral area running parallel to the angle of the nose. The upper lip was thin and the mouth downturned. The lower lip was everted and the chin small. Her chest was barrel-shaped with hypoplastic nipples. Her hands were small and blunt.
The term dysmorphic is derived from the Greek words “dys” (disordered, abnormal, painful) and “morph” (shape, form). Dysmorphology is a discipline of clinical genetics that studies and attempts to interpret the patterns of human growth and structural defects. These include malformation (an intrinsic developmental anomaly, e.g., spina bifida), disruption (an event disrupting intrinsically normal development, e.g., amniotic bands), deformation (an external force altering the shape of development, e.g., face shape due to severe oligohydramnios) and dysplasia (abnormal growth and maturation of cells, e.g., achondroplasia). However, the child with dysmorphic signs often does not have a major malformation, and he or she may simply have an appearance that is unusual compared with the general population and out of keeping with that of unaffected close relatives. A syndrome is simply a recognizable pattern of dysmorphic signs that have a common cause.
Why strive for a diagnosis for a child with dysmorphic signs?
Many children who are dysmorphic have significant internal or external malformations or developmental delay, or some combination of these. The inability to provide a definitive diagnosis and thus a presumptive cause, even when having a diagnosis does not imply that there will be a therapeutic intervention, can greatly frustrate a family. A precise diagnosis makes available all the accumulated knowledge and experience of that condition and generally provides a better estimate of the risk of recurrence. It informs prognosis and permits interventions that may prevent, anticipate or more successfully treat complications.2 In many jurisdictions it facilitates getting support, such as financial and educational aid, and it allows families to interact with specific support groups. Furthermore, an accurate diagnosis is key to research into the identification of causative genes, interventions and treatments.
How does one approach a diagnosis in dysmorphology?
The approach in dysmorphology is not fundamentally different from that of other medical disciplines. An enquiry into the family history should extend beyond the findings in the immediate patient to look for partial signs of a syndrome or other malformations that could result from different unbalanced products of a familial chromosome rearrangement. The 3-generation pedigree should include a careful search for consanguinity. The pregnancy history should include details of previous losses, pregnancy planning, parental occupation and health at the time of conception, exposure to drugs, medications and alcohol, and evidence of maternal fever, rash or illness, and details of fetal movement. A general question about unusual or untoward events during the pregnancy will often uncover parental beliefs about causation, and it is important to consider these during counselling. Events surrounding the delivery, including evidence of fetal distress, hydramnios or oligohydramnios, birth weight, length and head circumference, and perinatal behaviour of the child may be important and, if thought to be significant, original records should be sought. A child with dysmorphic signs may be at risk from the stress of birth, and later delay may be erroneously attributed to birth injury.3 A careful developmental history with an emphasis on milestones, formal assessments and behaviour is also required. Medical records should be sought to validate any diagnosis or treatment of a malformation.
The role of the dysmorphologist
A dysmorphologist will conduct a complete physical examination that includes all the body systems. Although major malformations will be noted, a great deal of the emphasis is devoted to the overall appearance (gestalt) and the presence of any minor anomalies (Table 1), defined as physical variations that occur in less than 5% of the population but are of no clinical significance, in the face (e.g., upslanting palpebrae, unusual ear helix, anteverted nares [nostrils]) and other areas including the hands, genitalia and skin. Where possible, subjective observation should be supplemented by objective measurement and photographic records.
