

Abstract
Despite unequivocal evidence that folate deficiency increases risk for human pathologies, and that folic acid intake among women of childbearing age markedly decreases risk for birth defects, definitive evidence for a causal biochemical pathway linking folate to disease and birth defect etiology remains elusive. The de novo and salvage pathways for thymidylate synthesis translocate to the nucleus of mammalian cells during S- and G2/M-phases of the cell cycle and associate with the DNA replication and repair machinery, which limits uracil misincorporation into DNA and genome instability. There is increasing evidence that impairments in nuclear de novo thymidylate synthesis occur in many pathologies resulting from impairments in one-carbon metabolism. Understanding the roles and regulation of nuclear de novo thymidylate synthesis and its relationship to genome stability will increase our understanding of the fundamental mechanisms underlying folate- and vitamin B12–associated pathologies.
Keywords: thymidylate, folate, replitase, neural tube defects, DNA synthesis
INTRODUCTION
Shortly following the discovery of folate, a water-soluble B vitamin, in the early 1940s as an essential nutrient required for cell proliferation (57), research rapidly led to the development of anticancer, antimicrobial, and anti-inflammatory folate antagonists, including methotrexate and pemetrexed, that are important and widely used pharmaceuticals in oncology and rheumatology. Meanwhile, advances in human nutrition research established the role of folate in health, as folate deficiency was demonstrated to increase risk for a variety of human pathologies, including certain cancers, anemia, neurodegeneration, and birth defects. Evidence that folic acid administration could markedly reduce a woman’s risk of having a neural tube defect (NTD)-affected pregnancy led to public health initiatives, including food fortification, to increase blood folate levels in women of reproductive age and thereby decrease rates of birth defects, most notably NTDs such as spina bifida and anencephaly (145).
Despite unequivocal evidence that folate deficiency increases risk for human pathologies, and that folic acid intake among women of child-bearing age markedly decreases risk for NTDs, definitive evidence for a causal biochemical pathway linking folate to disease and birth defect etiology remains elusive. Many molecular mechanisms underlying the linkage between folate metabolism and pathology have been proposed. Folate functions as an enzyme cofactor that chemically activates and carries single carbons for the synthesis of purines and thymidylate (dTMP) and for the remethylation of homocysteine to methionine (Figure 1) (44). Methionine is an essential amino acid required for protein synthesis as well as for cellular methylation reactions through its conversion to S-adenosylmethionine (AdoMet), a cofactor required for more than 100 methylation reactions (44). As such, folate cofactors are essential for DNA and RNA synthesis, chromatin methylation, phospholipid biosynthesis, genome stability, cell proliferation, and the synthesis and catabolism of numerous metabolites. These anabolic pathways function as a highly interconnected network known as folate-mediated one-carbon metabolism (FOCM), and folate deficiency and other factors that perturb the function of folate simultaneously influence all pathways within the network (Figure 2) (122). Therefore, identifying the causal pathways and molecular mechanisms for individual folate-associated pathologies has proven to be challenging (122).
Figure 1.
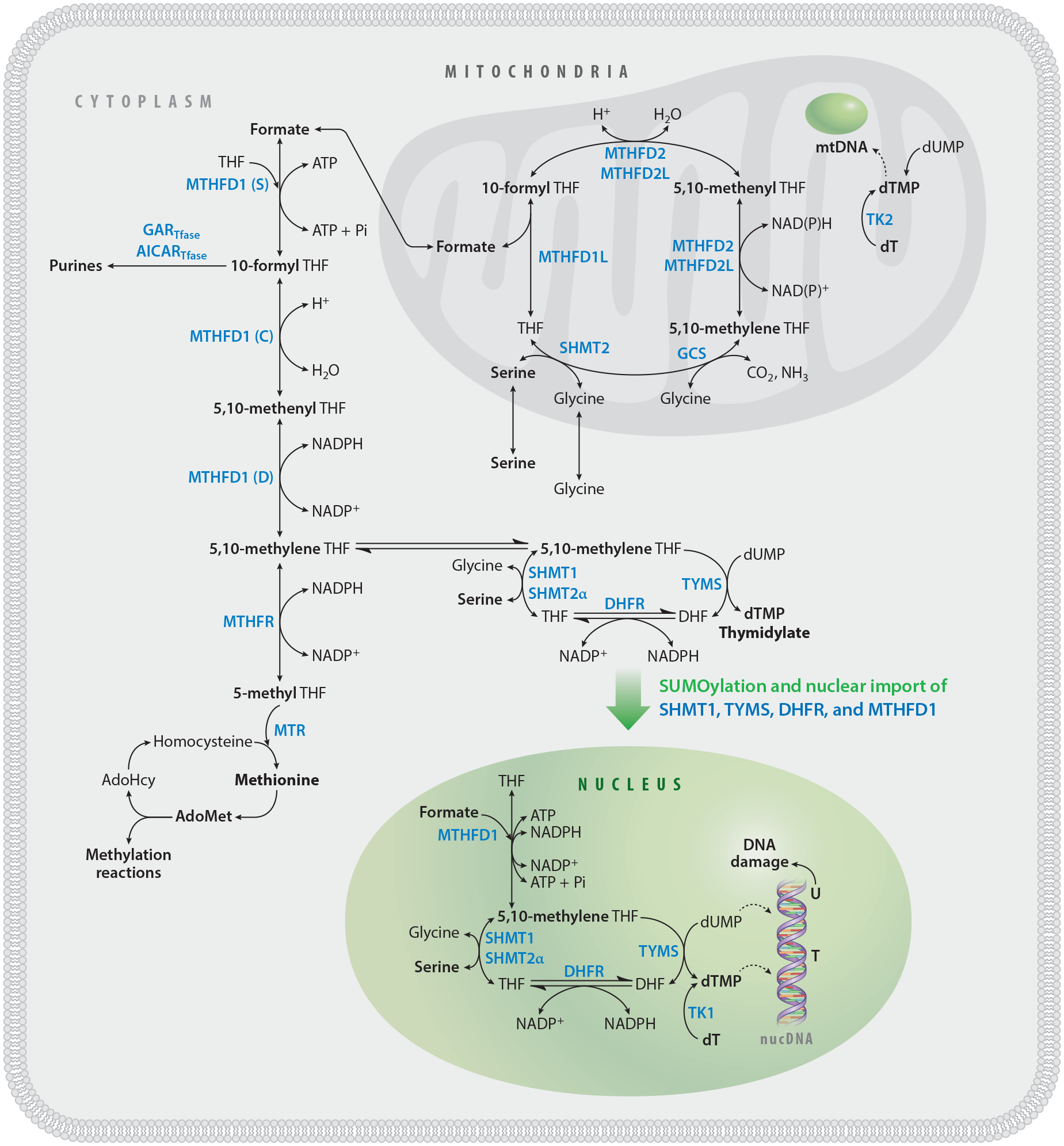
FOCM is compartmentalized within the cell. FOCM compartmentalization enables efficient metabolic flux of THF-activated one-carbon units to biosynthetic reactions and cycling of THF. One-carbon metabolism in the cytosol includes the remethylation of homocysteine to methionine and the de novo synthesis of purines and thymidylate. Nuclear de novo dTMP biosynthesis requires TYMS, DHFR, MTHFD1, SHMT1, and SHMT2α. These enzymes are SUMOylated and imported into the nucleus at the onset of S-phase. The Shmt2 gene is expressed as two transcripts: SHMT2, for mitochondrial one-carbon metabolism, and SHMT2α, which functions in the cytosol and nucleus. Formate and serine are the primary one-carbon sources for cytosolic and nuclear FOCM. The hydroxymethyl group of serine enters the pool of activated one-carbon units through the SHMT-catalyzed reaction in the cytosol and mitochondria. In mitochondria, serine and glycine are converted to formate, which traverses to the cytosol and nucleus, where it is condensed with THF by MTHFD1. MTHFD1 is a trifunctional enzyme possessing formylTHF synthetase (S), methenylTHF cyclohydrolase (C), and methyleneTHF dehydrogenase (D) activities. 5,10-MethyleneTHF is synthesized by SHMT or MTHFD1 and used for de novo dTMP synthesis. Thymidylate is also produced through the salvage of dT nucleoside by the action of TK1 in the cytosol and nucleus and of TK2 in mitochondria. Abbreviations: AdoHcy, S-adenosylhomocysteine; AdoMet, S-adenosylmethionine; AICARTfase, aminoimidazolecarboxamide ribonucleotide transformylase; DHFR, dihydrofolate reductase; dT, deoxythymidine; dTMP, deoxythymidine monophosphate; dUMP, deoxyuridine monophosphate; FOCM, folate-mediated one-carbon metabolism; GARTfase, glycinamide ribonucleotide tranformylase; MTHFD1, methylenetetrahydrofolate dehydrogenase 1; MTHFR, methylenetetrahydrofolate reductase; MTR, methionine synthase; mtDNA, mitochondrial DNA; SHMT, serine hydroxymethyltransferase; SUMO, small ubiquitin-like modifier; THF, tetrahydrofolate; TK, thymidine kinase; TYMS, thymidylate synthase.
Figure 2.
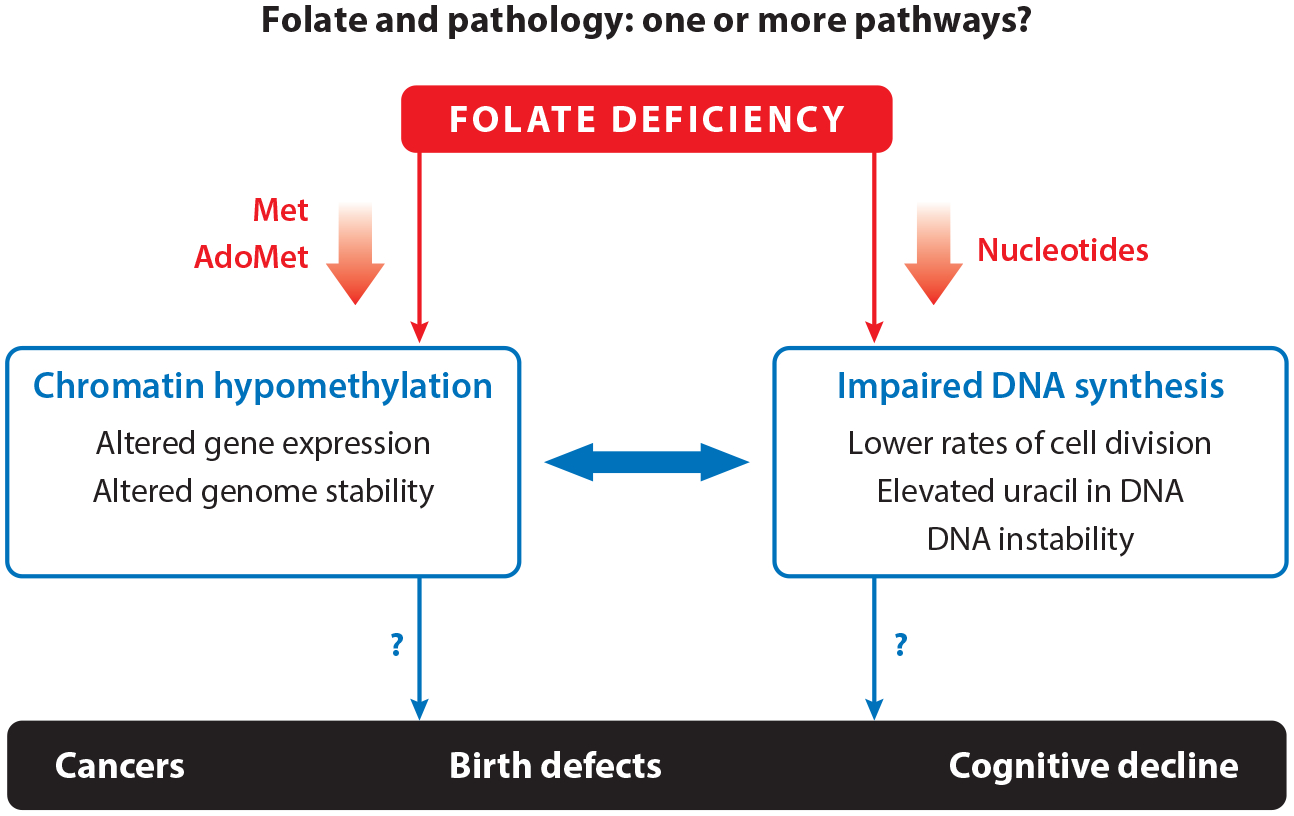
Folate and pathology. Impaired folate-mediated one-carbon metabolism is associated with biomarkers including decreased Met and AdoMet, which leads to chromatin hypomethylation, as well as decreased nucleotide synthesis, which leads to decreased cell division and increased uracil misincorporation into DNA. Impaired folate-mediated one-carbon metabolism is also associated with pathologies including cancer, neurodegenerative disease, and neural tube defects. All of these pathologies are complex traits, making it difficult to understand causal mechanisms that link impaired folate-mediated one-carbon metabolism to pathology. Abbreviations: AdoMet, S-adenosylmethionine; Met, methionine.
