

Abstract
Fluvoxamine (FXM) is a well-known selective serotonin reuptake inhibitor (SSRI) for treating depression and has recently been repurposed for efficacious treatment of coronavirus disease 2019. Although cyclodextrin (CD) encapsulation effectively improves the physicochemical properties of structurally diverse SSRIs, the molecular understanding of their associations is deficient. This comprehensive study used single-crystal X-ray diffraction integrated with density functional theory (DFT) calculation to provide deep insights into the conformationally flexible FXM and its inclusion complexation with β-CD. X-ray analysis revealed the first crystallographic evidence of the uncomplexed 3FXM–H+·3maleate− (1). Three FXM–H+ ions are counter-balanced by three planar maleate− ions to form a thin layer stabilized by infinite fused H-bond rings R44(12) and R64(16) and the interplay of π⋯π, CF⋯π and F⋯F interactions. For 2β-CD·2FXM–H+·maleate2−·23·2H2O (2), the tail-to-tail β-CD dimer encapsulates two FXM–H+ 4-(trifluoromethyl)phenyl moieties, which are charge-balanced by the rare non-planar maleate2− and stabilized by N/OH⋯O H-bonds and F⋯F interactions. This is a host–guest recognition pattern uniquely observed for all β-CD complexes with halogen (X)-bearing SSRIs, indicating the essence of X⋯X interactions and the shielding of X-containing moieties in the wall of the β-CD dimer. DFT calculations unveiled that the monomeric and dimeric β-CD–FXM complexes and FXM isomers are energetically stable, which alleviates the numbness and bitterness of the orally administered drug as previously patented. Additionally, an insightful conformational analysis of FXM emphasizes the importance of drug structural adaptation in pharmacological functions.
Keywords: Conformational flexibility, β-Cyclodextrin, Fluvoxamine maleate, SSRIs, X-ray analysis, DFT calculation
Graphical abstract

Highlights
-
•
First crystal structure of free fluvoxamine (FXM) maleate after the 40-year introduction.
-
•
Three FXM–H+ are counter-balanced by three planar maleate− in the uncomplexed form (1).
-
•
Two FXM–H+ B-rings enclosed in β-CD dimer are linked to the non-planar maleate2− (2).
-
•
Unique host–guest recognition pattern of all the 2:2 β-CD–SSRI inclusion complexes.
-
•
DFT calculations confirm the conformationally flexible and energetically stable FXM.
1. Introduction
Depression is a common mental health condition threatening several hundred million people worldwide according to estimates by the World Health Organization (WHO) in 2017 [1]. The WHO revealed that the coronavirus disease 2019 (COVID-19) pandemic caused a 25% global spike in anxiety and depression in 2020 [2]. In addition, 50% of patients who recovered from COVID-19 suffer from depression [3] and ∼10% of infected people (>65 million people worldwide) have post-acute sequelae of COVID-19 or long COVID [4]. Antidepressant drugs (ADDs) such as selective serotonin reuptake inhibitors (SSRIs) and tricyclic antidepressants (TCAs) have equivalent efficacy in treating depression as they increase the level of neurotransmitters (serotonin and/or norepinephrine) in the brain, although SSRIs have less side effects than TCAs [5]. The structurally diverse SSRIs including fluoxetine (FXT), sertraline (STL), paroxetine (PXT), citalopram (CTP), escitalopram (SCTP), and fluvoxamine (FXM) share a common selectivity mechanism for serotonin, whereas the structurally similar TCAs (e.g., amitriptyline, imipramine, and clomipramine (CPM)) exhibit weak inhibitors in the reuptake of both serotonin and norepinephrine [6].
The selectivity and potency of SSRIs for the uptake of serotonin (5-hydroxytryptamine (5-HT)) are mutually exclusive because of their different binding to the serotonin transporter (SERT). The SSRI selectivity order is SCTP > CTP > STL > FXM > PXT > FXT, whereas the SSRI potency order is PXT > SCTP ≈ STL > FXT > CTP > FXM [7]. Notably, FXM has an efficacy inferior to other SSRIs [8] and has more gastrointestinal side effects (e.g., vomiting/nausea, constipation, diarrhea) [9]. Conversely, accumulating evidence suggests that FXM is more promising in lowering both the severity of hospitalization [10] and the risk of long COVID [11]. FXM is the most effective repurposed ADD to treat COVID-19 because this controls inflammation through agonist activity at the sigma-1 receptor; thus reducing inflammation in COVID-19 [12]. FXM principally exists in two conformers, the pharmacologically active trans-isomer [(E)-isomer] and the inactive cis-isomer [(Z)-isomer]. Upon UV irradiation, the (E)-isomer undergoes photoisomerization to the (Z)-isomer [13]. The (E)-isomer is 2.40- and 1.80-kcal/mol more stable than the (Z)-isomer in gas and water phases, respectively [14] and the E → Z isomerization requires an activation energy of 55 kcal/mol [15], based on density functional theory (DFT) calculations.
Cyclodextrins (CDs) include α-, β-, and γ-CDs that contain 6, 7, and 8 d-glucose units respectively, and are effective encapsulating agents for various molecules, facilitating the improvement of molecular stability, water solubility and bioactivity (e.g., antioxidant property), and are therefore used in maintaining and enhancing the bioavailability of bioactive, pharmaceutical and food compounds. This is because of the remarkable inclusion property of the amphiphilic, toroidal-shaped CDs with a nonpolar nanocavity and hydrophilic rims. The plethora of literature on CD applications includes their uses in pharmacy, cosmetics, food, environment, and nanotechnology [16,17] (given here are examples of recent references). Over the past two decades, spectroscopic techniques, such as nuclear magnetic resonance (NMR), UV-visible, and fluorescence, have been used to investigate CD inclusion complexes with SSRIs mostly in aqueous solution at room temperature, providing complex stability constants and overall inclusion structures without atomic details. Recently, this research gap has been addressed and a thorough literature review has been completed by integrated studies of single-crystal X-ray diffraction and DFT calculation on β-CD inclusion complexes with STL, FXT, PXT, CTP, and SCTP (Fig. 1) [[18], [19], [20]]. Unlike the CD complexes with other SSRIs, the CD–FXM complexes are minimally understood. 1H NMR data indicate that both FXT HCl and FXM maleate contain a trifluoromethylbenzene (B-ring) embedded in the β-CD cavity [21]. A combined DFT and 19F NMR study suggests the unimolar complex with the FXM B-ring is encapsulated in the β-CD cavity [22]. FXM in complex with various CDs (α-, β-, and γ-CDs and hydroxypropyl-β-CD) has been formulated and orally administered to reduce side effects, i.e., alleviating the numbness and bitterness of FXM [23].
Fig. 1.

Chemical structures and atom numbering schemes of fluvoxamine (FXM; 2-[(E)-[5-methoxy-1-[4-(trifluoromethyl)phenyl]pentylidene]amino]oxyethanamine) and β-cyclodextrin (β-CD). Selective serotonin reuptake inhibitors (SSRIs) are commercially available in various salt forms, i.e., sertraline (STL) HCl, fluoxetine (FXT) HCl, paroxetine (PXT) HCl, citalopram (CTP) HBr, escitalopram (SCTP) oxalate, and FXM maleate. Clomipramine (CPM) HCl, a halogen-containing tricyclic antidepressant (TCA) commonly to SSRIs, is given for comparison. Different rings and chiral centers of SSRIs are marked. The SSRI selectivity-order is shown with orange arrowheads. The X-ray-derived inclusion modes of distinct antidepressant drug (ADD) moieties embedded with respect to O6–H and O2–H/O3–H sides of the β-CD cavity are highlighted in magenta [[18], [19], [20],39].
The atomic-level characteristics of β-CD complexes with the structurally dissimilar SSRIs (i.e., inclusion structures, host–guest stoichiometric ratios and intermolecular interactions) currently remain elusive. Therefore, we conducted a comprehensive structural investigation using X-ray diffraction combined with DFT calculation to validate the following three hypotheses. 1) Although FXM differs structurally from other SSRIs, the 4-(trifluoromethyl)phenyl moiety (B-ring) is expected to occupy the β-CD cavity; the unique inclusion mode with the aromatic ring-bearing halogen atoms has been crystallographically observed for previous β-CD–SSRI complexes [18,20] (see magenta highlights in Fig. 1). 2) Currently, FXM plausibly existing in the (E)- and (Z)-isomers has not been crystallized, either in the free or CD-bound form, and we envisage that the active (E)-FXM is being stabilized in the β-CD cavity, providing single crystals of the β-CD–(E)-FXM inclusion complex suitable for X-ray analysis. 3) Similar to other β-CD–SSRI complexes, β-CD–(E)-FXM is comparably energetically stable.