Table 1
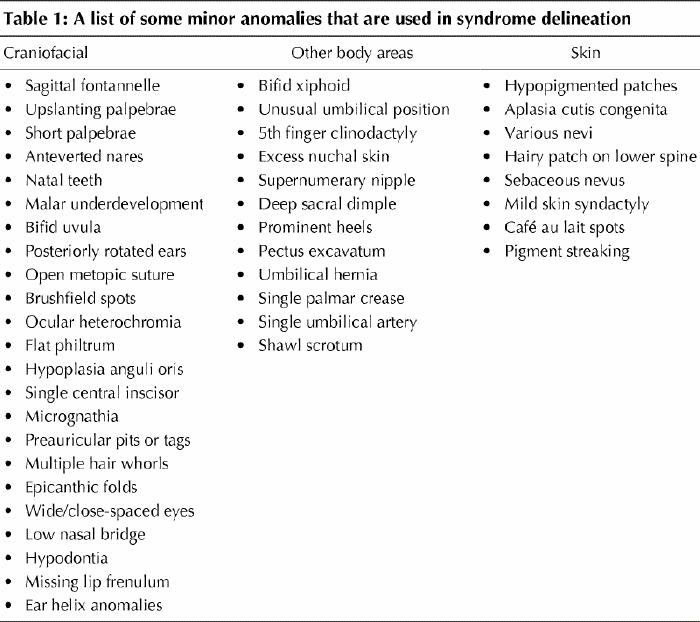
Most syndrome diagnoses are suggested by the gestalt of the patient, in the same way that most people would recognize a child with Down's syndrome. However, the dysmorphologist has more interest in and experience of diagnosis and has honed his or her skills in pattern recognition. Uncommon single or combinations of anomalies can be used to direct a computer search. Notwithstanding an initial impression, the dysmorphologist performs a detailed examination and compares the observations and history with those expected in the syndrome being considered or with the lead suggested by the database. It is important not to rush too early to a diagnosis because, once applied to a patient's condition, labels are hard to remove. Concerns about the subjectivity of some syndrome diagnoses, especially at the mild end of a syndrome's spectrum, have led some dysmorphologists to explore more objective diagnostic approaches such as photogrammetry and anthropometrics.4 Photogrammetry uses objective measurements from standardized photographs, and anthropometry from standardized physical landmarks, to assess patients objectively.
As is the case for the general population, the facial appearance of an individual with a syndrome is expected to change with growth and maturation. Sometimes the facial appearance will become more characteristic; in others it becomes less apparent with age (e.g., fetal alcohol syndrome, Beckwith-Wiedemann syndrome), in which case photographs from infancy or early childhood of the patient and parents may be helpful. Because of possible changes through time, and the continued description of new syndromes, follow-up of patients, especially newborns and young infants, may ultimately result in a diagnosis.
Unfortunately, for many individuals with malformations and/or an unusual appearance, with or without developmental delay, the history and examination do not suggest an immediate diagnosis. If there are objective (e.g., absent thumb) and not overly common (e.g., developmental delay is common and nonspecific) findings, the dysmorphologist can use computer software to elucidate possible diagnoses from the over 3000 reported syndromes.5,6 Often, photographs are available for comparison with the patient. However, these programs are systems for experts and not expert systems.6 Several books describe many of the best-defined syndromes.1,7,8 Dysmorphologists will often circulate photographs of their undiagnosed patients to colleagues, and telemedicine will likely expand this approach. A challenge for the dysmorphologist is the large number of single-family reports of “syndromes” consisting of developmental delay and a “characteristic,” but in fact very subtle, difference in appearance. Some may simply represent nonspecific developmental delay combined with family traits rather than a distinct syndrome. It should be noted that formerly used pejorative descriptors such as gargoylism, FLK, happy puppet and elfin face are unacceptable today.
Referral to a dysmorphologist for assessment
Most dysmorphologists are medical geneticists, increasingly with certification from the Royal College of Physicians and Surgeons of Canada, who are attached to genetics programs in major centres. The proportion of their time spent in dysmorphology will vary with the size of the centre. It is important for the general practitioner to inform the patient and family, and the genetics clinic, why the referral is being made (e.g., for diagnosis, for reproductive counselling, a suspected syndrome). Many genetic centres will send out a family history questionnaire and seek medical records before seeing a patient, and the process can be expedited if the relevant history and test results are included with the referral. When and whether to make a referral will depend upon the physician's own comfort in assessing a patient with developmental delay, dysmorphic signs and/or with a deteriorating medical condition, and whether there is a question to be answered or a problem to be solved. If a person looks somewhat unusual, but is otherwise completely healthy and developmentally normal, there is generally no reason to pursue matters further. A dysmorphologist is very unlikely to make a diagnosis9 in a developmentally delayed person unless there are some dysmorphic signs, but even in the absence of a diagnosis the medical geneticist may be able to answer a family's concern regarding future pregnancies by using empiric recurrence data.