FOCM is compartmentalized in the cell in the mitochondria, the cytosol, and the nucleus (Figure 1). The function of FOCM in mitochondria is to generate one-carbon units in the form of formate for one-carbon metabolism in the cytosol and to generate dTMP and N-formylmethionine tRNA for mitochondrial DNA and protein synthesis, respectively. The role of cellular compartmentation in the functioning of the network has been reviewed recently (137). This review focuses on recent advances in our understanding of nuclear de novo dTMP synthesis and nuclear FOCM, and their emerging roles in the etiology of folate-associated pathologies.
NUCLEAR FOLATE METABOLISM AND THE DISCOVERY OF THE REPLITASE
dTMP biosynthesis occurs in cells through both a nucleotide salvage pathway and a folate-dependent de novo synthesis pathway (Figure 1). The salvage pathway involves a single enzyme, thymidine kinase 1 (TK1), that phosphorylates thymidine to dTMP. The de novo synthesis of dTMP includes the enzymes serine hydroxymethyltransferase 1 and 2α (SHMT1 and SHMT2α, encoded by separate genes) and methylenetetrahydrofolate dehydrogenase 1 (MTHFD1), which catalyze the conversion of tetrahydrofolate (THF) to 5,10-methyleneTHF using serine and formate as formaldehyde donors, respectively. Thymidylate synthase (TYMS) requires 5,10-methyleneTHF to reductively methylate deoxyuridine monophosphate (dUMP) to dTMP, generating dihydrofolate (DHF). In this reaction, the folate cofactor serves not only as a formaldehyde donor, but also as a source of reducing equivalents. To complete the cycle, DHF is reduced to THF by the nicotinamide adenine dinucleotide phosphate (NADPH)-requiring enzyme dihydrofolate reductase (DHFR).
Early studies in 1965 demonstrated the presence of folate-dependent enzymes including SHMT and TYMS in nuclear fractions of rat liver (21), where the authors postulated, “It seems that the enzymatic synthesis of dTMP from deoxyuridylate is effected, in part at least, in the cell nucleus” (21, p. 36C). Previous to these findings, dTMP synthesis was assumed to occur exclusively in the cytosol, as for cytosine and purine nucleotide biosynthesis. Nucleotides synthesized in the cytosol can be shuttled to both the nucleus and the mitochondria in support of DNA synthesis for DNA replication and repair.
Another study found SHMT and enzymes that catabolize homocysteine in rat brain nuclei. Methionine synthase (MTR), which uses 5-methylTHF to convert homocysteine to methionine, was not identified in nuclei in this study (107). Mouse liver exposed to exogenous 14C-labeled folic acid contained labeled folate within nuclei 30 min following infusion, and nuclear levels of the label remained stable for up to 120 min. In contrast, mitochondrial 14C-folate levels continued to increase after 30 min in this study (114). In subsequent studies, 24-h perfusion of rat liver with labeled folic acid resulted in 32% of the labeled folate localizing to nuclei and 25% to mitochondria (160). Interestingly, almost all nuclear and mitochondrial folates presented as folate polyglutamates (126), indicating that they were able to function as metabolic cofactors. This is because folate enters cells as the monoglutamate form of the vitamin, but after entry into the cell it is converted to a polyglutamate form containing three to nine glutamate residues lined though unusual γ-glutamyl peptide bonds. The folate polyglutamate peptide aids in cellular retention of the vitamin and increases the affinity of folate for folate-dependent enzymes by as much as 1,000-fold (136, 144). The mechanism for folate transport into mitochondria is well established (84), whereas mechanisms for folate accretion in nuclei are unknown. During folate deficiency in mice, folate levels in the cytosol deplete markedly, whereas nuclear folate levels are resistant to depletion (40).
The first evidence that FOCM functioned in the nucleus was reported in 1980 in a landmark paper by Prem Veer Reddy & Pardee (106). This study identified six enzyme activities from S-phase nuclei from the Chinese hamster embryo fibroblast cell line CHEF/18 that cosedimented in sucrose density gradients (106). The multienzyme complex was termed the replitase and was present in S-phase nuclei but absent from nuclei in G1-phase. The replitase was found to contain the activities of DNA polymerase, TK1, DHFR, TYMS, nucleoside 5′-phosphate kinase, and ribonucleotide reductase (RNR) (96, 106, 158) (Table 1). Later, other proteins including c-myc (133), calmodulin, and calmodulin-binding protein (135) were identified as components of the replitase. Subsequent studies indicated that TYMS was active in live cells during S-phase only when it was present in the nucleus and only when it was associated with the replitase, even though the enzyme was present in the cytosol during all stages of the cell cycle (105). The observation that the replitase was present in S-phase cells was replicated in proliferating T cells by another research group (115). The replitase was purified as 20–30-nm spheres from isolated calf thymus nuclei in S-phase but again absent in nuclei from G1-phase cells. Curiously, both the de novo and the salvage pathway enzymes were present in the replitase.
Table 1.
Components of the replitase identified in 1980
| Component | Reference(s) |
|---|---|
| DNA polymerase | 96, 106 |
| Thymidine kinase | 96, 106 |
| Dihydrofolate reductase | 96, 106 |
| Nucleoside 5′-phosphate kinase | 96 |
| dTMP synthase | 106,158 |
| Ribonucleotide reductase | 96 |
| Topoisomerase | 96 |
| DNA methyltransferase | 96, 115 |
| c-Myc | 133 |
| Calmodulin-binding protein | 135 |
| Calmodulin | 135 |
Abbreviation: dTMP, deoxythymidine monophosphate.
The Pardee group provided evidence that the dTMP synthesis components of the replitase functioned as a coordinated unit that generated and channeled deoxyribonucleotides to DNA polymerase during DNA synthesis in living permeabilized cells. The replitase restricted the incorporation of exogenous deoxynucleotide triphosphates into DNA (106). Incorporation of exogenous deoxynucleotide triphosphates that entered permeabilized cells into DNA occurred only when RNR was inhibited with hydroxyurea.
The concept that the enzymes of the replitase functioned as a single unit, and that inhibition of a single enzyme within the replitase impaired all catalytic functions of the replitase, was controversial. Inhibition of DNA polymerase, RNR, or topoisomerase impaired TYMS activity in CHEF/18 cells (143), and inhibition of DNA repair in human foreskin fibroblasts resulted in decreased DNA synthesis activity as well as TYMS activity. The magnitude of the inhibition was the same for both enzymes (18). These findings led to the hypothesis that the replitase synthesizes and channels deoxythymidine triphosphate (dTTP) directly to DNA polymerase in CHEF/18 cells (19, 106). Chiba et al. (29) used a different approach to test this hypothesis in L1210 cells. Their study showed that inhibition of RNR by hydroxyurea, or of DNA polymerase by aphidicolin, impaired DNA synthesis up to 100% but with corresponding decreases in TYMS activity of only 65% and 60%, respectively. The authors concluded that TYMS activity was not completely dependent on or linked to RNR or DNA polymerase activity as previous studies suggested but nonetheless was sensitive to inhibition of enzymes within the replitase. The authors attributed the inhibition of TYMS to imbalances in deoxynucleotide triphosphate pools resulting from reductions in RNR or DNA polymerase activity. Rode et al. (112) also observed that reductions in TYMS activity were linked to reductions in DNA polymerase or RNR activity in mouse L1210 cells and human CCRF-CM and SKL7 tumor cells to varying degrees. Importantly, they also observed that impairments in TYMS activity were temporally delayed relative to RNR and DNA polymerase activity, supporting the hypothesis that decreased TYMS activity resulted from alterations in nucleotide pools rather than from allosteric interactions within the replitase multienzyme complex (112). This hypothesis that nucleotide imbalances impaired TYMS activity in whole cells was tested by examining the ability of nucleotide imbalances to inhibit replitase activity in CHEF/18 cells (102). This study not only replicated the finding that both RNR and DNA polymerase inhibitors impaired TYMS activity in cells, but also demonstrated that these inhibitors inhibited the dTMP salvage pathway, which is catalyzed by TK1. Furthermore, these changes in TYMS and TK1 enzyme activities occurred without alterations in nucleotide pools. This finding supported a mechanism of cross-inhibition in which an inhibitor of one enzyme in the replitase complex impaired the activity of other enzymes in the complex. This study was performed in cells that were almost completely synchronized in S-phase. Although this is a strength of the study, it did not allow for quantification of the temporal delay in TYMS or TK1 inhibition following inhibition of RNR or DNA polymerase activity.
Investigation into the role of the replitase in nuclear folate-dependent de novo dTMP biosynthesis lost momentum in the 1980s, potentially because studies of Saccharomyces cerevisiae demonstrated that TYMS localized to the nuclear periphery but not to the nucleus during S-phase (104). This finding indicated that nuclear localization of TYMS was not required for DNA synthesis in all eukaryotes. Rather, studies focused on understanding the functioning of the folate-dependent pathways in the cytosol, including de novo dTMP biosynthesis and the interaction of TYMS with other folate-dependent pathways. Empirical and mathematical modeling studies from Schirch and colleagues (118, 131, 132) and other research groups (136) revealed that the intracellular concentration of folate-utilizing enzymes was 2- to 10-fold higher than the intracellular concentration of folate, necessitating a competition among folate-utilizing enzymes for a limited pool of folate coenzyme cofactors (54). This competition is particularly pronounced for 5,10-methyleneTHF, which exists at a branch point in one-carbon metabolism. TYMS can utilize 5,10-methyleneTHF for dTMP synthesis or methylenetetrahydrofolate reductase (MTHFR) can irreversibly convert it to 5-methylTHF (Figure 1). Isotope tracer studies indicated that 5,10-methyleneTHF is directed toward de novo dTMP synthesis at the expense of homocysteine remethylation under folate-deficient conditions (50, 97) or as a result of increased SHMT1 expression (54). Initially, this observation was explained as a kinetic competition for 5,10-methyleneTHF cofactors between the enzymes MTHFR and TYMS and did not account for compartmentation of de novo dTMP biosynthesis in the nucleus. The roles of the salvage and de novo dTMP synthesis pathways in the nucleus remained unexplained.
SMALL UBIQUITIN-LIKE MODIFIER–DEPENDENT LOCALIZATION OF THE DE NOVO SYNTHESIS PATHWAY IN MAMMALIAN CELLS
Investigation into the role of nuclear dTMP biosynthesis was renewed following an exploratory study that sought to identify SHMT1-interacting proteins. Studies using a yeast two-hybrid system identified UBC9, an E3 small ubiquitin-like modifier (SUMO) ligase, as an SHMT1-interacting protein. Covalent posttranslational SUMO modification (SUMOylation) targets proteins for nuclear translocation, influences enzyme activity, and can be essential for enabling or enhancing protein-protein interactions through noncovalent interactions with SUMO-interacting motifs (53). The interaction between SHMT1 and UBC9 indicated that SHMT1, as well as other members of the de novo dTMP synthesis pathway, may be subject to SUMO modification leading to nuclear translocation. SHMT1, TYMS, and DHFR, as well as MTHFD1, contained consensus sites for SUMO modification, and the recombinant proteins were substrates for UBC9-mediated SUMOylation in vitro (8, 62, 152). Similarly, large-scale screens for SUMOylated proteins in human cells identified the enzymes of the de novo dTMP biosynthesis, and these data were comprehensively compiled and recently reviewed (53). However, large-scale proteomics screens have not identified SUMOylation of the dTMP salvage enzymes TK1 or thymidylate kinase, which converts dTMP to deoxythymidine diphosphate, in human cells (53), although they were identified as components of the nuclear replitase. This difference indicates that there could be more than one mechanism to translocate the enzymes of the replitase into the nucleus.