2. Materials and methods
2.1. Materials
FXM maleate (≥98%, code 46235) was supplied by Acros Organics (Geel, Belgium) (now Thermo Scientific Chemicals). β-CD (≥95%, code CY-2001) was purchased from Cyclolab (Budapest, Hungary). Absolute EtOH (≥99.8%) was provided by the Liquor Distillery Organization (Excise Department, Chachoengsao, Thailand). All chemicals were used as received. Ultrapure water was supplied by a Millipore Milli-Q Water Purification System (Millipore, Billerica, MA, USA).
2.2. Single-crystal structure determination
2.2.1. Crystallization
Powders of a 1:1 molar ratio mixture containing β-CD 50 mg (0.044 mmol) and FXM maleate 19 mg were dissolved in 1 mL of 50% (v/v) aqueous EtOH at 323 K. After two weeks of slow solvent evaporation in an air-conditioned room (298 K), single crystals of two different shapes, ribbons and rods, were harvested.
2.2.2. X-ray diffraction experiment
Briefly, two morphological crystals were formed, indicating the rare phenomenon of polymorphism in CD complexes. Therefore, several crystals of both habits were selected; each was mounted on a microloop (MiTeGen, NY, USA) and checked for the unit cell consistency and the scattering power for X-rays. Although the rod-like crystals were the desired inclusion complex of β-CD–FXM maleate (2) in the triclinic space group P1, the ribbon-like crystals were the uncomplexed FXM maleate (1) in the monoclinic space group C2/c (see the unit cell parameters below). Since the unit cell constants of 1 and 2 were different from those of various β-CD hydrates and relevant crystal data of FXM were not found in the Cambridge Crystallographic Data Center (CCDC), both 1 and 2 were new crystal structures. Diffraction data of 1 and 2 were collected at 296 K to 0.83 Å atomic resolution on a Bruker PROSPECTOR CCD area-detector diffractometer (Bruker AXS, Karlsruhe, Germany) with an IμS microfocus X-ray source, operating at 50 kV and 0.60 mA (CuKα radiation; λ = 1.54178 Å). Data were processed using the APEX2 software suite [24] by integrating the diffraction images with SAINT [25], scaling and applying for multiscan absorption correction with SADABS [24], and merging with XPREP [25]. This yielded numbers of reflections, completeness, and Rint of 73,262, 98.0%, 0.0960 for 2 and 49,951, 98.1%, 0.0552 for 1, respectively.
2.2.3. Structure solution and refinement
The intrinsic phasing method with SHELXTL XT [24] was used to solve the crystal structures of 1 and 2, giving all non-H atoms of β-CD, FXM and most water sites; except for the FXM CF3 group, maleate ion, and several water sites of 2 that were subsequently located by difference Fourier electron-density maps. The non-H atoms of both 1 and 2 were refined anisotropically, except for some water O atoms of 2 that were refined isotropically. In 2, the site-occupancy factors of both FXM molecules inside the β-CD dimeric cavity were refined to 1.0, yielding the 2:2 host–guest stoichiometric ratio, which was uniquely observed in all the β-CD–SSRI complexes [18,20], except the 2:1 and 1:1 β-CD–PXT complexes [19,26]. This was because 2 and other SSRI complexes crystallized in the triclinic space group P1 (no. 1) with similar unit-cell dimensions. The cell parameters of 2 were a = 15.1172(2) Å, b = 15.3647(2) Å, c = 19.3534(2) Å, α = 82.056(1)°, β = 69.942(1)°, γ = 76.189(1)°, Vol = 4092.7(1) Å3, and Z = 1, and were comparable with those of other SSRI complexes with unit-cell volumes of 4,200–4,700 Å3 [18,20]. The terminal NH2 group of each FXM was protonated, giving rise to two FXM–H+ embedded in the β-CD dimer, which were charge-balanced by the fully occupied dinegative maleate ion in the intermolecular spaces. One NH3+ group of FXM was coordinated with a maleate ion, β-CD O3–H group, and water site, whereas the other one donated H-bonds to four water sites and β-CD O2–H and O3–H groups (see Section 3.3).
In 1, the distinct crystal symmetries of the monoclinic, centrosymmetric space group C2/c (no. 15) had unit-cell constants of a = 58.7516(1) Å, b = 5.3346(1) Å, c = 43.1382(1) Å, α = γ = 90°, β = 100.004(1)°, Vol = 13314.6(3) Å3, and Z = 8, which suggested a complex structure of three independent FXM maleate in the asymmetric unit, yielding a total of 24 molecules per unit cell. The high thermal motions of the rather flexible FXM molecules resulted in a dozen atoms that were suggested to be split into two sites; particularly the C12–C13–C14 methylene side chains that were just in van der Waals contacts with neighboring molecules in the crystal lattice (see the 3D arrangement in Section 3.1). Attempts to improve the R-factor using the structure model with the split C12–C13–C14 chain and the aromatic B-ring were unsuccessful.
H atoms were geometrically positioned and handled with a riding model: C–H = 0.93 Å, Uiso = 1.2Ueq(C) (aromatic); C–H = 0.98 Å, Uiso = 1.2Ueq(C) (methine); C–H = 0.97 Å, Uiso = 1.2Ueq(C) (methylene); C–H = 0.96 Å, Uiso = 1.5Ueq(C) (methyl); and N–H = 0.89 Å, Uiso = 1.5Ueq(N) (4°-ammonium). Hydroxyl H atoms were placed and refined using “AFIX 147” or “AFIX 83” with restraints for O–H = 0.84 Å and Uiso = 1.5Ueq(O). For 2, 23.2 water molecules were spread over 30 sites with occupancy factors of 0.2–1.0 in the intermolecular interstices, outside the β-CD dimeric cavity. No ethanol molecule was cocrystallized, although this was used together with water as a crystallization solvent mixture. Water H atoms could not be located because of the restricted diffraction data quality. Only one O6–H group of glucose unit 1 of β-CD #2 was twofold disordered over sites A and B (denoted O61A/B_2; see the preceding paragraph of Section 3.1). For 1, the asymmetric unit comprised three independent FXM–H+ and three uninegative maleate ions, without solvent molecules. BUMP antibumping restraints were used throughout the refinement to prevent short intra- and inter-molecular H⋯H distances often observed in the macromolecular structures.
The refinement of the freely rotating CF3 group deserved further attention. The CF3 group of 4-(trifluoromethyl)benzene (B-ring) of FXT was found to be well ordered in various lattice environments [18]. Conversely, the twofold disorder phenomenon of the CF3 group was frequently observed in the relevant crystal structures, e.g., benzothiopyranone maleate salt [27] and diaryl telluroxide derivative [28]. Here, the three free FXM–H+ (1) and the two FXM–H+ embedded in the β-CD dimeric cavity (2) exhibited high thermal motions, making the structure refinements cumbersome. Further attempts were made to improve the refinements of 1 and 2, particularly of various FXM–H+. Several restraints were used to maintain and reflect the nature of the threefold symmetry of these freely rotating CF3 groups of all the five protonated FXM in 1 and 2. This produced five doubly disordered CF3 groups, each had six half-occupied F sites with elongated thermal ellipsoids (see Fig. 2). Although the structure model of FXM was improved and more realistic, the R factors were marginally decreased by 0.05%. The structure refinements of 2 and 1 converged at R1 = 0.0896 and 0.1003 for 13,574 and 6,203 reflections with F2 > 2σ(F2), respectively. Notably, the high R factors were due to the limited crystal quality and the subsequent weak diffraction data, the doubly disordered CF3 groups, disordered solvent molecules, and high thermal motions of FXM molecules. See Table S1 for details of data collection and refinement statistics.
Fig. 2.

Molecular structures and systematic numbering schemes of (E)-fluvoxamine (FXM) in free maleate salt form (1) and encapsulated in the β-cyclodextrin (β-CD) dimeric cavity (2). ORTEP plots are drawn at the 30% probability level (see also Fig. 1). Doubly disordered CF3 groups on the B-ring of five FXM–H+ in 1 and 2 and O61–H group of β-CD #2 in 2 are labeled as sites A and B. The connecting blue lines indicate the intramolecular O3–H⋯O2 H-bond stabilizing the three maleate− in 1; ions of the adjacent asymmetric units are in italics. Several intramolecular O3(n)⋯O2(n + 1) H-bonds maintaining the round CD conformation are absent because of the relevant OH groups H-bond with surrounding water sites. The intermolecular N–H⋯O H-bonds between various FXM–H+ and planar maleate−/non-planar maleate2− are shown with magenta lines.