How is the laboratory helpful in dysmorphology?
Cytogenetics
Cytogenetics is a mainstay of diagnosis in dysmorphology. However, chromosome studies are labour intensive and relatively expensive (> $400). Referral to a dysmorphologist may lead to the diagnosis of a single gene disorder or suggest a syndrome with a known microdeletion, that is, a chromosome deletion not visible by standard G-banding, thus obviating the need for a standard karyotype. To be visible, a chromosome deletion or duplication probably involves at least 3–4 kilobases of DNA10 (perhaps 15–30 genes, depending upon the location and the chromosome). Given the high proportion of genes involved in the development and functioning of the brain, major malformations in a developmentally normal child are not expected to have a cytogenetic cause. I have shown that a standard karyotype is very unlikely to be informative in a developmentally delayed child who is found by a dysmorphologist to lack dysmorphic signs. Indeed, the yield of abnormal chromosome results is proportional to the number of dysmorphic signs.9
Unless a nonchromosomal diagnosis is apparent, a neonate or young infant with an unusual appearance or major malformations, or both, should have a standard chromosome study. In the older infant or child, generally, one would also require some developmental delay. The earlier in the metaphase that chromosomes are examined, the longer and less condensed they will be, revealing more bands and theoretically allowing detection of smaller abnormalities. A repeat karyotype may be justified in a patient who has signs suggestive of a chromosome abnormality, if a negative karyotype was reported for fewer than 550–650 bands.
Fluorescence in situ hybridization (FISH) has caused a rebirth in cytogenetics, and 3 approaches or techniques are commonly applied in dysmorphology. First, probes that are specific to the locus, that is, the physical position of a gene on the chromosome, can bind to a segment of DNA, too small to be visible by light microscopy, on a specific chromosome. If the segment of DNA is deleted, the FISH probe will not bind, thus demonstrating the deletion. Hence the term microdeletion syndrome that is applied to a growing list of syndromes, including Prader-Willi, Angelman, Smith-Magenis, Miller-Dieker and velo-cardio-facial/DiGeorge, now often called del(22q11), several of which are quite common.1 In some syndromes, cases may result from either FISH-detectable deletions or point mutations (e.g., Alagille, Angelman). These tests should be requested because of clinical signs (dysmorphisms/behaviour) suggestive of the specific syndrome, and when used in this way the diagnostic yield can be high.9
Second, in whole chromosome painting (WCP), FISH probes are specific to a complete individual chromosome, rather than a single locus, and will paint the entire chromosome. Different approaches include combining fluorescent dyes to give each chromosome pair plus the X and Y chromosomes a different colour on a single metaphase spread and examining each chromosome separately, or a restricted group of chromosomes, in individual wells on a single slide. WCP is very useful for identifying the origin of additional chromosome material that is microscopically visible but not distinctive enough to be assigned to a specific chromosome. It can also be used to search for light microscopically invisible (cryptic) translocations where suspicion of a chromosome abnormality remains, despite a normal standard karyotype. The exchange of similarly sized and banded material between 2 chromosomes, which is not visible in a standard black and white G-banded study, becomes visible because of the exchange of different colours.
Third, an exciting new development is that of FISH probes specific to the subtelomeric region of the short (“p”) and long (“q”) arm of each chromosome, the telomeres being the ends of the chromosomes. The subtelomeric regions of chromosomes are gene rich, subject to frequent breaks and exchanges, and are notoriously difficult to see on a standard G-banded karyotype (chromosome study). The indications for the use of this technology are still evolving, but it appears that up to 8% of children with dysmorphic signs and moderate-to-severe delay will have a subtelomeric abnormality that was not apparent on a standard karyotype.11 Subtelomeric probes are likely to be superior to WCP techniques for uncovering cryptic translocations.12 Syndromes originally described as single gene disorders have now been shown to be the result of subtelomeric chromosome changes.13,14 WCP and subtelomeric FISH are expensive technologies that currently should be used at the discretion of dysmorphologists in consultation with a cytogeneticist.