Tandem affinity purification of SHMT1-binding partners from S-phase and UV-treated HeLa cells indicated that SHMT1 physically interacted with the enzymes of the de novo dTMP synthesis complex, as well as with numerous enzymes required for DNA replication and repair, including nuclear lamin proteins (7). Not surprisingly, many but not all proteins identified in 1980 as part of the replitase were directly or indirectly physically associated with SHMT1 (Table 1) (7, 106). Of interest, SHMT1-interacting proteins included proliferating cell nuclear antigen (which acts as a sliding clamp during nuclear DNA replication), UBC9, and lamin proteins (7). SHMT1 was confirmed to bind lamin A/C and B1 directly and tightly (7). UBC9 was also identified as an SHMT1-interacting protein. UBC9 is responsible for covalent modification of target proteins with the SUMO protein; target proteins are modified on conserved lysine residues with the consensus motif [IVL]KXE, where X can be any amino acid and the amino acids in brackets can be either isoleucine, valine, or leucine (53, 152). SUMOylation of SHMT1 is essential for its nuclear import and may be required for formation of the replitase multienzyme complex in the nucleus (7, 8, 152).
EVIDENCE FOR A LOCALIZATION OF THE DE NOVO dTMP BIOSYNTHESIS COMPLEX TO THE NUCLEAR LAMINA AND SITES OF DNA SYNTHESIS
SHMT (and likely SHMT2α), TYMS, DHFR, and MTHFD1 undergo SUMO-dependent translocation to the nucleus at the G1/S boundary in MCF-7 and HeLa cells and remain in the nucleus during S- and G2/M-phases when they have an intact SUMOylation site (152). MTHFD1, TYMS, and DHFR fluorescent fusion proteins colocalized with SHMT and nuclear lamin proteins during S- and G2/M-phases in HeLa cells (7). The de novo dTMP synthesis complex was enriched at origins of replication of an extrachromosomal plasmid, indicating de novo dTMP biosynthesis occurs at the replication fork (Figure 3) (7).
Figure 3.
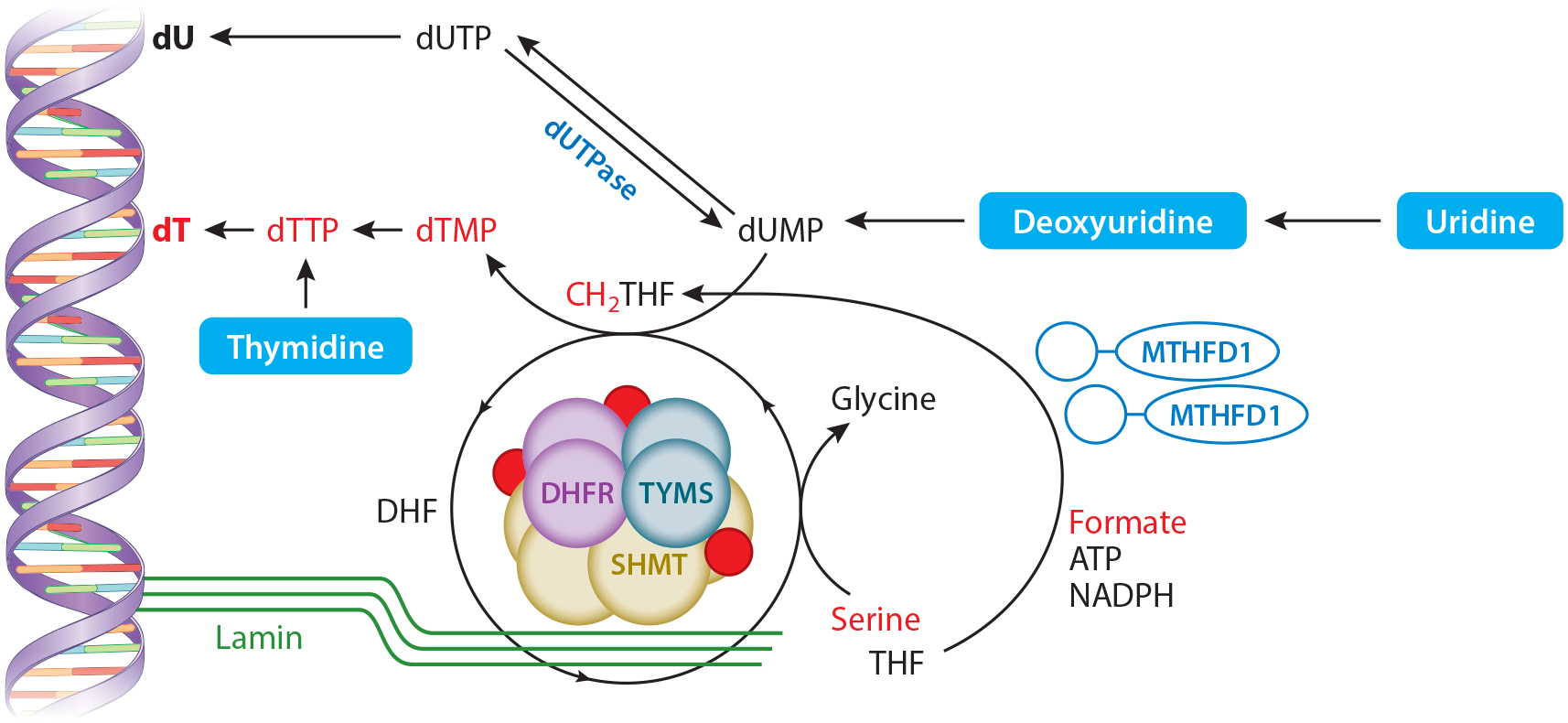
The de novo thymidylate synthesis pathway as a nuclear multienzyme complex at sites of DNA replication. SHMT, TYMS, and DHFR are SUMOylated covalently attached to the SUMO protein (red circles) at S-phase after which they translocate to the nucleus and form a multienzyme complex. This multienzyme complex is anchored to nuclear lamin proteins and DNA by SHMT (SHMT1 and/or SHMT2α isozymes). Both serine (through catalytic activity of SHMT1 and/or SHMT2α) and formate (through catalytic activity of MTHFD1) serve as one-carbon sources for the generation of CH2THF. TYMS catalyzes conversion of dUMP to dTMP, which is converted to dTTP and incorporated into DNA as dT (the T base in DNA). Alternatively, dUMP can be converted to dUTP, which is then incorporated into DNA as deoxyuridine (known as uracil misincorporation). The dUTPase enzyme converts dUTP to dUMP, which both limits dUTP accumulation and provides dUMP substrate for TYMS. Abbreviations: ATP, adenosine triphosphate; DHF, dihydrofolate; DHFR, dihydrofolate reductase; dT, deoxythymidine; dTMP, deoxythymidine monophosphate; dTTP, deoxythymidine triphosphate; dUMP, deoxyuridine monophosphate; dUTP, deoxyuridine triphosphate; MTHFD1, methylenetetrahydrofolate dehydrogenase 1; NADPH, nicotinamide adenine dinucleotide phosphate; SHMT1, cytoplasmic serine hydroxymethyltransferase 1; SUMO, small ubiquitin-like modifier; THF, tetrahydrofolate; TYMS, thymidylate synthase.
Importantly, expression of SHMT1 or SHMT2α, the nuclear isozyme of mitochondrial SHMT2 that provides functional redundancy with SHMT1 (6), is essential to tether the de novo dTMP synthesis enzymes TYMS and DHFR to DNA and the DNA replication machinery (7). Formation of the multienzyme complex responsible for nuclear de novo dTMP biosynthesis is also essential to prevent accumulation of uracil in DNA, as has been demonstrated in mice (73). Furthermore, expression of a catalytically inactive SHMT1 mutant also increases rates of de novo dTMP biosynthesis in cultured cells (97), emphasizing the importance of SHMT1 expression, localization, and function as a scaffold protein in dTMP synthesis independent of its catalytic activity. This finding is consistent with observations showing that formate, through MTHFD1 activity, provides most folate-activated one-carbon units for de novo dTMP synthesis (as opposed to serine through catalytic activity of SHMT) (54).
Knockout mouse models of de novo dTMP synthesis enzymes have helped further elucidate that SHMT serves as an essential scaffolding protein at sites of DNA synthesis, whereas MTHFD1 supplies the one-carbon units from formate for dTMP synthesis (7) (Figure 1). Mthfd1 is an essential gene in mice (77), whereas Shmt1 is not (75) likely owing to functional redundancy with SHMT2α (6). The primary phenotypes resulting from loss of SHMT1 activity in mice are a fourfold increase in uracil levels in liver and colon DNA and the development of folic acid–responsive NTDs (12, 75). Loss of Shmt1 expression has minimal impact on homocysteine remethylation (12, 75). Similarly, uracil in nuclear DNA is also elevated in a mouse model of increased SHMT1 expression; in this mouse model, overexpression of SHMT1 impaired its localization to the nucleus (73). In contrast, mice lacking one copy of Mthfd1 and consuming a folate-deficient diet were protected from uracil accumulation in colon DNA and did not develop NTDs (13, 78). As described below, both observations are likely explained by the preferential translocation and enrichment of MTHFD1 protein to liver nuclei in Mthfd1gt/+ mice consuming a folate-deficient diet (40).
The MTHFD1 protein is partitioned between the nucleus and the cytosol to support nuclear de novo dTMP synthesis and homocysteine remethylation, respectively. The partitioning of MTHFD1 between these two compartments appears to be regulated robustly. Human MTHFD1 single-nucleotide polymorphisms are associated with adverse pregnancy outcomes such as NTDs and congenital heart defects (20, 100, 101). Identification of a proband with severe MTHFD1 deficiency by exome capture sequencing has further advanced our understanding of the role of MTHFD1 in nuclear one-carbon metabolism and de novo dTMP synthesis (41, 64, 148). The proband presented with classical signs of impaired vitamin B12 metabolism (e.g., elevated plasma homocysteine, decreased plasma methionine, and megaloblastic anemia) as well as severe combined immunodeficiency (SCID) (64, 148). The proband inherited deleterious mutations in both MTHFD1 alleles, resulting in an early stop codon in the cyclohydrolase/dehydrogenase domain and an R173C mutation in the NADP+ binding site in the other allele. Levels of full-length MTHFD1 protein were reduced by 50% in patient fibroblasts, which exhibited decreased methionine and de novo dTMP synthesis, whereas de novo purine synthesis was unaffected. However, the MTHFD1-catalyzed incorporation of formate into methionine in protein was reduced by 90% in patient fibroblasts, whereas formate incorporation into dTMP was reduced by only 50% (41). Subsequent studies indicated that MTHFD1 was preferentially partitioned into the nucleus of patient fibroblasts compared with control fibroblasts, thereby protecting nuclear de novo dTMP synthesis at the expense of methionine synthesis in the cytoplasm. Interestingly, the mitochondrial MTHFD2 protein colocalized with newly synthesized DNA in the nuclei of U251, HeLa, and HCT116 cells (125). MTHFD2 overexpression also increased HCT116 cell proliferation, even when MTHFD2 lacking functional dehydrogenase activity was overexpressed (125). This observation suggests that the function of MTHFD2 within the nucleus is distinct from its catalytic activity.
In summary, SHMT1 partitioning between the cytoplasm and nucleus impacts dTMP synthesis but has little impact on homocysteine remethylation capacity (73). This is not the case for MTHFD1 partitioning. Impaired MTHFD1 activity reduces rates of homocysteine remethylation and de novo dTMP synthesis (16), resulting in uracil incorporation and accumulation in DNA (41). MTHFD1 protein is preferentially enriched in the nucleus during folate deficiency (40), without changes in MTHFD1 expression. Loss of MTHFD1 activity also results in increased MTHFD1 protein partitioning into the nucleus of human fibroblasts (41), such that nuclear enrichment of MTHFD1 supports nuclear de novo dTMP synthesis at the expense of suppling one-carbon units for homocysteine remethylation in the cytosol (40, 41). Nuclear localization of MTHFD1 also accounts for the observation that even when SHMT1 is expressed, formate remains the major source of one-carbon units required for nuclear de novo dTMP synthesis (40, 54). More recent studies have demonstrated that 5-formylTHF (a potent, slowly binding inhibitor of SHMT1) is enriched in the nucleus, indicating that SHMT1 activity is inhibited mostly within the nuclear compartment (99) (Figure 1).