2.3. DFT full-geometry optimization
Crystal structure determination via X-ray diffraction provides a space-and-time averaged structure, i.e., a structural snapshot of inclusion complexation in the solid state. Therefore, to derive meaningful intermolecular interactions of the actual host–guest structures, starting the energy minimization from X-ray-derived structures, if available, is suitable for reasonable computation cost. DFT full-geometry optimization performed in the gas phase is sufficient for reproducing the complex structures deduced by X-ray analysis. The relevant structures are superimposable with root mean square (RMS) fits of 0.6–0.7 Å, as demonstrated in recent examples of β-CD dimer encapsulating CTP and SCTP molecules [20]. The neutral form of FXM was chosen for the calculation because the protonated species (FXM–H+) embedded in the β-CD cavity was slightly influenced by the maleate salt outside the cavity (see Section 3.3), as observed previously for the DFT calculations of β-CD complexes with TCAs nortriptyline HCl and amitriptyline HCl [29]. Additionally, the FXM freebase in complex with severe acute respiratory syndrome coronavirus 2 targets was investigated for conformational changes and binding free energies using molecular docking and molecular dynamics simulation [30].
As noted in Section 3.3, in the solid state the pseudo-twofold symmetric β-CD–FXM dimeric inclusion complex comprised two different monomers, particularly the two side chains of FXM. Therefore, the dimeric and monomeric β-CD–FXM complexes were considered for DFT full-geometry optimization. Since both the geometrically positioned and electron-density-map-located H atoms have rather short H distances (C–H = 0.93–0.98 Å, N–H = 0.89 Å, and O–H = 0.82 Å), the initial molecular structures from X-ray analysis were normalized to neutron H distances (C–H, 1.083 Å; N–H, 1.009 Å; and O–H, 0.983 Å) [31]. Corrected structures were first optimized by a semiempirical PM3 method and then fully re-optimized by DFT calculation using the B3LYP functional in the gas phase with mixed basis sets 6-31+G(d) for H, N, O, F and 4-31G for C. All calculations were conducted using GAUSSIAN09 [32] on a DELL PowerEdge T430 server. The stabilization and interaction energies of the monomeric and dimeric complexes (ΔEstb and ΔEint) were calculated using eqs. Table S6.
| ΔEstb = Ecpx − (Eβ-CD_opt + ED_opt) | (1) |
| ΔEint = Ecpx − (Eβ-CD_sp + ED_sp) | (2) |
where Ecpx, Eβ-CD_opt and ED_opt are the total molecular energies from full-geometry optimization of complex, host β-CD and drug FXM, respectively; Eβ-CD_sp and ED_sp are the corresponding single-point energies in the complexed states.
DFT calculations for non-covalent interactions in CD inclusion complexes were improved in two ways. 1) ΔEints were corrected for dispersion interactions using the B97D functional and for the basis set superposition error (BSSE) using counterpoise correction [33] by single-point calculations in the gas phase with a larger basis set 6-31+G(d,p) for all atomsTable S7. Notably, because Eβ-CD_opt and ED_opt were respectively derived from the geometry optimization of free host and free guest molecules, the resulting ΔEstb could not be corrected for BSSE. 2) The geometry optimization along with the calculation of ΔEstb and ΔEint were conducted using the M06-2X exchange-correlation functional in vacuum and implicit water solvent (polarizable continuum model (PCM)).Table S8
In addition to the three uncomplexed FXM and the two enclosed FXM in the β-CD dimeric cavity, three different FXM–protein complexes were found via further structural surveys in two databases including the Cambridge Structural Database (CSD; www.ccdc.cam.ac.uk) and the Protein Data Bank (PDB; www.rcsb.org). Conformational analysis disclosed that the two side chains of (E)- and (Z)-isomers of FXM are differently oriented in space (see Section 3.5). Therefore, the total molecular energies (Etots) of eight FXM existing in distinct lattice environments were evaluated in neutral form in vacuum at the B3LYP/6-31+G(d,p) level of theory and compared in Table 1.
Table 1.
Structural parameters and inclusion geometries of fluvoxamine (FXM) maleate in free form (1) and in complex with β-cyclodextrin (β-CD) dimer (2) and proteins.
| Structural parameters /inclusion geometries | 3FXM–H+·3maleate− (1) | 2β-CD·2FXM–H+·maleate2−·(2) | FXM–protein | |||||
|---|---|---|---|---|---|---|---|---|
| ||||||||
| FXM | Mol 1 (X)h | Mol 2 (Y)h | Mol 3 (Z)h | Mol 1 (M)h | Mol 2 (N)h | 4ENHi | 4MM9i | 6AWPi |
| Conformation | E | E | E | E | E | E | Z | E |
| Torsion angle C1–C8 N1–O1 (τ1) | −177.9 | −176.8 | 178.7 | 176.9 | −178.7 | 153.6 | 45.7 | −179.8 |
| Torsion angle C1–C8–C11–C12 (τ2) | 72.6 | 63.0 | −72.0 | 97.2 | −94.5 | −87.0 | −67.4 | −113.2 |
| B-ring centroid–N2 distance (d1) | 7.688 | 7.710 | 7.673 | 7.247 | 7.462 | 8.537 | 6.783 | 8.124 |
| B-ring centroid–C15 distance (d2) | 8.365 | 8.103 | 8.396 | 8.078 | 7.913 | 7.602 | 7.504 | 6.919 |
| B-ring plane vs. C1–C8 N1–O1 plane | 6.6 | 8.5 | 6.9 | 10.3 | 9.5 | 64.3 | 60.1 | 8.2 |
| RMS fitb | 0 | 0.150 | 1.147 | 0.876 | 1.142 | 1.383 | 1.878 | 1.642 |
| ΔEtot (kcal/mol)c | 1.33 | 2.19 | 1.37 | 0 | 0.26 | 3.79 | 5.90 | 4.11 |
| Maleate | Mol 1 (I)h | Mol 2 (J)h | Mol 3 (K)h | Mol 1 (L)h | ||||
| Charge | −1 | −1 | −1 | −2 | ||||
| RMS deviations of C1–C2 C3–C4d | 0.002 | 0.001 | 0.001 | 0.006 | ||||
| RMS deviations of all atoms | 0.053 | 0.054 | 0.058 | 0.516 | ||||
| C1–C2 C3–C4 plane vs. O1–C1–O2 plane | 6.6 | 6.7 | 7.3 | 69.6 | ||||
| C1–C2 C3–C4 plane vs. O3–C4–O4 plane | 3.6 | 3.4 | 3.2 | 13.0 | ||||
| RMS fite |
0 |
0.110 |
0.010 |
0.641 |
||||
| ||||||||
| Moiety protrudes from O6-side | 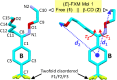 |
CF3 | 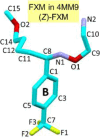 |
|||||
| Moiety enclosed in β-CD cavity | B-ring and C11–C8–N1 | |||||||
| B-ring plane vs. β-CD O4 plane | 67.0 | 66.9 | ||||||
| B-ring–β-CD O4 plane dist. (diagonal)f | 0.605 | 0.513 | ||||||
| B-ring–β-CD O4 plane dist. (vertical) | 0.557 | 0.472 | ||||||
| β-CD dimer | ||||||||
| O4 plane vs. O4 planeg | 2.2 | |||||||
| O4 centroid–O4 centroid distance | 9.046 | |||||||
Atom numbering of the X-ray-derived structures of (E)- and (Z)-FXM, see insets.
All non-H atoms of FXM molecule 1, excluding CF3 (1) is a reference structure used for the calculations of RMS fits. If only the B-ring and C11–C8 N1 moiety are used, the RMS fits = 0.021–0.590 Å.
Relative total molecular energies (ΔEtot) of FXM neutral form derived from DFT full-optimization in the gas phase at the B3LYP/6-31+G(d,p) level of theory. The fully-optimized structure of FXM molecule 1 embedded in β-CD cavity has the lowest Etot of −1143.73938 Hartree (H); 1 H = 627.5 kcal/mol.
RMS deviations from the planarity of the involved atoms.
All non-H atoms of maleate 1 (1) is a reference structure used for the calculations of RMS fits.
When the β-CD O6-side points upwards, the positive (negative) centroid–centroid distances indicate that the relevant position of the embedded guest molecule is above (beneath) the O4 plane.
Interplanar angle of the tail-to-tail (T2T) dimeric β-CD in complex with FXM (2).
Three FXM–H+ and two FXM–H+ are counter-balanced by three maleate−and one maleate2−in the respective asymmetric units of 1 and 2.
3. Results and discussion
For simplicity, we adopted the atom numbering of β-CD based on the conventional nomenclature of carbohydrates, i.e., atoms C61–O61A(B)_2 stand for the methylene C6–H2 linked to the doubly disordered hydroxyl O6–H (sites A and B) of glucose residue 1 (G1) of β-CD #2 in the dimeric motif (2), Fig. 2 and Table S2. The asymmetric unit contents of 2 and 1 are 2β-CD·2FXM–H+·maleate2−·23·2H2O and 3FXM–H+·3maleate−, respectively. In 2 and 1, five different FXM–H+ are present: two FXM–H+ are enclosed in a β-CD dimer (labeled as M and N) with three uncomplexed FXM–H+ (labeled as I, J, and K), which are counter-balanced by one dinegative maleate (labeled as L) and three uninegative maleate (labeled as X, Y, and Z), respectively (see Table 1).