Molecular (DNA) diagnostics
Technologies and information from the human genome project have accelerated the discovery of genes responsible for many genetic syndromes. Increasingly, mutation analysis can confirm a suspected syndrome diagnosis. At present, this is most practical for small genes and/or those that show recurrent mutations at a small number of specific codons (a codon is one of 61 triplet combinations of DNA bases that specifies one of the 20 amino acids or one of the 3 nonsense [stop] codons). For larger genes, and those with mutations that occur at many different sites, current technology remains slow, labour intensive, expensive and unable to find all mutations. This situation will improve with the further development of computer-driven chips and microarrays.15 Availability is not a good reason to order mutation analysis. An unambiguous clinical diagnosis does not require molecular confirmation. Molecular diagnosis may be indicated if the parents are at risk of having a second affected child and plan prenatal diagnosis, where there is some doubt as to the diagnosis, or in cases where predictive testing for a familial genetic disease may be of benefit. In all cases, the practitioner must be aware of the limitations of the testing, such as causal heterogeneity of a syndrome or the frequent inability to find all mutations in a gene, and the broader implications surrounding DNA diagnostics.16
Biochemical laboratory testing
The traditional association between the biochemical laboratory and dysmorphology has occurred with the storage disorders such as the mucopolysaccharidoses (e.g., Hurler syndrome). However, there is a growing list of inherited biochemical diseases that are associated with dysmorphic signs. Thus, biochemical genetic disease should not be dismissed simply because of the presence of malformations or a dysmorphic appearance. Examples include disorders of peroxisomes (Zellweger syndrome, chondrodysplasia punctata), a number of abnormalities in the energy pathways that are associated with brain cell migrational defects17 and disorders of cholesterol metabolism (Smith-Lemli-Opitz syndrome).18
The cases revisited
Case 1
The eating history, bifrontal narrowness and small mouth raise the possibility of Prader-Willi syndrome, but the height, head circumference and hand size are against the diagnosis. Although Brushfield spots are associated with Down's syndrome, they are common in the general population, there were no other findings of Down's syndrome mentioned and the G-banded karyotype was normal. The clue was the initial karyotype that was reported as being normal, but with a 14p+ variant not found in the parents. True variants tend to be stable and transmitted from a parent. The geneticist ordered a repeat karyotype. The 14p+ was not typical of the usual normal variant; whole chromosome painting was requested and showed that the large “p” arm of chromosome 14 was caused by translocated material from chromosome 20 (Fig. 1A). Specific subtelomeric probes showed its origin was the short arm of chromosome 20 (20p) (Fig. 1B), and the child was thus trisomic for part of chromosome 20p, which accounted for his mild dysmorphic signs and moderate developmental delay. The parents were relieved to finally have an explanation and the extended family was informed that other family members were not at risk of having similarly affected children.
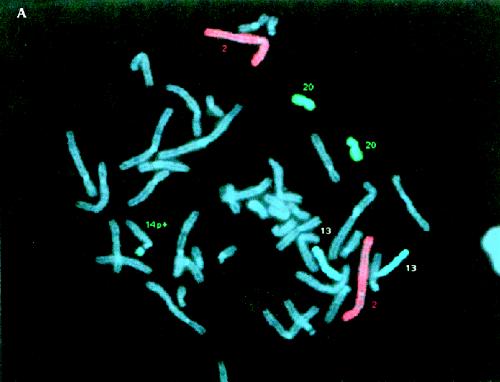
Fig. 1: (A) Partial fluorescence in situ hybridization (FISH) metaphase from patient 1 showing the differentially coloured chromosomes 20 (turquoise) with a similar coloured segment attached to the chromosome 14 labelled 14p+. (B) Partial FISH metaphase from patient 1 showing the chromosomes 20 with their short (p) arms labelled red, with a similar coloured segment attached to the chromosome 14 labelled 14p+.