PRESENCE OF OTHER FOLATE-RELATED PATHWAYS IN THE NUCLEUS
To date, there is no strong evidence that either folate-dependent de novo purine biosynthesis or homocysteine remethylation occurs outside the cytosol. Enzymes required for de novo purine biosynthesis form a multienzyme complex in mammalian cells cultured under purine-deficient conditions, but these enzymes and the complex localize exclusively to the cytosol (3, 39). Likewise, homocysteine remethylation to methionine also appears to be a cytosolic process, as the enzymes that compose this cycle that converts homocysteine to methionine, including MTHFR, MTR, and methionine synthase reductase, are cytosolic (Figure 1) (47, 85, 107). Methionine is a precursor of AdoMet, the methyl group donor required for methylation of both DNA and histones within the nucleus.
In contrast to methionine synthesis, AdoMet synthesis occurs in both cytosolic and nuclear compartments. Methionine adenosyltransferase (MAT) isozymes, which synthesize AdoMet from methionine and adenosine triphosphate, localize to the nuclear compartment in rat liver in response to acute liver injury (36) and in Chinese hamster ovary cells (111). In Chinese hamster ovary cells, MAT1 overexpression also selectively increases H3K27 trimethylation, suggesting a coupling between AdoMet synthesis and histone methylation (60, 111). In the process of serving as a methyl donor, AdoMet is converted to S-adenosylhomocysteine (AdoHcy), which is then recycled to homocysteine by the enzyme S-adenosylhomocysteine hydrolase (SAHH) (Figure 1) (83). AdoHcy binds more tightly to many AdoMet-requiring enzymes than AdoMet does, therefore acting as an inhibitor of the methyltransferase enzymes (28). SAHH also translocates to the nucleus (36), presumably to limit nuclear AdoHcy levels and prevent methyltransferase inhibition (60).
Glycine N-methyltransferase (GNMT) localizes to the nucleus. It is a cytosolic protein that primarily regulates the cellular methylation potential (also known as the AdoMet-to-AdoHcy ratio) by catalyzing the conversion of AdoMet and glycine to N-methylglycine (sarcosine) and AdoHcy. GNMT expression is highest in human liver, kidney, and pancreas, but GNMT is also expressed in heart, brain, lung, testes, and ovary (35). In rat liver, it composes as much as 3% of total cytosolic liver protein (159) and is present in rat liver nuclei (68, 159). Its primary function is to limit AdoMet accumulation by catabolizing AdoMet, thereby suppressing and regulating intracellular AdoMet levels and, by extension, the AdoMet-to-AdoHcy ratio and the cellular methylation potential (35, 72). GNMT is also a tight, 5-methylTHF-binding protein, and 5-methylTHF binding inhibits GNMT catalytic activity. GNMT is found in nuclei of normal and transformed human cells (35,68). In cancer cells, nuclear localization of GNMT induces apoptosis independent of GNMT catalytic activity (35), indicating that its 5-methylTHF-binding activity may play an important physiological role. Interestingly, partitioning of GNMT to the nucleus increases during folate deficiency in HepG2 cells. Furthermore, GNMT overexpression limits uracil accumulation in DNA (147). The metabolic role of GNMT in the nucleus is not clear, and GNMT has not been identified as a component of the replitase. But mice lacking GNMT expression exhibit elevated uracil in DNA, indicating impaired de novo dTMP synthesis, and the levels of uracil in DNA in this model are not responsive to dietary folate (147). 5-MethylTHF is a major folate form in the nucleus during vitamin B12 deficiency (99). Accumulation of 5-methylTHF impairs de novo dTMP biosynthesis through a methyl trap (an accumulation of 5-methylTHF at the expense of other folate cofactors), resulting in inhibition of de novo dTMP biosynthesis (124). This suggest that GNMT may play a critical role in sequestering nuclear 5-methylTHF, which is an inhibitor of SHMT1, but further experiments are required to elucidate the role of GNMT in nuclear de novo dTMP synthesis.
URACIL IN DNA AS A BIOMARKER OF IMPAIRED NUCLEAR FOLATE-MEDIATED ONE-CARBON METABOLISM
Serum and tissue biomarkers of impaired FOCM include depressed AdoMet levels and elevated homocysteine and AdoHcy levels (32, 54), leading to hypomethylated DNA and protein (including histones), which affects gene expression and DNA stability (45, 46, 59, 61). Impaired FOCM also depresses de novo dTMP synthesis (16), resulting in uracil misincorporation and accumulation in nuclear DNA (Figure 4). The relationship between B-vitamin nutrition and DNA uracil content is not fully understood. Uracil has been suggested to be a biomarker of folate (16) and vitamin B12 status (63), but not all studies agree. Uracil accumulated in DNA of proliferating cells threefold higher than in quiescent cells, indicating that misincorporation during replication is the major source of DNA uracil (4). Uracil levels in white blood cells did not correlate with folate status in females 20–23 years old but did correlate inversely with vitamin B12 status in folate-replete individuals (63). In a study of 86 human subjects with a previous colorectal adenoma, high levels of folic acid (5 mg/day) and vitamin B12 (1.25 mg/day) supplementation increased levels of uracil DNA by 5.37 fmol/μg DNA in the rectal mucosa (141). Levels of uracil accumulation in DNA differ by tissue in mice (95). Homozygous deletion of the uracil excision repair enzyme uracil-DNA N-glycosylase (UNG) elevates nuclear DNA uracil levels by approximately twofold in liver and kidney but over sixfold in testes and spleen during folate deficiency (95). Ung encodes both nuclear and mitochondrial UNG isozymes, and folate-deficient Ung−/− mice, but not folate-deficient wild-type mice, exhibit increased mitochondrial DNA content in brain (67). However, it is not established whether AdoMet-, methylation-, or uracil-related biomarkers are causally related to the initiation or progression of FOCM-associated pathologies. Needed in the field are predictive biomarkers that directly co-report on nutritional status and disease initiation or progression along causal pathways.
Figure 4.
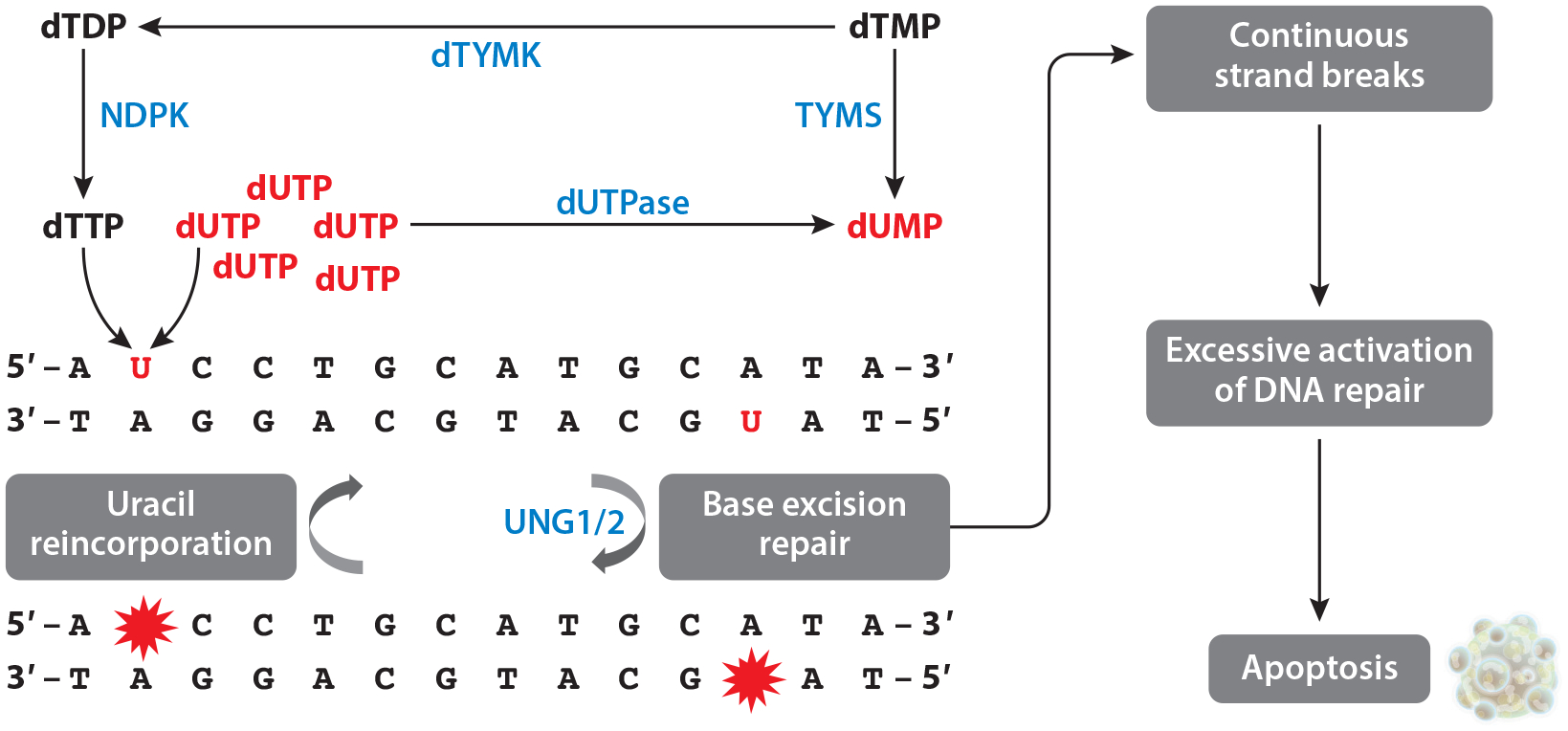
Causes and consequences of uracil in DNA. Either dTTP or dUTP can be incorporated into DNA. dUTPase dephosphorylates dUTP into dUMP. TYMS transfers a one-carbon unit onto dUMP, generating dTMP. dTYMK and NDPK subsequently phosphorylate dTMP and dTDP, respectively, generating dTTP. Impaired de novo thymidylate biosynthesis can lead to dUTP incorporation into DNA. Uracil in DNA is excised primarily by the base excision repair pathway, initiated primarily by UNG1 in the mitochondria and UNG2 in the nucleus. Base excision repair generates strand breaks as a repair intermediate. However, if dTTP pools are relatively limited, uracil will be incorporated into DNA again. This constant generation of strand breaks leads to continuous activation of double-strand break repair machinery, triggering apoptosis. Abbreviations: dTDP, deoxythymidine diphosphate; dTMP, deoxythymidine monophosphate; dTTP, deoxythymidine triphosphate; dTYMK, deoxythymidylate kinase; dUMP, deoxyuracil monophosphate; dUTP, deoxyuracil triphosphate; dUTPase, dUTP phosphorylase; NDPK, nucleoside diphosphate kinase; TYMS, thymidylate synthase; UNG1/2, uracil DNA N-glycosylase 1/2.
ROLE OF NUCLEAR dTMP SYNTHESIS IN FOLATE-ASSOCIATED PATHOLOGIES
Nutrition and genetic epidemiological studies, as well as randomized controlled trials, implicate impaired FOCM in several pathologies, including NTDs (153), neurodegenerative disease (67), and cancer (2, 16, 30, 65, 103). Folic acid supplementation alone is not sufficient to reduce or eliminate risk of pathologies associated with impaired FOCM, especially pathologies resulting from secondary nutrient deficiencies (e.g., vitamin B12) or those associated with genetic subpopulations involving gene-nutrient interactions (58).
Elucidating the causal pathways underpinning folate-related pathologies has proved to be challenging because both folate-dependent pathways and their associated biomarkers are tightly interconnected (Figures 1 and 2) (130). The complexity of the FOCM network is characterized by (a) a limited pool of folate cofactors that necessitate competition among the folate-dependent pathways (54); (b) cross talk among the folate-dependent pathways that include long-range and indirect regulatory interactions; (c) multienzyme complex formation and substrate channeling that create microenvironments for metabolic activity; (d) cellular compartmentation of pathways in the nucleus and mitochondria that are interconnected through the shuttling of serine, glycine, and formate; (e) feedback and feedforward interactions with other metabolic pathways that are connected to FOCM through upstream and downstream interactions; (f) dependence of the pathways on other B-vitamin cofactors including vitamin B6, vitamin B12, riboflavin, and niacin; and (g) penetrant genetic variants (44, 122). Because folate-dependent pathways generate purine and thymidine nucleotides for DNA synthesis, and AdoMet for methylation of both DNA and chromatin, the functioning of this metabolic network influences the expression and stability of the genome. Therefore, the genome is a reservoir of biomarkers that report on the functioning of folate metabolism (Figure 2). The illustrative examples below provide evidence for the role of nuclear de novo dTMP biosynthesis in the etiology of folate-responsive pathologies.