3.1. First crystal structure of FXM maleate after 40 years of its launch in 1983
FXM was one of the first SSRIs introduced in Switzerland in 1983 and was marketed as Luvox in the US in 1994. FXM has high flexibility and instability, and therefore it is challenging to obtain single crystals of X-ray quality. Herein, the atomic resolution crystal structures of three FXM molecules in the uncomplexed form (1) and two FXM molecules embedded in the β-CD dimeric cavity (2) have been reported. All five FXM molecules are in the (E)-form, confirming the greater thermodynamic stability of the (E)-form over the (Z)-form [14] (see Section 3.5 for the relative stabilities of FXM existing in varied lattice environments). The restricted rotation of double bond C8 N1 and the coplanarity of conjugated C8 N1 and the aromatic B-ring produced the comparable torsion angles C1–C8 N1–O1 ∼±180° and the interplanar angle close to 0° (Fig. 2 and Table 1). For more details of structural comparison, see Section 3.2.
An interesting question arises: “Why a maleate salt and not a hydrochloride salt for FXM?” Drugs in the HCl salt form for improved water solubility are commonly used in the pharmaceutical industry, as evidenced by most TCAs and SSRIs [5]. However, carboxylic counterions including fumarate and maleate are popular alternatives in pharmaceutical formulations, such as FXM maleate [37]. A maleate salt helps to improve not only water solubility of drugs, but also their chemical stability in the crystalline state, as observed in 1 and 2. Maleate is an optimum salt form of FXM as this has a better biopharmaceutical profile compared with that of the free base [37]. In the solid state, most free ADDs in HCl form exist as ADD–H+⋯Cl− with the protonated amine of ADD directly linked by Cl− [38], which are interrupted by CD encapsulation (e.g., crystal structures of β-CD–CPM HCl [39] and β-CD–STL HCl complexes [18]). Conversely, the five FXM–H+ in uncomplexed form (1) and in complex with β-CD (2) are essentially coordinated by maleate− and maleate2− (Fig. 2, Fig. 3 and Table 1).
Fig. 3.

Various intermolecular interactions contribute to the stability of 5.3-Å-layer stacking along the b-axis in the ribbon-like crystals of 3FXM–H+·3maleate− (1). ORTEP plots are drawn at the 30 % probability level. (A) Infinite fused H-bond rings R44(12) and R64(16) along the a-axis; the asymmetric unit is in a ball-and-stick model with atoms labeled in red. (B) Interactions of adjacent 4-(trifluoromethyl)phenyl moieties include face-to-face (F2F; along the b-axis) and edge-to-face (E2F; along the a–c-plane) π⋯π, CF⋯π, and F⋯F interactions. FXM: fluvoxamine.
The crucial role of maleate salt deserves further discussion. Maleate acts not only as a space filler, but also as a H-bonding mediator in stabilizing the crystal structures of both 1 and 2. As illustrated in Fig. 3, if maleate− changed to maleate2− (lacking the COOH group), the crystal structure of 1 without ample intramolecular O3–H⋯O2 H-bonds would collapse. The complex H-bond networks coordinated by maleate− primarily contribute to the crystal stability and cannot be compensated by the substitution of Cl−. The lattice of the ribbon-shaped crystal (1) is constructed by a 5.3 Å-layer (i.e., the b-axis length) and is stabilized by two key non-covalent interactions. 1) The infinite fused H-bond rings R44(12) and R64(16), which are formed by FXM–H+ aminoethyl side chains and maleate ions along the a-axis at c = 0, 0.5 and 1. 2) Various types of intermolecular interactions of the adjacent 4-(trifluoromethyl)phenyl moieties including face-to-face (stacking along the b-axis) and edge-to-face (packing along the a–c-plane) π⋯π, CF⋯π, and F⋯F interactions also contribute to crystal stabilization (Fig. 3, and Tables 2 and S3). The structure and function of maleate2− in 2 are discussed below.
Table 2.
Comparison of N/O–H⋯O hydrogen bond interactions of fluvoxamine (FXM) maleate in free form and in complex with β-cyclodextrin (β-CD) dimer (Å, °).
| D−H⋯A | D−H | H⋯A | D⋯A | ∠(DHA) | D−H⋯A | D−H | H⋯A | D⋯A | ∠(DHA) |
|---|---|---|---|---|---|---|---|---|---|
| Free FXM (1)a | β-CD–FXM (2)a | ||||||||
| N2X–H1⋯O1Ii,b | 0.89 | 1.92 | 2.804(4) | 170.5 | N2M–H1⋯O21_1 | 0.89 | 2.27 | 2.99(2) | 138.1 |
| N2X–H2⋯O2Iii | 0.89 | 1.99 | 2.855(4) | 162.6 | N2M–H1⋯O31_1 | 0.89 | 2.29 | 2.99(2) | 135.4 |
| N2X–H3⋯O4J | 0.89 | 1.93 | 2.800(4) | 166.6 | N2M–H2⋯O4Wiv | 0.89 | 2.14 | 2.86(3) | 136.9 |
| N2Y–H1⋯O1J | 0.89 | 1.90 | 2.790(4) | 176.7 | N2M–H3⋯O5WC | 0.89 | 2.07 | 2.95(2) | 169.1 |
| N2Y–H2⋯O2K | 0.89 | 2.00 | 2.847(4) | 157.8 | N2N–H2⋯O34_1 | 0.89 | 1.95 | 2.68(2) | 137.9 |
| N2Y–H3⋯O4Kiii | 0.89 | 1.92 | 2.795(4) | 168.7 | N2N–H2⋯O1Lv | 0.89 | 2.17 | 2.90(3) | 139.0 |
| N2Z–H1⋯O4Iii | 0.89 | 1.93 | 2.805(4) | 166.6 | N2N–H2⋯O5WA | 0.89 | 2.14 | 2.91(3) | 144.3 |
| N2Z–H2⋯O3J | 0.89 | 2.00 | 2.859(4) | 163.1 | O34_2–H⋯O2N | 0.82 | 2.02 | 2.72(2) | 142.6 |
| N2Z–H3⋯O1K | 0.89 | 1.91 | 2.796(4) | 172.4 | |||||
| O3I–H⋯O2I | 0.82 | 1.65 | 2.468(3) | 174.1 | |||||
| O3J–H⋯O2J | 0.82 | 1.66 | 2.476(3) | 177.0 | |||||
| O3K–H⋯O2K | 0.82 | 1.66 | 2.465(3) | 165.1 |
3.2. All SSRIs share a unique inclusion mode in the tail-to-tail β-CD dimer
SSRIs (excluding STL) share the phenylpropylamine motif and analog, and halogen atoms although their overall structures are more diverse with 2–3 C-length alkylamine side chains connected to 1–4 non-aromatic and aromatic rings (Fig. 1). The inclusion mode with the aromatic moiety of SSRI embedded in the hydrophobic β-CD cavity is uniquely observed for all SSRI complexes [[18], [19], [20]]. The CD encapsulation secures the aromatic ring carrying halogens (CF3/F/Cl) on the para-position (Ar–X moiety) that is responsible for drug specificity to SERT [40]. This is analogous to the binding of SSRI halogens to the same halogen-binding pocket (HBP) in SERT [40], emphasizing the use of CDs as a model system for elucidating protein–drug interactions. Therefore, CD encapsulation helps to lessen the side effects of drugs for efficient delivery into cells [23,41]. Although multimode inclusions of SSRIs in the β-CD cavity are frequently observed in solution as previously reviewed, the lesser flexible β-CD macrocycle with cavity dimensions of a 7.9-Å height and 6.0–6.5-Å diameter gives the best fit to the SSRI aromatic ring moiety (magenta highlights in Fig. 1) [[18], [19], [20],39]. This section compares the β-CD conformational changes upon inclusion of the structurally diverse SSRIs.