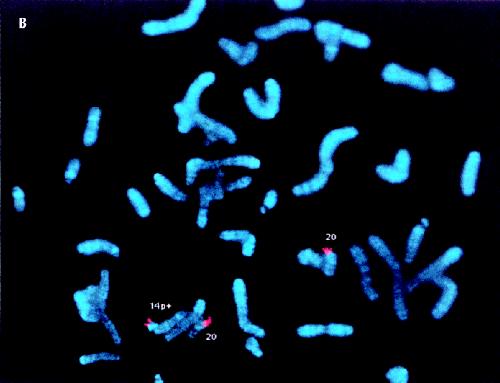
Fig. 1: (A) Partial fluorescence in situ hybridization (FISH) metaphase from patient 1 showing the differentially coloured chromosomes 20 (turquoise) with a similar coloured segment attached to the chromosome 14 labelled 14p+. (B) Partial FISH metaphase from patient 1 showing the chromosomes 20 with their short (p) arms labelled red, with a similar coloured segment attached to the chromosome 14 labelled 14p+.
Case 2
The geneticist had not seen a patient with Smith- Magenis syndrome, but the behavioural pattern and subtle facial features (Fig. 2) in this 13-year-old girl suggested the diagnosis. This chromosome microdeletion syndrome was confirmed by FISH studies with a probe for 17p11.2 (Fig. 3) that showed this region to be missing from one chromosome 17. The diagnosis allows the possibility of providing a more specific intervention. In the younger child, this includes an intensive program of speech and signing, early intervention to manage maladaptive behaviours, and help with feeding and swallowing difficulties.19 Later education can focus on relative strengths such as receptive language, long-term memory and interest in computers, while working around major deficits in short-term memory and sequential processing. Furthermore, there is growing evidence that melatonin and β1-adrenergic antagonists may aid sleep by altering the abnormal circadian melatonin secretion.19,20

Fig. 2: Partial lateral photograph of patient 2 showing the underdeveloped malar region, the philtrum running parallel to the nose and an everted lower lip, which are all characteristic of Smith-Magenis syndrome.
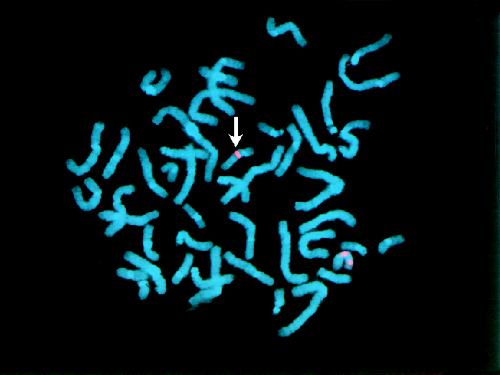
Fig. 3: Partial FISH metaphase from patient 2 showing the 2 chromosome 17s highlighted by a pink fluorescent stain. The chromosome marked with the white arrow is missing the stain for the Smith-Magenis locus and has only the control fluorescence. The normal chromosome has 2 fluorescent points.
Articles to date in this series .
Polifka JE, Friedman JM. Medical genetics: 1. Clinical teratology in the age of genomics. CMAJ 2002;167(3):265-73.
Additional resources .
The following resources, which are included in the reference list, are excellent general texts to aid in the diagnosis of syndromes and to provide some basic information on each.
Gorlin RJ, Cohen MM Jr, Hennekam RCM, editors. Syndromes of the head and neck. 4th ed. New York: Oxford University Press; 2001.
Winter RB, Donnai D, editors. Congenital malformation syndromes. London: Chapman & Hall Medical; 1995.
Hunter AGW. Outcome of the routine assessment of patients with mental retardation in a genetics clinic. Am J Med Genet 2000;90:60-8.
The following reference places a useful emphasis upon the management of some of the more common syndromes.
Cassidy SB, Allanson JE, editors. Management of genetic syndromes. New York: John Wiley & Sons; 2001.
Web site resources
· Online Mendelian Inheritance in Man (OMIM) provides a catalogue of human single gene disorders: www3.ncbi.nlm.gov/Omim/
· GeneTests·GeneClinics provides excellent reviews of a broad range of genetic diseases and syndromes: www.geneclinics.org
· National Organization for Rare Diseases (NORD) provides information and links to support groups: www.rarediseases.org
β See related article page 373
Footnotes
This article has been peer reviewed.
Competing interests: None declared.