CANCER AND CHEMOTHERAPY
Almost immediately following the discovery of folate as an essential metabolic cofactor for DNA synthesis, folate analogs, otherwise referred to as antifolates, were developed to inhibit dTMP and purine nucleotide biosynthesis and have proved to be effective antimicrobial and antineoplastic agents that are still widely used (142). More recently, human epidemiological, genetic, and biochemical studies and animal studies have conclusively demonstrated that impairments in FOCM, resulting from dietary deficiencies or genetic manipulations, contribute to the initiation or progression of epithelial and potentially other cancers, especially colon cancer (80, 81). Maintenance of adequate folate status is essential for cancer prevention (82, 146), but elevated cellular folate concentrations have been proposed to promote carcinogenesis or tumor growth; however, the evidence base is limited. Meta-analyses have not supported increased cancer risk associated with dietary folic acid (43).
Pharmaceuticals targeting folate-dependent de novo dTMP synthesis enzymes include aminopterin, methotrexate, and nucleoside analogs (49). Methotrexate binds to DHFR, inhibiting its activity (1). DHFR binds its own messenger RNA, which prevents translation such that DHFR represses its own production (1, 9, 24). Resistance to methotrexate can develop when methotrexate prevents DHFR from binding to its own messenger RNA, interfering with the repressed translation (15). The 19-bp deletion polymorphism in intron-1 of DHFR affects DHFR transcription and translation. This polymorphism increases risk for several folate-associated pathologies, but risk seems to vary among populations (for a review, see Reference 1). DHFR activity is important both for de novo dTMP synthesis and for the conversion of dietary folic acid to THF, which is believed to occur in the cytosol. The effect of methotrexate on nuclear de novo dTMP synthesis has not been explored.
5-Fluorouracil (5-FU) inhibits TYMS, which leads to impaired dTMP production and cell death. More specifically, the 5-FU metabolite, 5-fluorodeoxyuridine monophosphate (FdUMP), forms a stable ternary complex with TYMS and 5,10-methyleneTHF, leading to formation of a covalent bond between FdUMP and TYMS. This covalent bond inactivates TYMS (116, 128), leading to dUMP accumulation and dTTP depletion. 5-FU is widely used in the treatment of breast, colorectal, epithelial, esophageal, and pancreatic cancers (71). Antifolates such as pemetrexed and raltitrexed also inhibit TYMS and are currently used to treat lung and colorectal cancers (49). Antifolate-induced impairments in dTMP biosynthesis increase genomic uracil incorporation and levels (73, 79) and increase DNA repair rates (119, 120), resulting in futile cycles of uracil misincorporation and base excision. These futile cycles ultimately lead to accumulation of DNA strand breaks and cell death owing to inadequate dTTP. FdUMP can also be converted to 5-fluorodeoxyuridine triphosphate (FdUTP) by thymidylate kinase, which converts FdUMP to 5-fluorodeoxyuridine diphosphate (FdUDP), and by nucleoside diphosphate kinase, which then converts FdUDP to FdUTP. FdUTP incorporation into DNA also contributes to 5-FU-mediated toxicity. In mammalian cells, 5-FU treatment can lead to higher levels of 5-FdU incorporation into DNA than of uracil incorporation into DNA. Conversely, in yeast cells exposed to 5-FU, uracil levels in DNA exceed those of 5-FdUTP (123). Yeast synthesize dTMP exclusively in the cytosol, raising the possibility that nuclear localization of TYMS may be a key modifier of the mechanism of action of 5-FU. In fact, evidence is emerging that nuclear compartmentation of the de novo dTMP synthesis pathway alters 5-FU efficacy in mammalian cells. Increased nuclear TYMS levels are associated with poorer response to 5-FU and with decreased survival in colorectal carcinoma (154). The common SHMT1 variant, SHMT1 C1420T (rs 1979277), results in an amino acid substitution (Leu474Phe) located near the site where SHMT1 interacts with the SUMO ligase enzymes (152). The SHMT1 C1420T polymorphism impairs SHMT1 SUMOylation and nuclear localization (5) and may decrease nuclear de novo dTMP synthesis capacity. Interestingly, the SHMT1 C1420T polymorphism is associated with better response and longer progression-free survival in colorectal cancer patients treated with a 5-FU combination therapy (5-FU, leucovorin, irinotecan) (22). Taken together, these studies suggest that nuclear localization of the de novo dTMP biosynthesis pathway may be a critical modifier of toxicity induced by 5-FU and other antifolates. This raises the possibility that accounting for nuclear localization of the de novo dTMP synthesis pathway and modifiers thereof may be useful in improving antifolate efficacy (31).
NEURAL TUBE DEFECTS
NTDs are a class of birth defects that result from failure of the neural folds to close during neurulation. Even the least severe NTDs can result in lifelong disability. Several genetic and environmental factors increase risk for NTDs, but the strongest among these is blood folate levels (140). Clinical observations in the 1960s indicated that reduced maternal folate status was associated with an elevated risk for NTDs (55, 56, 127). Accordingly, further studies indicated elevated maternal homocysteine as a risk factor for NTDs (86, 129). Folic acid supplementation decreased NTD occurrence (34) and recurrence (93) rates by as much as 70% in randomized controlled trials and population-wide fortification initiatives (27, 87, 117). The metabolic mechanisms linking folic acid supplementation or FOCM to NTD pathogenesis are an active area of investigation, and most women with an NTD-affected pregnancy do not exhibit overt folate deficiency (66, 88, 90). In addition to inadequate dietary folate intake, genetic variations that affect cellular folate accumulation including folate absorption, transport, metabolic processing, intracellular retention, and catabolism may also impair folate status (74). Genetic variations that affect the activity, stability, or intracellular trafficking of folate-dependent metabolic enzymes, as well as deficiencies of other required nutrients for adequate FOCM functioning (such as vitamin B12 and choline), also affect folate status. In fact, case-control studies demonstrate an association between low maternal vitamin B12 status and NTD risk (38, 48, 51, 52, 66, 91, 109, 134, 151, 162), but causal relationships remain to be elucidated.
It is hypothesized that increased folic acid intake may decrease NTD incidence by compensating for genetically linked impairments in folate utilization or secondary nutrient deficiencies (14). The MTHFR C677T polymorphism, which varies widely among human populations, has been associated with NTD risk in some but not all epidemiologic studies (155–157, 161). This polymorphism causes a single amino acid substitution, which decreases the stability of the MTHFR protein, effectively decreasing MTHFR activity and 5-methylTHF levels, leading to elevated homocysteine. However, because the MTHFR C677T polymorphism effectively alters the distribution of folate-activated one-carbon forms to less chemically stable forms (e.g., 10-formylTHF levels rise at the expense of 5-methylTHF levels), the polymorphism also results in lower folate status. Recent studies have shown that the association of the MTHFR C677T polymorphism with NTD risk is due to its effect on lowering folate levels in red blood cells (33, 139). In support of this conclusion, mouse models with decreased Mthfr expression exhibit elevated homocysteine but do not develop NTDs (70). Similarly, polymorphisms in genes that regulate homocysteine catabolism through the transsulfuration pathway are sometimes associated with high levels of plasma homocysteine but are not commonly associated with increased NTD risk (70).
Formate has been linked to folic acid–resistant NTDs. Mitochondrial FOCM produces formate from serine or glycine, and formate is the primary source of one-carbon units for all cytoplasmic FOCM reactions, including de novo dTMP synthesis, de novo purine synthesis, and homocysteine remethylation (54, 137). Mouse models with disrupted mitochondrial FOCM, including Mthfd1L(17) and glycine cleavage system knockout models, develop NTDs that are rescued with formate supplementation. NTDs in these models are not responsive to folic acid (92, 94, 98). De novo purine biosynthesis has not been associated with NTD risk either in human single-nucleotide polymorphism association studies or in mouse models, but in general, data are lacking for mouse models of impaired de novo purine synthesis. In summary, there is little evidence that homocysteine remethylation to methionine or de novo purine biosynthesis is directly involved in NTD pathogenesis and that the supply of formate for FOCM is critical for the prevention of NTDs.
To date, Shmt1+/− and Shmt1−/− mouse models represent the only models of reduced folate-dependent enzyme activity that develop folic acid–responsive NTDs (11, 12). The Shmt1+/− and Shmt1−/− mouse models reflect human NTD pathogenesis in that NTDs are low penetrance and folic acid responsive. The lack of lethality in Shmt1−/− mice and the incomplete penetrance of the NTDs in this model may be due in part to functional redundancy provided by SHMT2α (6). The primary phenotype of the Shmt1+/− and Shmt1−/− mice is elevated uracil in genomic DNA: These mice do not exhibit elevated homocysteine (75). SHMT1 nuclear localization is required to prevent uracil misincorporation in mouse liver and colon (75, 76, 79), and in a mouse model in which SHMT1 expression is restricted mostly to the cytosol, uracil levels in DNA are also elevated (73). Taken together, these data suggest that inadequate SHMT1 nuclear localization or inadequate de novo dTMP nuclear complex formation causes folic acid–responsive NTDs in mice. These data also suggest that associations of elevated homocysteine with NTDs reflect NTD-associated impairments in folate status, but that elevated homocysteine is not on the causal pathway of NTD pathogenesis. The SHMT1 C1420T variant is associated with increased risk for NTDs in humans (110). Further studies are required to determine whether depressed rates of de novo dTMP synthesis or uracil accumulation in the nuclear genome is causally related to NTD incidence.
FOLATE-MEDIATED ONE-CARBON METABOLISM IN HEMATOPOIESIS
Megaloblastic anemia can result from drug-induced impairments in de novo dTMP synthesis (121). Folate or vitamin B12 deficiency also leads to megaloblastic anemia owing to inadequate nucleotide synthesis, resulting in impaired DNA replication and cell division in red blood cell precursors. In addition to overt folate deficiency, inborn errors of metabolism (IEMs) involved in folate absorption or metabolism are also associated with megaloblastic anemia. Hereditary folate malabsorption results from an IEM in the proton-coupled folate transporter (PCFT) and presents with whole-body folate deficiency and the onset of macrocytic anemia, among other complications early in life (163). PCFT expression is highest at the apical brush border membrane of the small intestine, and PCFT transport activity is optimal in the low-pH environment of the duodenum and jejunum (164). Patients with a DHFR IEM also frequently present with megaloblastic anemia, and other complications, such as peripheral neuropathy (10, 25), also arise. As indicated above, multiple mechanisms ensure nuclear de novo dTMP synthesis is protected during folate deficiency and other impairments in FOCM. Hence, severe nutritional, genetic, and other insults (e.g., chemotherapeutic agents) to FOCM are required to compromise nuclear de novo dTMP synthesis, leading to megaloblastic anemia.
Megaloblastic anemia is a diagnostic indicator of vitamin B12 deficiency resulting from an accumulation of folate cofactors such as 5-methylTHF due to lack of MTR activity. MTR catalyzes remethylation of homocysteine to methionine by using 5-methylTHF as a cofactor (Figure 1). The inhibition of MTR and subsequent accumulation of 5-methylTHF, at the expense of other folate cofactors, is known as the methyl trap. The methyl trap is believed to cause megaloblastic anemia owing to impairments in dTMP synthesis, leading to uracil accumulation in DNA because the cells are starved for THF (124). The MTR-catalyzed conversion of homocysteine to methionine occurs exclusively in the cytosol, but the accumulation of 5-methylTHF cofactors during vitamin B12 deficiency occurs in both the cytosol and the nucleus (99). Hence, the nuclear compartment is sensitive to the methyl trap, implicating impairment of nuclear de novo dTMP synthesis as a cause of megaloblastic anemia.
Interestingly, an IEM in dUTPase, the enzyme that degrades dUTP and thereby limits its accumulation and incorporation into DNA (Figure 4), causes megaloblastic anemia. This observation further highlights the importance of nuclear de novo dTMP biosynthesis complex formation, which is essential to limit uracil in DNA (73), in the pathophysiology of megaloblastic anemia. Because dUTPase is a source of dUMP, the precursor of dTMP, this IEM may impair de novo dTMP synthesis. Alternatively, this may also suggest that uracil in DNA, as opposed to impaired de novo dTMP synthesis in isolation, may be causal in the development of megaloblastic anemia.