The inclusion complexation conforms to the “induced-fit” process [42], which is accomplished by the mutual host–guest structural adaptation. Although SSRIs have large degree of freedoms and their conformations are more flexible (see Section 3.5), β-CD is rather rigid and adapts its structure to a lesser extent. This is indicated by the small spans of structural parameters depicted as radar plots in Figs. 4A and B and listed in Table S2 for the glucose tilt angles and O3(n)⋯O2(n + 1) distances of β-CD #1 and #2 (2) compared with those of the uncomplexed β-CD dodecahydrate (iii [43]). The corresponding values are 1.9–15.9° and 2.679–2.836 Å for the pseudo-twofold symmetric β-CD dimer (2) and 6.4–26.2° and 2.770–2.957 Å for iii (Figs. 4A and B, and Table S2), suggesting that the two complexed β-CDs are conformationally round, like the β-CD hydrate [43]. This is also true for β-CDs encapsulating other SSRIs (STL, FXT [18], PXT [19,26], and CTP, SCTP [20]) because of the intramolecular, interglucose O2/O3⋯O2/O3 H-bonds, securing the CD annular conformation (Figs. 4A–D). Conversely, in other complexes with TCA CPM [39] and polyphenol (–)-epicatechin(EC) [44], β-CDs become more distorted from a round structure to better fit the embedded aromatic rings, as shown with the increased tilt angles and O3(n)⋯O2(n + 1) distances of several glucose residues that reach 26.5° and 2.917 Å and 33.7° and 3.346 Å, respectively.
Fig. 4.

Radar plots depicting the variation of key structural parameters in the glucose units (G1–G7) of β-cyclodextrin (β-CD) #1, #2 (dimeric motif) upon encapsulating fluvoxamine (FXM) (2), fluoxetine (FXT) (i [18]), and citalopram (CTP) (ii [20]). (A) Glucose tilt angles. (B) Intramolecular, interglucose O3(n)⋯O2(n + 1) distances. For comparison, data of the 1:1 β-CD–(–)-epicatechin (EC) ([44], β-CD–clomipramine (CPM) [39], and β-CD·12H2O (iii [43]) are also incorporated; angles and distances are in ° and Å. Structure overlays of β-CD monomers and dimers. (C) Side and (D) top views of seven β-CD monomers. (E) Three tail-to-tail β-CD dimers in 2, i and ii. (F) Summary of the RMS fits, see also Fig. 5 and Table S2.
Other structural parameters indicating the CD roundness include the normal glucose puckering parameters Q, θ [45]: 0.0–6.0°, 0.536–0.581 Å (2) vs. 1.4–7.6°, 0.559–0.596 Å (iii), Table S2. The parameters involving the glycosidic O4, i.e., 1) the average of the O4(n)⋯O4(n − 1)/O4(n)⋯centroid distance ratio, and 2) the sum of averages of the endocyclic torsion angles ϕ [O5(n + 1)–C1(n + 1)–O4(n)–C4(n)], ψ [C1(n + 1)–O4(n)–C4(n)–C5(n)]. The corresponding values are 1) 0.868, 0.867 (2) vs. 0.870 (iii) (0.868 for an ideal heptagon of the linked seven O4 atoms), and 2) −0.5°, −0.7° (2) vs. −1.3° (iii) (value close to zero indicates the CD roundness), respectively (Table S2). Note that comparing each CD structural parameter as discussed above is less convenient than the global indication of the RMS deviation of structure superposition. The six β-CD monomers in the 2:2 β-CD–FXM (2), β-CD–FXT (i [18]), and β-CD–CTP (ii [20]) are similarly round, as indicated by the RMS fits in the range of 0.099–0.404 Å. They are comparatively distorted from the β-CD hydrate [43] with RMS fits of 0.271–0.454 Å (Figs. 4C and D). Only non-H atoms excluding the rotatable O6 atoms are used for the RMS fit calculations.
In the crystals of the β-CD–SSRI inclusion complexes, β-CDs form a dimer with the adjacent O6–H rims facing each other, namely the tail-to-tail (T2T) motif. The dimers are stacked like a row of coins with the facing O2–H/O3–H rims, yielding an infinite head-to-head (H2H) channel threaded by SSRI molecules and stabilized by the interplay of intermolecular halogen⋯halogen and π⋯π interactions (Fig. 5A). The T2T β-CD dimers are similar with comparable angles of O4 planes (close to 0°) and O4 centroid–O4 centroid distances (∼9.0 Å) (Table 1). They are superimposable with RMS fits of 0.177–0.482 Å (Figs. 4E and F). Additionally, the T2T β-CD dimer is further stabilized by OH⋯O H-bonds of the facing O6–H rims and of O6–H groups with 23.2 water molecules spread over 30 sites in the intermolecular spaces (Fig. 5B, and Table S4). This is indicated by the exocyclic torsion angles χ [C4–C5–C6–O6] and ω [O5–C5–C6–O6] in the ranges of 52.4–61.7° and −56.6 to −69.4°. The exception is the disordered O61–H (site B) that directs toward the β-CD cavity with torsion angles χ, 172.9° and ω, 49.7° (Table S2).
Fig. 5.

Dimeric nature of β-cyclodextrin (β-CD) inclusion complexes with selective serotonin reuptake inhibitors (SSRIs). (A) 3D-arrangements in channel structures are built from T2T β-CD dimers encapsulating fluvoxamine (FXM) and sertraline (STL) molecules and stabilized by intermolecular face-to-face π⋯π and halogen⋯halogen interactions. The primary O6–H side (tail; T) and secondary O2–H/O3–H side (head; H) of β-CD are marked. (B) ORTEP plots of the tail-to-tail (T2T) β-CD–FXM dimeric complex (30% probability level). For clarity, FXM molecules are shown in a space-filling model.
3.3. Characteristics of 2β-CD·2FXM–H+·maleate2−·23·2H2O (2)
Maleate ions as salts in the crystal structures of active pharmaceutical ingredients usually exist in the planar, uninegative form, not in the dinegative form because this is stabilized by the intramolecular OH⋯O H-bond [38]. Examples include an antihypertensive and antiglaucoma drug timolol maleate [46] and an antihistamine drug chlorpheniramine maleate [47]. This is demonstrated from the existence of one COO− and one COOH (1) with corresponding bond distances: C1–O1 = 1.228(4)–1.231(4) Å; C1–O2 = 1.254(5)–1.260(4) Å; C4–O4 = 1.208(4)–1.216(4) Å; and C4–O3 = 1.283(5)–1.293(4) Å (see atom numbering in Fig. 2).
The situation is different for the maleate salt in the dimeric β-CD–FXM complex (2). The unique dinegative maleate bearing two COO− groups is indicated by the comparable bond distances of C1L–O1L = 1.25(1) Å and C1L–O2L = C4L–O3L = C4L–O3L = 1.26(1) Å. One maleate2− is counter-balanced by two encapsulated FXM–H+ and adopts a non-planar structure, facilitating the carboxylate O1L and O2L atoms to fully accept H-bonds from O32–H (β-CD #1), O23–H (β-CD #2), N2N–H2 (FXM–H+), and water sites O5WC/D and O6W (Figs. 2 and 5B and Table S4). This is indicated by the structural parameters of maleate2− including the RMS deviations of all atoms (0.516 Å) and the interplanar angle of C1–C2–C3–C4 and O1–C1–O2 (69.6°) (Table 1). Not only does one maleate2− (2) differ from three maleate− (1) but two FXM–H+ (2) and three FXM–H+ (1) also show greater differences as indicated by the RMS fits up to 0.641 and 1.147 Å, respectively (Table 1). The five (E)-FXM–H+ in 2 and 1 have comparable structural parameters including the torsion angles C1–C8 N1–O1 and C1–C8–C11–C12 and the angle between planes of the B-ring and the C1–C8 N1–O1 moiety (see τ1 and τ2 in Table 1). The restricted β-CD dimeric cavity limits the motion of FXM–H+ (2), producing shorter distances of the B-ring centroid to the side-chain terminals (atoms N2 and C15) compared with those of the free salt (1): 7.247–7.462 vs. 7.673–7.710 Å and 7.913–8.078 vs. 8.103–8.396 Å (see d1 and d2 in Table 1).
Theoretically, FXM comprising three structural moieties (2-aminoethyloxime and methoxybutyl side chains and one 4-(trifluoromethyl)phenyl B-ring) are possibly included in the β-CD cavity. For the host–guest spatial fit, the B-ring is more favorable than the two side chains. This is the case in solution as previously predicted via NMR [21,22] and is confirmed here. Single-crystal X-ray diffraction uniquely provided detailed inclusion geometries of two different FXM embedded in the pseudo-twofold symmetric T2T β-CD dimer and the intermolecular interactions stabilizing the molecular and crystal structures of 2, which cannot be deduced by other techniques. Notably, the β-CD encapsulation shreds the complex H-bond network and other intermolecular interactions in 1 to enhance the solubility of FXM in water. To attain the energetically favorable structure of dimeric β-CD–FXM complex (see Section 3.4), each of the two non-superimposable FXM–H+ (RMS fit = 1.043 Å for all non-H atoms, excluding F atoms) deeply inserts the B-ring from the wider O2–H/O3–H rim, across the β-CD cavity until the twofold disordered CF3 group ais proximal to the edge of the narrower O6–H rim. The optimal inclusion geometries are such that both FXM B-rings are inclined by 67.0° and the ring centroids are 0.559 Å above the β-CD O4-plane (average values), which are stabilized by intermolecular N2–HFXM⋯O2/O3CD, N2–HFXM⋯O1maleate/Ow H-bonds (Fig. 4 and Table 2). In the crystal, the dimeric complex is further maintained at the T2T interface by O6CD⋯Ow⋯(Ow)⋯O6CD and F⋯F interactions and at the H2H interface by O2/O3CD⋯Ow⋯(Ow)⋯O2/O3CD and N2–HFXM⋯OCD H-bonds (Fig. 5B and Table S4).