Correspondence to: Dr. Alasdair Hunter, Eastern Ontario Genetics Program, Children's Hospital of Eastern Ontario, 401 Smyth Rd., Ottawa ON K1H 8L1; fax 613 738-4822; heddalasdair@aol.com
References
- 1.Jones KL, editor. Smith's recognizable patterns of human malformation. 5th ed. Philadelphia: WB Saunders; 1997.
- 2.Cassidy SB, Allanson JE, editors. Management of genetic syndromes. New York: John Wiley & Sons; 2001.
- 3.Hall DMB. Birth asphyxia and cerebral palsy: birth asphyxia is hard to define but is rarely the cause of cerebral palsy. BMJ 1989;299:279-82. [DOI] [PMC free article] [PubMed]
- 4.Allanson JE. Objective techniques for craniofacial assessment: What are the choices? Am J Med Genet 1997;70:1-5. [DOI] [PubMed]
- 5.POSSUM. Version 5.2. Melbourne (Australia): Murdoch Institute; 2000.
- 6.Winter RM, Baraitser M. Dysmorphology photo library on CD-ROM. Version 2.2. Oxford Medical Databases. Oxford: Oxford University Press; 2000.
- 7.Gorlin RJ, Cohen MM Jr, Hennekam RCM, editors. Syndromes of the head and neck. 4th ed. New York: Oxford University Press; 2001.
- 8.Winter RB, Donnai D, editors. Congenital malformation syndromes. London: Chapman & Hall Medical; 1995.
- 9.Hunter AGW. Outcome of the routine assessment of patients with mental retardation in a genetics clinic. Am J Med Genet 2000;90:60-8. [DOI] [PubMed]
- 10.Gardner RJM, Sutherland GR, editors. Chromosome abnormalities in genetic counselling. 2nd ed. New York: Oxford University Press; 1996. p. 269.
- 11.Knight SJL, Regan R, Nicod A, Horsley SW, Kearney L, Homfray T, et al. Subtle chromosomal rearangements in children with unexplained mental retardation. Lancet 1999;354:1676-81. [DOI] [PubMed]
- 12.Schaffer LG, Kashork CD, Bejjani BA. What FISH methodology should I order? [lecture]. Symposium on Cytogenetic Diagnostic Dilemmas, annual clinical genetics meeting of the American Board of Medical Genetics; 2001 Mar 1-4; Miami.
- 13.Herens C, Jamar M, Alvarez-Gonzales ML, Lesenfants S, Lombet J, Bonnivert J, et al. Private multiple congenital anomaly syndromes may result from unbalanced subtle translocations: t(2q;4p) explains the Lambotte syndrome. Am J Med Genet 1997;73:127-31. [DOI] [PubMed]
- 14.Verloes A, Lesenfants S, Jamar M, Dideberg V, Herens C. GOMBO syndrome: another “pseudorecessive” disorder due to a cryptic translocation. Am J Med Genet 2000;95:185-6. [PubMed]
- 15.Sinclair A. Genetics 101: detecting mutations in human genes. CMAJ 2002; 167(3):275-9. [PMC free article] [PubMed]
- 16.Baird PA. Identifying people's genes: ethical aspects of DNA sampling in populations. Perspect Biol Med 1995;38:159-66. [DOI] [PubMed]
- 17.Hunter AGW. The brain. In: Stevenson RE, Hall JG, Goodman RM, editors. Human malformations and related anomalies. Vol 2. New York: Oxford University Press; 1993. p. 38-51.
- 18.Nowaczyk MJM, Whelan DT, Heshka T, Hill RE. Smith-Lemli-Opitz syndrome: a treatable inherited error of metabolism causing mental retardation. CMAJ 1999;161(2):165-70. [PMC free article] [PubMed]
- 19.Smith ACM, Gropman A. Smith-Magenis syndrome. In: Cassidy SB, Allanson JE, editors. Management of genetic syndromes. New York: John Wiley & Sons; 2001. p. 363-87.
- 20.De Leersnyder H, de Blois MC, Vekemans M, Sidi D, Villain E, Kindermans C, et al. β1-adrenergic antagonists improve sleep and behavioural disturbances in a circadian disorder, Smith-Magenis syndrome. J Med Genet 2001; 38:586-90. [DOI] [PMC free article] [PubMed]