SCID results from inadequate T and B cell function, as well as from mitochondrial disorders, and is associated with several metabolic defects including IEMs in nucleotide, carbohydrate, amino acid, and lipid metabolism (69). Adenosine deaminase (ADA) deficiency is a common IEM that causes SCID. ADA is required for purine catabolism, and ADA deficiency results in accumulation of deoxyadenosine. Deoxyadenosine accumulation inhibits RNR, which results in inadequate synthesis of deoxyribonucleotides for DNA replication, leading to impaired DNA synthesis (138). Human mutations in MTHFD1 are associated with SCID (23), highlighting the importance of nuclear de novo dTMP synthesis, in addition to impaired purine synthesis (as in ADA deficiency), for lymphocyte proliferation and effective immune response. Furthermore, the lack of association between IEMs that elevate homocysteine (cystathionine β-synthase or MTHFR deficiency) and SCID suggests that the presentation of SCID in MTHFD1 deficiency is likely caused by impaired nuclear de novo dTMP synthesis. MTHFD1-deficient patient fibroblasts exhibited increased levels of phosphorylated histone H2AX (γH2AX), which is a marker of DNA strand breaks that result from uracil excision repair pathways. Interestingly, fibroblasts from patients with SCID resulting from ADA deficiency did not exhibit either impaired de novo dTMP synthesis or increased nuclear γH2AX levels (41, 42). This finding suggests that MTHFD1 and ADA deficiencies cause SCID by distinct mechanisms.
NEURODEGENERATIVE DISEASES
Folate or vitamin B12 deficiency in the absence of low dietary intake of these vitamins can be attributable to malabsorption due to inflammatory bowel disease and bariatric surgery, alcoholism, autoimmune disorders, or other conditions that impair cellular import. Pernicious anemia, an autoimmune condition that targets parietal cells, results in vitamin B12 deficiency, leading to myelin degeneration in the spinal cord and subsequently to a condition known as subacute combined degeneration, which consists of both myelin degeneration in the spinal cord and peripheral neuropathy (degeneration of the peripheral nerves, which are external to the spinal cord). The clinical signs of these conditions include impaired balance and coordination, muscle weakness, and sensory abnormalities. Nerve cells connecting the central nervous system to voluntary muscles must communicate across long ranges using electrical impulses. Myelin establishes efficiency in this communication by preventing leakage of electrical charges, thereby increasing the speed of conduction. Case studies have shown that intramuscular injection of vitamin B12 resolves the anemia and peripheral neuropathy, indicating that correcting this disease-induced nutritional deficiency results in clinical improvement.
The mechanisms underlying vitamin B12–related neurodegeneration remain unestablished. Vitamin B12 may support myelin synthesis within the cell by providing methyl groups for phosphatidylcholine synthesis in the Schwann cell membranes that compose the myelin sheath (37). Vitamin B12 is also required for synthesis of neurotransmitters (37). Studies of pigs exposed to nitrous oxide, which oxidizes the vitamin B12 cofactor and thereby inactivates MTR, suggested that methionine supplementation rescued demyelination in some but not all nitrous oxide–treated animals (149), implying that additional mechanisms may be involved in pathogenesis of vitamin B12 deficiency-induced subacute combined degeneration and peripheral neuropathy.
Peripheral neuropathy is a complex disease whose risk is associated with interacting environmental, pathophysiological, and genetic factors. Genetic causes of peripheral neuropathy include both highly penetrant genetic mutations associated with IEMs and more common genetic polymorphisms that interact with environmental and disease factors that contribute to risk. Chemotherapeutic agents, some of which act as antifolate compounds that impair de novo dTMP and DNA synthesis and decrease genome stability, can cause peripheral neuropathy (26, 113, 150). Similarly, environmental toxins such as arsenic have been associated with development of peripheral neuropathy in humans (108). Arsenic impairs de novo dTMP synthesis by causing excessive SUMOylation and degradation of MTHFD1 and SHMT1, leading to decreased genome stability (62). Hence, folate-associated neurological disorders may result from impairments in nuclear de novo dTMP synthesis, leading to DNA damage and loss of Schwann cells.
SUMMARY
In mammals there is increasing evidence that de novo dTMP synthesis must occur in the nucleus to provide sufficient dTTP for DNA replication (89). It is unclear why nuclear compartmentation of the de novo dTMP synthesis pathway is unique to mammals as lower eukaryotes conduct all deoxyribonucleotide metabolism in the cytosol. The provision of dTTP directly at sites of DNA synthesis may allow cells to limit uracil incorporation into DNA, genome instability, and subsequent cell death. SHMT1 is the most critical enzyme for efficient nuclear de novo dTMP synthesis because it is the scaffold protein that connects this pathway to the nuclear lamina; hence, changes in SHMT1 expression directly impact de novo dTMP synthesis capacity. However, it is the partitioning of MTHFD1 between the cytosol and the nucleus that appears to mediate the balance between the provision of one-carbon units between de novo dTMP synthesis and homocysteine remethylation. The mechanisms whereby changes in MTHFD1 activity and cellular folate levels regulate partitioning of MTHFD1 between these two compartments remain to be elucidated. Understanding the roles and regulation of nuclear de novo dTMP synthesis will likely increase our understanding of numerous folate- and vitamin B12–associated pathologies.
Folate:
a water-soluble B vitamin (vitamin B9) that carries and chemically activates one-carbon units for biosynthetic reactions
Neural tube defect (NTD):
a common congenital abnormality that results from failure of neural tube closure during neurogenesis that affects 0.5 to 60 in 10,000 births worldwide
dTMP:
deoxythymidine monophosphate or thymidylate
AdoMet:
S-adenosylmethionine
FOCM:
folate-mediated one-carbon metabolism
TK1:
thymidine kinase 1
SHMT:
serine hydroxymethyltransferase
MTHFD1:
methylenetetrahydrofolate dehydrogenase 1
THF:
tetrahydrofolate
TYMS:
thymidylate synthase
DHFR:
dihydrofolate reductase
MTR:
methionine synthase
Replitase:
a multienzyme complex containing enzymes responsible for deoxyribonucleotide synthesis and DNA synthesis including, DNA polymerase, TK1, DHFR, TYMS, nucleoside 5′-phosphate kinase, and RNR
RNR:
ribonucleotide reductase
MTHFR:
methylenetetrahydrofolate reductase
SUMO:
small ubiquitin-like modifier
SUMOylation:
covalent modification of enzymes with the SUMO protein, which regulates nuclear import of enzymes, multienzyme complex formation, and protein turnover
SCID:
severe combined immunodeficiency
AdoHcy:
S-adenosylhomocysteine
GNMT:
glycine N-methyltransferase
Uracil misincorporation:
incorporation of deoxyuridylate into DNA during DNA replication opposite an A base on the template strand
5-FU:
5-fluorouracil
ADA:
adenosine deaminase
ACKNOWLEDGMENTS
This work was supported by grants DK58144 and HD059120 to P.J.S. from the US Public Health Service.
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Abali EE, Skacel NE, Celikkaya H, Hsieh YC. 2008. Regulation of human dihydrofolate reductase activity and expression. Vitam. Horm 79:267–92 [DOI] [PubMed] [Google Scholar]
- 2.Ames BN. 2001. DNA damage from micronutrient deficiencies is likely to be a major cause of cancer. Mutat. Res 475:7–20 [DOI] [PubMed] [Google Scholar]
- 3.An S, Kumar R, Sheets ED, Benkovic SJ. 2008. Reversible compartmentalization of de novo purine biosynthetic complexes in living cells. Science 320:103–6 [DOI] [PubMed] [Google Scholar]
- 4.Andersen S, Heine T, Sneve R, Konig I, Krokan HE, et al. 2005. Incorporation of dUMP into DNA is a major source of spontaneous DNA damage, while excision of uracil is not required for cytotoxicity of fluoropyrimidines in mouse embryonic fibroblasts. Carcinogenesis 26:547–55 [DOI] [PubMed] [Google Scholar]
- 5.Anderson DD, Eom JY, Stover PJ. 2012. Competition between sumoylation and ubiquitination of serine hydroxymethyltransferase 1 determines its nuclear localization and its accumulation in the nucleus. J. Biol. Chem 287:4790–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson DD, Stover PJ. 2009. SHMT1 and SHMT2 are functionally redundant in nuclear de novo thymidylate biosynthesis. PLOS ONE 4:e5839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anderson DD, Woeller CF, Chiang EP, Shane B, Stover PJ. 2012. Serine hydroxymethyltransferase anchors de novo thymidylate synthesis pathway to nuclear lamina for DNA synthesis. J. Biol. Chem 287:7051–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anderson DD, Woeller CF, Stover PJ. 2007. Small ubiquitin-like modifier-1 (SUMO-1) modification of thymidylate synthase and dihydrofolate reductase. Clin. Chem. Lab. Med 45:1760–63 [DOI] [PubMed] [Google Scholar]
- 9.Appleman JR, Prendergast N, Delcamp TJ, Freisheim JH, Blakley RL. 1988. Kinetics of the formation and isomerization of methotrexate complexes of recombinant human dihydrofolate reductase. J. Biol. Chem 263:10304–13 [PubMed] [Google Scholar]
- 10.Banka S, Blom HJ, Walter J, Aziz M, Urquhart J, et al. 2011. Identification and characterization of an inborn error of metabolism caused by dihydrofolate reductase deficiency. Am. J. Hum. Genet 88:216–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beaudin AE, Abarinov EV, Malysheva O, Perry CA, Caudill M, Stover PJ. 2012. Dietary folate, but not choline, modifies neural tube defect risk in Shmt1 knockout mice. Am. J. Clin. Nutr 95:109–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beaudin AE, Abarinov EV, Noden DM, Perry CA, Chu S, et al. 2011. Shmt1 and de novo thymidylate biosynthesis underlie folate-responsive neural tube defects in mice. Am. J. Clin. Nutr 93:789–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beaudin AE, Perry CA, Stabler SP, Allen RH, Stover PJ. 2012. Maternal Mthfd1 disruption impairs fetal growth but does not cause neural tube defects in mice. Am. J. Clin. Nutr 95:882–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beaudin AE, Stover PJ. 2009. Insights into metabolic mechanisms underlying folate-responsive neural tube defects: a minireview. Birth Defects Res. A Clin. Mol. Teratol 85:274–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bertino JR, Donohue DR, Gabrio BW, Silber R, Alenty A, et al. 1962. Increased level of dihydrofolic reductase in leucocytes of patients treated with amethopterin. Nature 193:140–42 [DOI] [PubMed] [Google Scholar]
- 16.Blount BC, Mack MM, Wehr CM, MacGregor JT, Hiatt RA, et al. 1997. Folate deficiency causes uracil misincorporation into human DNA and chromosome breakage: implications for cancer and neuronal damage. PNAS 94:3290–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bolusani S, Young BA, Cole NA, Tibbetts AS, Momb J, et al. 2011. Mammalian MTHFD2L encodes a mitochondrial methylenetetrahydrofolate dehydrogenase isozyme expressed in adult tissues. J. Biol. Chem 286:5166–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boorstein RJ, Pardee AB. 1983. Coordinate inhibition of DNA synthesis and thymidylate synthase activity following DNA damage and repair. Biochem. Biophys. Res. Commun 117:30–36 [DOI] [PubMed] [Google Scholar]