3.4. Thermodynamic stabilities of β-CD–FXM inclusion complexes and FXM isomers
Crystallographic evidence for the conformational flexibility of FXM and the pseudo-twofold symmetric dimeric β-CD–FXM inclusion complexation suggests two questions that deserve addressing from a theoretical viewpoint. The DFT/B3LYP fully-optimized and X-ray-derived structures of the monomeric and dimeric β-CD–FXM complexes and the FXM isomers existing in different lattices (see Section 3.5) can be superimposed with RMS fits of 0.647–0.678, 0.356 and 0.236–0.605 Å, respectively (Fig. S1); all non-H atoms, excluding F atoms, are considered for the calculations. This indicates that DFT energy minimization gave a meaningful structure–energy relationship.
In the two DFT/B3LYP-optimized monomeric complexes, the 4-(trifluoromethyl)phenyl B-ring and 2-aminoethyloxime, methoxybutyl side chains of both (E)-FXM molecules adapt differently to fit the β-CD cavity and to establish intermolecular N/O–H⋯O H-bonds, respectively (Fig. 6). The round β-CD macrocycles are stabilized by intramolecular O2/O3⋯O2/O3 H-bonds. Additionally, the T2T β-CD dimeric structure in the gas phase is maintained by three O6–H⋯O6 H-bonds, compensating the interactions with surrounding water molecules in the solid state (Fig. 5, Fig. 6). However, the monomeric β-CD–FXM complexes (#1 and #2) are stabilized by N2M–H2⋯O21 and bifurcated O23–H⋯O2M⋯H–O33 H-bonds, and the dimeric complex is maintained by N2M–H2⋯O31_1, O33_1–H⋯O2M, and O34_2–H⋯O2N H-bonds (Table S5); consequently, the corresponding stabilization and [interaction] energies, ΔEstbs and [ΔEints] are −20.15 [−26.06], −13.48 [−16.57], and −24.14 [−38.14] kcal/mol, respectively (Table S6), which agree with those of other β-CD–SSRI complexes [[18], [19], [20]]. Furthermore, attempts to improve the DFT calculation accuracy by considering two corrections of the dispersion forces and BSSE provide ΔEint(B97D+BSSE)s and BSSE energies [ΔEBSSEs] of −42.38, [5.51] and −32.96, [4.99] kcal/mol for monomers #1 and #2, respectively (Table S7). This suggests that the larger basis set 6-31 + G(d,p) significantly decreases the effects of ΔEBSSEs on ΔEint(B97D)s to 11.5% and 13.2% for respective monomers 1 and 2 (Table S7) compared with ∼25% for the β-CD–CTP complexes at the B97D/6-31 + G(d)/4-31G level of theory [20].
Fig. 6.

Illustration of the summary of the key density functional theory (DFT) results. (A, C) Monomeric and (B) dimeric inclusion complexes of β-cyclodextrin (β-CD)–fluvoxamine (FXM) base from DFT full-geometry optimization using B3LYP and M06-2X functionals in vacuum and polarizable continuum model (PCM) water. For better comparison, the energy of complex (Ecpx) and stabilization energy (ΔEstb) are given in respective units of Hartree (H) and kcal/mol; see also Fig. S1 and Tables S5, S6, and S8. The complexes are stabilized by intramolecular O–H⋯O H-bonds (maintaining the round β-CD macrocycles; blue lines) and intermolecular N/O–H⋯O H-bonds (maintaining the β-CD dimeric structure and the FXM inclusion geometries; magenta lines).
The effects of water solvation and electron exchange and correlation on the structures and complexation energies are demonstrated for the two distinct β-CD–FXM monomers using the implicit PCM water and the M06-2X functional, respectively. Notably, the M06-2X-optimized geometries in vacuum and PCM water of β-CD–FXM monomers #1 and #2 are similar and agree with the corresponding X-ray structures, as indicated by the average RMS fits of 0.331, 0.452 and 0.457 Å, respectively (Fig. S1C), which are better than those derived from the B3LYP functional in vacuum with the mean RMS fit of 0.663 Å (Fig. S1A). The energy minimization of monomers #1 and #2 using the M06-2X/PCM method gives rise to lowering of Ecpxs by 53.08–54.41 kcal/mol and to raising of ΔEstbs and ΔEints by 10.53–19.77 kcal/mol, compared to the calculations in vacuum (Table S8). Clearly, the M06-2X functional used for the β-CD–FXM complexes much better distinguishes ΔEstbs and ΔEints in PCM water from those in vacuum when compared with the B3LYP functional used for the β-CD–nortriptyline/amitriptyline complexes with the corresponding values of 0.68–2.45 kcal/mol [29]. The geometry optimization using the M06-2X/PCM method gives ΔEstbs and {ΔEints} of −26.86, {−28.53} and −25.04, {−29.26} kcal/mol for monomeric complexes #1 and #2, respectively (Table S8). This indicates that both monomers are comparatively energetically stable by intermolecular N/OH⋯O H-bonds, which are similarly observed when using the B3LYP gas-phase calculation (Figs. 6A and C, and Tables S5, S6 and S8).
In this study, the standard B3LYP functional was used for the energy minimization of CD complexes with a compromise to computational cost and accuracy, affording optimized structures in good agreement with the X-ray-derived ones. The obtained interaction energies were subsequently corrected for the dispersion forces and basis set errors. However, when the exchange-correlation functionals (e.g., M06-2X, ωB97X-D) and the effect of water solvation were considered for the geometry optimization of non-covalent CD supramolecular complexes, more accurate estimates of complexation energies and more reliable structures could be obtained [48].
3.5. FXM conformation is highly adapted for its pharmacological functions
Among SSRIs, FXM is the most flexible drug because of the two long side-chain arms connected to the aromatic B-ring. Although the pharmacologically active (E)-FXM is marginally more stable than the (Z)-FXM by ∼2 kcal/mol [14], the E → Z isomerization energy of 55 kcal/mol is considerably higher [15]. This suggests that the (E)-isomer of FXM is more naturally abundant than the (Z)-isomer (Table 1). The conformational analysis of FXM is accomplished by combining snapshots of the drug existing in three distinct lattice environments 1) from the origin, the free maleate salt form; 2) along the way, the drug embedded in the CD cavity (carrier) during delivery; and 3) to the destination, the drug in action while optimally interacting with the surrounding flexible amino acids around the protein-binding pocket. The larger RMS fits of drugs (greater structural differences) present crystallographic evidence for their high conformational flexibility. Note that the isomeric types of FXM and the presence of maleate salt are not mentioned in the reported crystal structures of FXM in complex with various proteins: a mutant of biogenic amine transporter (PDB code: 4MM9 [34]); cytochrome P450 46A1 (CYP46A1) (cholesterol 24-hydroxylase), a key enzyme of cholesterol turnover in the brain (PDB code: 4ENH [35]); and SERT (PDB code: 6AWP [36]).
Clearly, seven of eight FXM structures exist in the (E)-form and only one (Z)-FXM in complex with a mutant of biogenic amine transporter [34]. The two geometrical isomers, (E)- and (Z)-forms, are differentiated by torsion angles C1–C8 N1–O1 ∼±180° and ∼0°, respectively; see τ1 in Table 1. Expectedly, the three uncomplexed (E)-FXM tend to be more relaxed than the four (E)-FXM confined in the β-CD dimeric cavity and bound to protein pocket. This is indicated by the distances from the B-ring centroid to the two side-chain terminals, atoms N2 and C15: 7.673–7.710 and 8.103–8.396 Å, 7.913–8.078 and 7.247–7.462 Å, and 8.124–8.537 and 6.919–7.602 Å, respectively (see d1 and d2 in Table 1). The corresponding values for the (Z)-isomer are 6.783 and 7.504 Å. Consequently, the entire FXM molecules in distinct lattices are normally non-superimposable as indicated by the large RMS fits of 0.876–1.878 Å. The exceptions are the free FXM molecules 1 and 2 that are highly similar and well overlayed with an RMS fit of 0.150 Å. All non-H atoms of FXM molecule 1, excluding CF3 (1) as a reference structure, were used for the calculations of RMS fits (Table 1). If only the B-ring and C11–C8 N1 moiety are considered, the RMS fits fall in a smaller range of 0.021–0.590 Å (Fig. 7).
Fig. 7.