- 19.Bower C, Stanley FJ. 1989. Dietary folate as a risk factor for neural-tube defects: evidence from a case-control study in Western Australia. Med. J. Aust 150:613–19 [DOI] [PubMed] [Google Scholar]
- 20.Brody LC, Conley M, Cox C, Kirke PN, McKeever MP, et al. 2002. A polymorphism, R653Q, in the trifunctional enzyme methylenetetrahydrofolate dehydrogenase/methenyltetrahydrofolate cyclohydrolase/formyltetrahydrofolate synthetase is a maternal genetic risk factor for neural tube defects: report of the Birth Defects Research Group. Am. J. Hum. Genet 71:1207–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown SS, Neal GE, Williams DC. 1965. Subcellular distribution of some folic acid-linked enzymes in rat liver. Biochem. J 97:34C–36C [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Budai B, Komlosi V, Adleff V, Pap E, Reti A, et al. 2012. Impact of SHMT1 polymorphism on the clinical outcome of patients with metastatic colorectal cancer treated with first-line FOLFIRI+bevacizumab. Pharmacogenet. Genom 22:69–72 [DOI] [PubMed] [Google Scholar]
- 23.Burda P, Kuster A, Hjalmarson O, Suormala T, Burer C, et al. 2015. Characterization and review of MTHFD1 deficiency: four new patients, cellular delineation and response to folic and folinic acid treatment. J. Inherit. Metab. Dis 38:863–72 [DOI] [PubMed] [Google Scholar]
- 24.Bystroff C, Kraut J. 1991. Crystal structure of unliganded Escherichia coli dihydrofolate reductase. Ligand-induced conformational changes and cooperativity in binding. Biochemistry 30:2227–39 [DOI] [PubMed] [Google Scholar]
- 25.Cario H, Smith DE, Blom H, Blau N, Bode H, et al. 2011. Dihydrofolate reductase deficiency due to a homozygous DHFR mutation causes megaloblastic anemia and cerebral folate deficiency leading to severe neurologic disease. Am. J. Hum. Genet 88:226–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carozzi VA, Canta A, Chiorazzi A. 2015. Chemotherapy-induced peripheral neuropathy: What do we know about mechanisms? Neurosci. Lett 596:90–107 [DOI] [PubMed] [Google Scholar]
- 27.Castilla EE, Orioli IM, Lopez-Camelo JS, da Graça Dutra M, Nazer-Herrera J. 2003. Preliminary data on changes in neural tube defect prevalence rates after folic acid fortification in South America. Am. J. Med. Genet 123A:123–28 [DOI] [PubMed] [Google Scholar]
- 28.Chiang PK, Gordon RK, Tal J, Zeng GC, Doctor BP, et al. 1996. S-adenosylmethionine and methylation. FASEB J 10:471–80 [PubMed] [Google Scholar]
- 29.Chiba P, Bacon PE, Cory JG. 1984. Studies directed toward testing the “channeling” hypothesis–ribonucleotides—DNA in leukemia L1210 cells. Biochem. Biophys. Res. Commun 123:656–62 [DOI] [PubMed] [Google Scholar]
- 30.Choi SW, Mason JB. 2000. Folate and carcinogenesis: an integrated scheme. J. Nutr 130:129–32 [DOI] [PubMed] [Google Scholar]
- 31.Chon J, Stover PJ, Field MS. 2017. Targeting nuclear thymidylate biosynthesis. Mol. Aspects Med 53:48–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clarke S, Banfield K. 2001. S-adenosylmethionine-dependent methyltransferases. In Homocysteine in Health and Disease, ed. Carmel R, Jacobson DW, pp. 63–78. Cambridge, UK: Cambridge Univ. Press [Google Scholar]
- 33.Crider KS, Devine O, Hao L, Dowling NF, Li S, et al. 2014. Population red blood cell folate concentrations for prevention of neural tube defects: Bayesian model. BMJ 349:g4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Czeizel AE, Dudas I. 1992. Prevention of the first occurrence of neural-tube defects by periconceptional vitamin supplementation. N. Engl. J. Med 327:1832–35 [DOI] [PubMed] [Google Scholar]
- 35.DebRoy S, Kramarenko II, Ghose S, Oleinik NV, Krupenko SA, Krupenko NI. 2013. A novel tumor suppressor function of glycine N-methyltransferase is independent of its catalytic activity but requires nuclear localization. PLOS ONE 8:e70062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Delgado M, Garrido F, Pérez-Miguelsanz J, Pacheco M, Partearroyo T, et al. 2014. Acute liver injury induces nucleocytoplasmic redistribution of hepatic methionine metabolism enzymes. Antioxid. Redox Signal 20:2541–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dror DK, Allen LH. 2012. Interventions with vitamins B6, B12 and C in pregnancy. Paediatr. Perinat. Epidemiol 26(Suppl. 1):55–74 [DOI] [PubMed] [Google Scholar]
- 38.Felkner M, Suarez L, Canfield MA, Brender JD, Sun Q. 2009. Maternal serum homocysteine and risk for neural tube defects in a Texas-Mexico border population. Birth Defects Res. A Clin. Mol. Teratol 85:574–81 [DOI] [PubMed] [Google Scholar]
- 39.Field MS, Anderson DD, Stover PJ. 2011. Mthfs is an essential gene in mice and a component of the purinosome. Front. Genet 2:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Field MS, Kamynina E, Agunloye OC, Liebenthal RP, Lamarre SG, et al. 2014. Nuclear enrichment of folate cofactors and methylenetetrahydrofolate dehydrogenase 1 (MTHFD1) protect de novo thymidylate biosynthesis during folate deficiency. J. Biol. Chem 289:29642–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Field MS, Kamynina E, Watkins D, Rosenblatt DS, Stover PJ. 2015. Human mutations in methylenetetrahydrofolate dehydrogenase 1 impair nuclear de novo thymidylate biosynthesis. PNAS 112:400–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Field MS, Kamynina E, Watkins D, Rosenblatt DS, Stover PJ. 2015. New insights into the metabolic and nutritional determinants of severe combined immunodeficiency. Rare Dis 3:e1112479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Field MS, Stover PJ. 2018. Safety of folic acid. Ann. N. Y. Acad. Sci 1414:59–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fox JT, Stover PJ. 2008. Folate-mediated one-carbon metabolism. Vitam. Horm 79:1–44 [DOI] [PubMed] [Google Scholar]
- 45.Friso S, Choi SW, Dolnikowski GG, Selhub J. 2002. A method to assess genomic DNA methylation using high-performance liquid chromatography/electrospray ionization mass spectrometry. Anal. Chem 74:4526–31 [DOI] [PubMed] [Google Scholar]
- 46.Friso S, Choi SW, Girelli D, Mason JB, Dolnikowski GG, et al. 2002. A common mutation in the 5,10-methylenetetrahydrofolate reductase gene affects genomic DNA methylation through an interaction with folate status. PNAS 99:5606–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Froese DS, Wu X, Zhang J, Dumas R, Schoel WM, et al. 2008. Restricted role for methionine synthase reductase defined by subcellular localization. Mol. Genet. Metab 94:68–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gaber KR, Farang MK, Soliman SE, El-Bassyouni HT, El-Kamah G. 2007. Maternal vitamin B12 and the risk of fetal neural tube defects in Egyptian patients. Clin. Lab 53:69–75 [PubMed] [Google Scholar]
- 49.Gonen N, Assaraf YG. 2012. Antifolates in cancer therapy: structure, activity and mechanisms of drug resistance. Drug Resist. Updates 15:183–210 [DOI] [PubMed] [Google Scholar]
- 50.Green JM, MacKenzie RE, Matthews RG. 1988. Substrate flux through methylenetetrahydrofolate dehydrogenase: predicted effects of the concentration of methylenetetrahydrofolate on its partitioning into pathways leading to nucleotide biosynthesis or methionine regeneration. Biochemistry 27:8014–22 [DOI] [PubMed] [Google Scholar]
- 51.Groenen PM, van Rooij IA, Peer PG, Gooskens RH, Zielhuis GA, Steegers-Theunissen RP. 2004. Marginal maternal vitamin B12 status increases the risk of offspring with spina bifida. Am. J. Obstet. Gynecol 191:11–17 [DOI] [PubMed] [Google Scholar]
- 52.Gu Q, Li Y, Cui ZL, Luo XP. 2012. Homocysteine, folate, vitamin B12 and B6 in mothers of children with neural tube defects in Xinjiang, China. Acta Paediatr 101:e486–90 [DOI] [PubMed] [Google Scholar]
- 53.Hendriks IA, Vertegaal AC. 2016. A comprehensive compilation of SUMO proteomics. Nat. Rev. Mol. Cell Biol 17:581–95 [DOI] [PubMed] [Google Scholar]
- 54.Herbig K, Chiang EP, Lee LR, Hills J, Shane B, Stover PJ. 2002. Cytoplasmic serine hydroxymethyltransferase mediates competition between folate-dependent deoxyribonucleotide and S-adenosylmethionine biosyntheses. J. Biol. Chem 277:38381–89 [DOI] [PubMed] [Google Scholar]
- 55.Hibbard BM. 1964. The role of folic acid in pregnancy; with particular reference to anaemia, abruption and abortion. J. Obstet. Gynaecol. Br. Commonw 71:529–42 [DOI] [PubMed] [Google Scholar]
- 56.Hibbard BM. 1967. Defective folate metabolism in pathological conditions of pregnancy. Acta Obstet. Gynecol. Scand 46(Suppl. 7):47–59 [DOI] [PubMed] [Google Scholar]
- 57.Hoffbrand AV, Weir DG. 2001. The history of folic acid. Br. J. Haematol 113:579–89 [DOI] [PubMed] [Google Scholar]
- 58.Holmes MV, Newcombe P, Hubacek JA, Sofat R, Ricketts SL, et al. 2011. Effect modification by population dietary folate on the association between MTHFR genotype, homocysteine, and stroke risk: a meta-analysis of genetic studies and randomised trials. Lancet 378:584–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang C, Sloan EA, Boerkoel CF. 2003. Chromatin remodeling and human disease. Curr. Opin. Genet. Dev 13:246–52 [DOI] [PubMed] [Google Scholar]
- 60.Igarashi K, Katoh Y. 2013. Metabolic aspects of epigenome: coupling of S-adenosylmethionine synthesis and gene regulation on chromatin by SAMIT module. Subcell. Biochem 61:105–18 [DOI] [PubMed] [Google Scholar]
- 61.Jaenisch R, Bird A. 2003. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet 33(Suppl.):245–54 [DOI] [PubMed] [Google Scholar]
- 62.Kamynina E, Lachenauer ER, DiRisio AC, Liebenthal RP, Field MS, Stover PJ. 2017. Arsenic trioxide targets MTHFD1 and SUMO-dependent nuclear de novo thymidylate biosynthesis. PNAS 114:E2319–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kapiszewska M, Kalemba M, Wojciech U, Milewicz T. 2005. Uracil misincorporation into DNA of leukocytes of young women with positive folate balance depends on plasma vitamin B12 concentrations and methylenetetrahydrofolate reductase polymorphisms. A pilot study. J. Nutr. Biochem 16:467–78 [DOI] [PubMed] [Google Scholar]
- 64.Keller MD, Ganesh J, Heltzer M, Paessler M, Bergqvist AG, et al. 2013. Severe combined immunodeficiency resulting from mutations in MTHFD1. Pediatrics 131:e629–34 [DOI] [PubMed] [Google Scholar]
- 65.Kim YI. 1999. Folate and cancer prevention: a new medical application of folate beyond hyperhomocysteinemia and neural tube defects. Nutr. Rev 57:314–21 [DOI] [PubMed] [Google Scholar]
- 66.Kirke PN, Molloy AM, Daly LE, Burke H, Weir DG, Scott JM. 1993. Maternal plasma folate and vitamin B12 are independent risk factors for neural tube defects. Q. J. Med 86:703–8 [PubMed] [Google Scholar]
- 67.Kronenberg G, Gertz K, Overall RW, Harms C, Klein J, et al. 2011. Folate deficiency increases mtDNA and D-1 mtDNA deletion in aged brain of mice lacking uracil-DNA glycosylase. Exp. Neurol 228:253–58 [DOI] [PubMed] [Google Scholar]
- 68.Krupenko NI, Wagner C. 1997. Transport of rat liver glycine N-methyltransferase into rat liver nuclei. J. Biol. Chem 272:27140–46 [DOI] [PubMed] [Google Scholar]
- 69.Kwan A, Abraham RS, Currier R, Brower A, Andruszewski K, et al. 2014. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA 312:729–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li D, Pickell L, Liu Y, Wu Q, Cohn JS, Rozen R. 2005. Maternal methylenetetrahydrofolate reductase deficiency and low dietary folate lead to adverse reproductive outcomes and congenital heart defects in mice. Am. J. Clin. Nutr 82:188–95 [DOI] [PubMed] [Google Scholar]