Partial structure overlays of fluvoxamine (FXM) in distinct lattice environments. (A) 3FXM–H+·3maleate− (1). (B) 2β-CD·2FXM–H+·maleate2−·23·2H2O (2). (C) Different FXM molecules in three protein-binding pockets indicated by PDB codes 4MM9 [34], 4ENH [35], and 6AWP [36]; for more details, see Table 1, footnote (i). (D) From free salt form, on the way during transport in β-cyclodextrin (β-CD) cavity until to the target-binding protein of FXM. Reference structures are shown in blue wireframes with their names marked in gray areas and only non-H atoms of the rigid B-ring linked with C11–C8 N1 group are considered for the calculation of RMS fits. The corresponding RMS fit for each structure pair is indicated by nearby distance. Only site A of the twofold disordered CF3 group in 1 and 2 is shown.
The predominant (E)-form of FXM is also demonstrated by the DFT fully-optimized structures of FXM from different lattices. The two (E)-FXM enclosed in the β-CD cavity are most energetically favorable and slightly more stable than the three free (E)-FXM and two (E)-FXM bound to proteins by 1.33–2.19 and 3.79–4.11 kcal/mol (see ΔEtot in Table 1). The (Z)-FXM is less stable than various (E)-FXM by 1.79–4.57 kcal/mol, in agreement with published results [14,15]. Furthermore, molecular dynamics simulations recently indicate that whereas two similar (E)-FXM are strongly bound to nonstructural protein 14 (NSP14) and main protease (Mpro) with respective binding free energies of −25.14 and −19.78 kcal/mol, one (Z)-FXM is moderately bound to papain-like protease (PLpro) with a binding free energy of −9.21 kcal/mol [30]. DFT combined with X-ray crystallography highlights the thermodynamically stable active (E)-FXM and the importance of conformational flexibility in its optimal binding with proteins.
FXM is rather flexible through the delivery process (origin–destination) (Fig. 7D). Overall, FXM and other SSRIs (STL, FXT and PXT [18,19]) exhibit conformational flexibilities, facilitating the optimal binding of their halogen(s) on the aromatic ring to the same HBP in SERT [40]. This resonates with the importance of the mutually induced conformational adaptation of SSRIs and SERT [49] as shown by crystal structures of biogenic leucine transporter (LeuBAT)–STL/FXT/PXT complexes [34].
4. Conclusions
The incidence of depression was observed to worsen in the post-COVID-19 era [3], triggering the efficient use of antidepressants, particularly SSRIs. Our comprehensive study used single-crystal X-ray diffraction integrated with DFT calculation to provide atomistic insight into the conformationally flexible SSRIs and their inclusion complexation with β-CD, which reduces side effects, masks bitterness, and improves the water solubility, molecular stability, and bioavailability of drugs. Herein, we address the atomic interactions of FXM with β-CD, which has become the most promising repurposed antidepressant for treating COVID-19 [12].
Surprisingly, the ethanol aqueous solution of a unimolar mixture of β-CD and FXM maleate in the same vial provided single crystals of two distinct morphologies. Although the rod-shaped crystals belong to the triclinic space group P1 with the asymmetric unit of the dimeric complex as 2β-CD·2FXM–H+·maleate2−·23·2H2O (2), the ribbon-shaped crystals are in the monoclinic space group C2/c with the asymmetric unit of the drug in salt form as 3FXM–H+·3maleate−·(1). In the first X-ray structure of the uncomplexed FXM maleate (1), three FXM–H+ counter-balanced by three planar maleate− form a thin layer stabilized by infinite fused H-bond rings R44(12) and R64(16) and the interplay of π⋯π, CF⋯π, and F⋯F interactions. In 2, two similarly round β-CDs form a T2T dimer enclosing the aromatic B-rings of two different FXM–H+, which are charge-balanced by the rare non-planar maleate2−. The dimeric complex is stabilized by N/OH⋯O H-bonds and F⋯F interactions. This is a host–guest recognition pattern uniquely observed for all SSRI complexes [[18], [19], [20]], which secures the aromatic ring carrying halogens (CF3/F/Cl) that is responsible for the drug selectivity for inhibition of serotonin reuptake. Therefore, CD encapsulation helps improve the physicochemical properties of the drug for efficient delivery into cells [23,41].
DFT calculations revealed that the thermodynamic stabilities of the monomeric and dimeric β-CD–FXM complexes stem from weak intermolecular interactions with stabilization energies of −25.04 to −26.86 kcal/mol, and for the conformationally flexible FXM existing in various lattice circumstances, the distinct (E)-isomers are more energetically stable than the (Z)-isomer. Conformational analysis showed that the two side chains of FXM shrink in the β-CD cavity (during transport) compared with the more relaxed molecules in the free salt form and in protein-binding pockets, emphasizing the importance of conformational adaptation of drugs in their pharmacological functions. However, although FXM has numerous benefits to humans, ADDs are emerging aquatic contaminants, which can also be remedied by CDs as universal encapsulating agents [50].
Data availability
Data are available after acceptance. CCDC: 2242694; 2242695.
CRediT authorship contribution statement
Thammarat Aree: Writing – review & editing, Writing – original draft, Project administration, Methodology, Funding acquisition, Formal analysis, Conceptualization.
Declaration of competing interest
The authors declare that there are no conflicts of interest.
Acknowledgments
This work was supported by the Ratchadapisek Sompoch Endowment Fund, Chulalongkorn University, Thailand (Grant No.: RCU_67_023_010).
Footnotes
Peer review under responsibility of Xi'an Jiaotong University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jpha.2024.101040.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.World Health Organization (WHO) Depression and Other Common Mental Disorders: Global Health Estimates. 2017. http://apps.who.int/iris/bitstream/10665/254610/1/WHO-MSD-MER-2017.2-eng.pdf
- 2.WHO COVID-19 pandemic triggers 25% increase in prevalence of anxiety and depression worldwide. 2022. https://www.who.int/news/item/02-03-2022-covid-19-pandemic-triggers-25-increase-in-prevalence-of-anxiety-and-depression-worldwide [PMC free article] [PubMed]
- 3.Perlis R.H., Ognyanova K., Santillana M., et al. Association of acute symptoms of COVID-19 and symptoms of depression in adults. JAMA Netw. Open. 2021;4 doi: 10.1001/jamanetworkopen.2021.3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davis H.E., McCorkell L., Vogel J.M., et al. Long COVID: Major findings, mechanisms and recommendations. Nat. Rev. Microbiol. 2023;21 doi: 10.1038/s41579-022-00846-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brunton L., Knollman B., editors. Goodman and Gilman’s the Pharmacological Basis of Therapeutics. 14th ed. McGraw-Hill; 2023. [Google Scholar]
- 6.Stahl S.M. 5th ed. Cambridge University Press; 2021. Stahl’s Essential Psychopharmacology: Neuroscientific Basis and Practical Applications. [Google Scholar]
- 7.Owens M.J., Knight D.L., Nemeroff C.B. Second-generation SSRIs: Human monoamine transporter binding profile of escitalopram and R-fluoxetine. Biol. Psychiatry. 2001;50:345–350. doi: 10.1016/s0006-3223(01)01145-3. [DOI] [PubMed] [Google Scholar]
- 8.MacKay F.J., Dunn N.R., Wilton L.V., et al. A comparison of fluvoxamine, fluoxetine, sertraline and paroxetine examined by observational cohort studies, Pharmacoepidemiol. Drug Saf. 1997;6:235–246. doi: 10.1002/(SICI)1099-1557(199707)6:4<235::AID-PDS293>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 9.Omori I.M., Watanabe N., Nakagawa A., et al. Fluvoxamine versus other anti-depressive agents for depression. Cochrane Database Syst. Rev. 2010 doi: 10.1002/14651858.CD006114.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reis G., Dos Santos Moreira-Silva E.A., Silva D.C.M., et al. Effect of early treatment with fluvoxamine on risk of emergency care and hospitalisation among patients with COVID-19: The TOGETHER randomised, platform clinical trial. Lancet Glob. Health. 2022;10:e42–e51. doi: 10.1016/S2214-109X(21)00448-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sidky H., Sahner D.K., Girvin A.T., et al. Assessing the effect of selective serotonin reuptake inhibitors in the prevention of post-acute sequelae of COVID-19. medRxiv. 2023 doi: 10.1016/j.csbj.2023.12.045. 2022.11.09.22282142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hashimoto Y., Suzuki T., Hashimoto K. Mechanisms of action of fluvoxamine for COVID-19: A historical review. Mol. Psychiatry. 2022;27:1898–1907. doi: 10.1038/s41380-021-01432-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kwon J.W., Armbrust K.L. Photo-isomerization of fluvoxamine in aqueous solutions. J. Pharm. Biomed. Anal. 2005;37:643–648. doi: 10.1016/j.jpba.2004.09.057. [DOI] [PubMed] [Google Scholar]
- 14.Dadkhah S., Bagheri Novir S., Balali E. Computational investigation of structural and electronic properties of cis and trans structures of fluvoxamine as a nano-drug. Comput. Theor. Chem. 2017;1105:33–45. [Google Scholar]
- 15.Odai K., Sugimoto T., Ito E. Quantum chemical calculation of intrinsic reaction coordinates from trans to cis structure of fluvoxamine. Comput. Theor. Chem. 2020;1192 [Google Scholar]
- 16.Crini G., Aleya L. Cyclodextrin applications in pharmacy, biology, medicine, and environment. Environ. Sci. Pollut. Res. 2022;29:167–170. doi: 10.1007/s11356-021-16871-2. [DOI] [PubMed] [Google Scholar]
- 17.Fourmentin S., Crini G., Lichtfouse E., editors. Cyclodextrin Applications in Medicine, Food, Environment and Liquid Crystals. Springer International Publishing; Cham: 2018. [Google Scholar]
- 18.Aree T. Advancing insights on β-cyclodextrin inclusion complexes with SSRIs through lens of X-ray diffraction and DFT calculation. Int. J. Pharm. 2021;609 doi: 10.1016/j.ijpharm.2021.121113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aree T. Inclusion scenarios and conformational flexibility of the SSRI paroxetine as perceived from polymorphism of β-cyclodextrin–paroxetine complex. Pharmaceuticals (Basel) 2022;15 doi: 10.3390/ph15010098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aree T. Supramolecular assemblies of citalopram and escitalopram in β-cyclodextrin dimeric cavity: Crystallographic and theoretical insights. Carbohydr. Polym. 2024;329 doi: 10.1016/j.carbpol.2023.121771. [DOI] [PubMed] [Google Scholar]
- 21.Trefi S., Gilard V., Balayssac S., et al. Quality assessment of fluoxetine and fluvoxamine pharmaceutical formulations purchased in different countries or via the Internet by 19F and 2D DOSY 1H NMR. J. Pharm. Biomed. Anal. 2008;46:707–722. doi: 10.1016/j.jpba.2007.11.038. [DOI] [PubMed] [Google Scholar]
- 22.Dastjerdi L.S., Shamsipur M. 19F-nuclear magnetic resonance spectroscopy as a tool to investigate host-guest complexation of some antidepressant drugs with natural and modified cyclodextrins. Trop. J. Pharm. Res. 2017;15:2675–2682. [Google Scholar]
- 23.Y. Yokoi, Inventor; Oral pharmaceutical compositions containing fluvoxamine maleate and cyclodextrins, Japan patent JP2011088858A, 6 May 2011.
- 24.Bruker . 2014. APEX2, SADABS and SHELXTL. [Google Scholar]
- 25.Bruker . 2008. SAINT and XPREP. [Google Scholar]
- 26.Caira M.R., De Vries E., Nassimbeni L.R., et al. Inclusion of the antidepressant paroxetine in β-cyclodextrin. J. Incl. Phenom. Macrocycl. Chem. 2003;46:37–42. [Google Scholar]
- 27.Li P., Guo K., Fu L., et al. Solubility-driven optimization of benzothiopyranone salts leading to a preclinical candidate with improved pharmacokinetic properties and activity against Mycobacterium tuberculosis. Eur. J. Med. Chem. 2023;246 doi: 10.1016/j.ejmech.2022.114993. [DOI] [PubMed] [Google Scholar]
- 28.Docker A., Johnson T.G., Kuhn H., et al. Multistate redox-switchable ion transport using chalcogen-bonding anionophores. J. Am. Chem. Soc. 2023;145:2661–2668. doi: 10.1021/jacs.2c12892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aree T. β-Cyclodextrin encapsulation of nortriptyline HCl and amitriptyline HCl: Molecular insights from single-crystal X-ray diffraction and DFT calculation. Int. J. Pharm. 2020;575 doi: 10.1016/j.ijpharm.2019.118899. [DOI] [PubMed] [Google Scholar]
- 30.Panda S.K., Gupta P.S.S., Rana M.K. Potential targets of severe acute respiratory syndrome coronavirus 2 of clinical drug fluvoxamine: Docking and molecular dynamics studies to elucidate viral action. Cell Biochem. Funct. 2023;41:98–111. doi: 10.1002/cbf.3766. [DOI] [PubMed] [Google Scholar]
- 31.Allen F.H., Bruno I.J. Bond lengths in organic and metal-organic compounds revisited: X-H bond lengths from neutron diffraction data. Acta Crystallogr. B. 2010;66:380–386. doi: 10.1107/S0108768110012048. [DOI] [PubMed] [Google Scholar]
- 32.Frisch M.J., Trucks G.W., Schlegel H.B., et al. 2009. GAUSSIAN09. [Google Scholar]
- 33.Boys S.F., Bernardi F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970;19:553–566. [Google Scholar]
- 34.Wang H., Goehring A., Wang K.H., et al. Structural basis for action by diverse antidepressants on biogenic amine transporters. Nature. 2013;503:141–145. doi: 10.1038/nature12648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mast N., Linger M., Clark M., et al. In silico and intuitive predictions of CYP46A1 inhibition by marketed drugs with subsequent enzyme crystallization in complex with fluvoxamine. Mol. Pharmacol. 2012;82:824–834. doi: 10.1124/mol.112.080424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coleman J.A., Gouaux E. Structural basis for recognition of diverse antidepressants by the human serotonin transporter. Nat. Struct. Mol. Biol. 2018;25:170–175. doi: 10.1038/s41594-018-0026-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bharate S.S. Carboxylic acid counterions in FDA-approved pharmaceutical salts. Pharm. Res. 2021;38:1307–1326. doi: 10.1007/s11095-021-03080-2. [DOI] [PubMed] [Google Scholar]
- 38.Groom C.R., Bruno I.J., Lightfoot M.P., et al. The Cambridge structural database. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2016;72:171–179. doi: 10.1107/S2052520616003954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aree T. Supramolecular complexes of β-cyclodextrin with clomipramine and doxepin: Effect of the ring substituent and component of drugs on their inclusion topologies and structural flexibilities. Pharmaceuticals (Basel) 2020;13 doi: 10.3390/ph13100278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou Z., Zhen J., Karpowich N.K., et al. Antidepressant specificity of serotonin transporter suggested by three LeuT-SSRI structures. Nat. Struct. Mol. Biol. 2009;16:652–657. doi: 10.1038/nsmb.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abouhussein D.M.N., El Nabarawi M.A., Shalaby S.H., et al. Sertraline-cyclodextrin complex orodispersible sublingual tablet: Optimization, stability, and pharmacokinetics. J. Pharm. Innov. 2021;16:53–66. [Google Scholar]
- 42.Koshland D.E., Jr. Protein and biological control. Sci. Am. 1973;229:52–64. doi: 10.1038/scientificamerican1073-52. [DOI] [PubMed] [Google Scholar]
- 43.Lindner K., Saenger W. Crystal and molecular structure of cyclohepta-amylose dodecahydrate. Carbohydr. Res. 1982;99:103–115. [Google Scholar]
- 44.Aree T., Jongrungruangchok S. Crystallographic evidence for β-cyclodextrin inclusion complexation facilitating the improvement of antioxidant activity of tea (+)-catechin and (−)-epicatechin. Carbohydr. Polym. 2016;140:362–373. doi: 10.1016/j.carbpol.2015.12.066. [DOI] [PubMed] [Google Scholar]
- 45.Cremer D., Pople J.A. General definition of ring puckering coordinates. J. Am. Chem. Soc. 1975;97:1354–1358. [Google Scholar]
- 46.Bredikhin A.A., Bredikhina Z.A., Zakharychev D.V., et al. Chiral drug timolol maleate as a continuous solid solution: Thermochemical and single crystal X-ray evidence. CrystEngComm. 2012;14:648–655. [Google Scholar]
- 47.Parvez M. Structure of an anhistaminic drug, racemic chlorpheniramine hydrogen maleate. Acta Crystallogr. C. 1990;46:943–945. [Google Scholar]
- 48.Mazurek A.H., Szeleszczuk Ł. Current status of quantum chemical studies of cyclodextrin host-guest complexes. Molecules. 2022;27 doi: 10.3390/molecules27123874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tavoulari S., Forrest L.R., Rudnick G. Fluoxetine (Prozac) binding to serotonin transporter is modulated by chloride and conformational changes. J. Neurosci. 2009;29:9635–9643. doi: 10.1523/JNEUROSCI.0440-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang W., Gong W., Zhou S., et al. β-cyclodextrin improve the tolerant of freshwater algal Spiny Scenedesmus to chiral drugs venlafaxine and its metabolite. J. Hazard. Mater. 2020;399 doi: 10.1016/j.jhazmat.2020.123076. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available after acceptance. CCDC: 2242694; 2242695.