- 71.Longley DB, Harkin DP, Johnston PG. 2003. 5-Fluorouracil: mechanisms of action and clinical strategies. Nat. Rev. Cancer 3:330–38 [DOI] [PubMed] [Google Scholar]
- 72.Luka Z, Mudd SH, Wagner C. 2009. Glycine N-methyltransferase and regulation of S-adenosylmethionine levels. J. Biol. Chem 284:22507–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.MacFarlane AJ, Anderson DD, Flodby P, Perry CA, Allen RH, et al. 2011. Nuclear localization of de novo thymidylate biosynthesis pathway is required to prevent uracil accumulation in DNA. J. Biol. Chem 286:44015–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.MacFarlane AJ, Greene-Finestone LS, Shi Y. 2011. Vitamin B-12 and homocysteine status in a folate-replete population: results from the Canadian Health Measures Survey. Am. J. Clin. Nutr 94:1079–87 [DOI] [PubMed] [Google Scholar]
- 75.MacFarlane AJ, Liu X, Perry CA, Flodby P, Allen RH, et al. 2008. Cytoplasmic serine hydroxymethyltransferase regulates the metabolic partitioning of methylenetetrahydrofolate but is not essential in mice. J. Biol. Chem 283:25846–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.MacFarlane AJ, McEntee MF, Stover PJ. 2014. Azoxymethane-induced colon carcinogenesis in mice occurs independently of de novo thymidylate synthesis capacity. J. Nutr 144:419–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.MacFarlane AJ, Perry CA, Girnary HH, Gao D, Allen RH, et al. 2009. Mthfd1 is an essential gene in mice and alters biomarkers of impaired one-carbon metabolism. J. Biol. Chem 284:1533–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.MacFarlane AJ, Perry CA, McEntee MF, Lin DM, Stover PJ. 2011. Mthfd1 is a modifier of chemically induced intestinal carcinogenesis. Carcinogenesis 32:427–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.MacFarlane AJ, Perry CA, McEntee MF, Lin DM, Stover PJ. 2011. Shmt1 heterozygosity impairs folate-dependent thymidylate synthesis capacity and modifies risk of Apcmin-mediated intestinal cancer risk. Cancer Res 71:2098–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.MacFarlane AJ, Stover PJ. 2007. Convergence of genetic, nutritional and inflammatory factors in gas-trointestinal cancers. Nutr. Rev 65:S157–66 [DOI] [PubMed] [Google Scholar]
- 81.Mason JB. 2011. Unraveling the complex relationship between folate and cancer risk. Biofactors 37:253–60 [DOI] [PubMed] [Google Scholar]
- 82.Mason JB, Choi SW. 2000. The mechanisms by which folate depletion enhances colorectal carcinogenesis: a unified scheme. Nestle Nutr. Workshop Ser. Clin. Perform. Programme 4:87–99 [DOI] [PubMed] [Google Scholar]
- 83.Mato JM, Martínez-Chantar ML, Lu SC. 2008. Methionine metabolism and liver disease. Annu. Rev. Nutr 28:273–93 [DOI] [PubMed] [Google Scholar]
- 84.McCarthy EA, Titus SA, Taylor SM, Jackson-Cook C, Moran RG. 2004. A mutation inactivating the mitochondrial inner membrane folate transporter creates a glycine requirement for survival of Chinese hamster cells. J. Biol. Chem 279:33829–36 [DOI] [PubMed] [Google Scholar]
- 85.Mellman I, Willard HF, Rosenberg LE. 1978. Cobalamin binding and cobalamin-dependent enzyme activity in normal and mutant human fibroblasts. J. Clin. Investig 62:952–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mills JL, McPartlin JM, Kirke PN, Lee YJ, Conley MR, et al. 1995. Homocysteine metabolism in pregnancies complicated by neural-tube defects. Lancet 345:149–51 [DOI] [PubMed] [Google Scholar]
- 87.Mills JL, Signore C. 2004. Neural tube defect rates before and after food fortification with folic acid. Birth Defects Res. A Clin. Mol. Teratol 70:844–45 [DOI] [PubMed] [Google Scholar]
- 88.Mills JL, Tuomilehto J, Yu KF, Colman N, Blaner WS, et al. 1992. Maternal vitamin levels during pregnancies producing infants with neural tube defects. J. Pediatr 120:863–71 [DOI] [PubMed] [Google Scholar]
- 89.Misselbeck K, Marchetti L, Field MS, Scotti M, Priami C, Stover PJ. 2017. A hybrid stochastic model of folate-mediated one-carbon metabolism: effect of the common C677T MTHFR variant on de novo thymidylate biosynthesis. Sci. Rep 7:797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Molloy AM, Kirke P, Hillary I, Weir DG, Scott JM. 1985. Maternal serum folate and vitamin B12 concentrations in pregnancies associated with neural tube defects. Arch. Dis. Child 60:660–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Molloy AM, Kirke PN, Troendle JF, Burke H, Sutton M, et al. 2009. Maternal vitamin B12 status and risk of neural tube defects in a population with high neural tube defect prevalence and no folic acid fortification. Pediatrics 123:917–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Momb J, Lewandowski JP, Bryant JD, Fitch R, Surman DR, et al. 2013. Deletion of Mthfd1l causes embryonic lethality and neural tube and craniofacial defects in mice. PNAS 110:549–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.MRC Vitamin Study Research Group. 1991. Prevention of neural tube defects: results of the Medical Research Council Vitamin Study. Lancet 338:131–37 [PubMed] [Google Scholar]
- 94.Narisawa A, Komatsuzaki S, Kikuchi A, Niihori T, Aoki Y, et al. 2012. Mutations in genes encoding the glycine cleavage system predispose to neural tube defects in mice and humans. Hum. Mol. Genet 21:1496–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nilsen H, Stamp G, Andersen S, Hrivnak G, Krokan HE, et al. 2003. Gene-targeted mice lacking the Ung uracil-DNA glycosylase develop B-cell lymphomas. Oncogene 22:5381–86 [DOI] [PubMed] [Google Scholar]
- 96.Noguchi H, Prem Veer Reddy G, Pardee AB. 1983. Rapid incorporation of label from ribonucleoside disphosphates into DNA by a cell-free high molecular weight fraction from animal cell nuclei. Cell 32:443–51 [DOI] [PubMed] [Google Scholar]
- 97.Oppenheim EW, Adelman C, Liu X, Stover PJ. 2001. Heavy chain ferritin enhances serine hydroxymethyltransferase expression and de novo thymidine biosynthesis. J. Biol. Chem 276:19855–61 [DOI] [PubMed] [Google Scholar]
- 98.Pai YJ, Leung KY, Savery D, Hutchin T, Prunty H, et al. 2015. Glycine decarboxylase deficiency causes neural tube defects and features of non-ketotic hyperglycinemia in mice. Nat. Commun 6:6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Palmer AM, Kamynina E, Field MS, Stover PJ. 2017. Folate rescues vitamin B12 depletion-induced inhibition of nuclear thymidylate biosynthesis and genome instability. PNAS 114:E4095–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Parle-McDermott A, Kirke PN, Mills JL, Molloy AM, Cox C, et al. 2006. Confirmation of the R653Q polymorphism of the trifunctional C1-synthase enzyme as a maternal risk for neural tube defects in the Irish population. Eur. J. Hum. Genet 14:768–72 [DOI] [PubMed] [Google Scholar]
- 101.Parle-McDermott A, Pangilinan F, Mills JL, Signore CC, Molloy AM, et al. 2005. A polymorphism in the MTHFD1 gene increases a mother’s risk of having an unexplained second trimester pregnancy loss. Mol. Hum. Reprod 11:477–80 [DOI] [PubMed] [Google Scholar]
- 102.Plucinski TM, Fager RS, Reddy GP. 1990. Allosteric interaction of components of the replitase complex is responsible for enzyme cross-inhibition. Mol. Pharmacol 38:114–20 [PubMed] [Google Scholar]
- 103.Pogribny IP, Basnakian AG, Miller BJ, Lopatina NG, Poirier LA, James SJ. 1995. Breaks in genomic DNA and within the p53 gene are associated with hypomethylation in livers of folate/methyl-deficient rats. Cancer Res 55:1894–901 [PubMed] [Google Scholar]
- 104.Poon PP, Storms RK. 1994. Thymidylate synthase is localized to the nuclear periphery in the yeast Saccharomyces cerevisiae. J. Biol. Chem 269:8341–47 [PubMed] [Google Scholar]
- 105.Prem Veer Reddy G 1982. Catalytic function of thymidylate synthase is confined to S phase due to its association with replitase. Biochem. Biophys. Res. Commun 109:908–15 [DOI] [PubMed] [Google Scholar]
- 106.Prem Veer Reddy G, Pardee AB. 1980. Multienzyme complex for metabolic channeling in mammalian DNA replication. PNAS 77:3312–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rassin DK, Gaull GE. 1975. Subcellular distribution of enzymes of transmethylation and transsulphuration in rat brain. J. Neurochem 24:969–78 [DOI] [PubMed] [Google Scholar]
- 108.Ratnaike RN. 2003. Acute and chronic arsenic toxicity. Postgrad. Med. J 79:391–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ray JG, Wyatt PR, Thompson MD, Vermeulen MJ, Meier C, et al. 2007. Vitamin B12 and the risk of neural tube defects in a folic-acid-fortified population. Epidemiology 18:362–66 [DOI] [PubMed] [Google Scholar]
- 110.Rebekah PK, Tella S, Buragadda S, Tiruvatturu MK, Akka J. 2017. Interaction between maternal and paternal SHMT1 C1420T predisposes to neural tube defects in the fetus: evidence from case-control and family-based triad approaches. Birth Defects Res 109:1020–29 [DOI] [PubMed] [Google Scholar]
- 111.Reytor E, Pérez-Miguelsanz J, Alvarez L, Perez-Sala D, Pajares MA. 2009. Conformational signals in the C-terminal domain of methionine adenosyltransferase I/III determine its nucleocytoplasmic distribution. FASEB J 23:3347–60 [DOI] [PubMed] [Google Scholar]
- 112.Rode W, Jastreboff MM, Bertino JR. 1985. Thymidylate synthase inhibition in cells with arrested DNA synthesis is not due to an allosteric interaction in the replitase complex. Biochem. Biophys. Res. Commun 128:345–51 [DOI] [PubMed] [Google Scholar]
- 113.Saif MW, Wilson RH, Harold N, Keith B, Dougherty DS, Grem JL. 2001. Peripheral neuropathy associated with weekly oral 5-fluorouracil, leucovorin and eniluracil. Anti-Cancer Drugs 12:525–31 [DOI] [PubMed] [Google Scholar]
- 114.Sankar DV, Geisler A, Rozsa PW. 1969. Intracellular distribution of folic acid in mouse liver. Experientia 25:691–92 [DOI] [PubMed] [Google Scholar]
- 115.Sano H, Noguchi H, Kodama I, Itoh T. 1988. Rapid methylation of genomic DNA in proliferating T-cells from bovine thymocytes. Biochim. Biophys. Acta 950:274–81 [DOI] [PubMed] [Google Scholar]
- 116.Santi DV, McHenry CS, Sommer H. 1974. Mechanism of interaction of thymidylate synthetase with 5-fluorodeoxyuridylate. Biochemistry 13:471–81 [DOI] [PubMed] [Google Scholar]
- 117.Sayed AR, Bourne D, Pattinson R, Nixon J, Henderson B. 2008. Decline in the prevalence of neural tube defects following folic acid fortification and its cost-benefit in South Africa. Birth Defects Res. A Clin. Mol. Teratol 82:211–16 [DOI] [PubMed] [Google Scholar]
- 118.Schirch V, Strong WB. 1989. Interaction of folylpolyglutamates with enzymes in one-carbon metabolism. Arch. Biochem. Biophys 269:371–80 [DOI] [PubMed] [Google Scholar]
- 119.Schormann N, Ricciardi R, Chattopadhyay D. 2014. Uracil-DNA glycosylases-structural and functional perspectives on an essential family of DNA repair enzymes. Protein Sci 23:1667–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Schrader CE, Guikema JE, Wu X, Stavnezer J. 2009. The roles of APE1, APE2, DNA polymerase β and mismatch repair in creating S region DNA breaks during antibody class switch. Philos. Trans. R. Soc. B 364:645–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Scott JM, Weir DG. 1980. Drug-induced megaloblastic change. Clin. Haematol 9:587–606 [PubMed] [Google Scholar]
- 122.Scotti M, Stella L, Shearer EJ, Stover PJ. 2013. Modeling cellular compartmentation in one-carbon metabolism. Wiley Interdiscip. Rev. Syst. Biol. Med 5:343–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Seiple L, Jaruga P, Dizdaroglu M, Stivers JT. 2006. Linking uracil base excision repair and 5-fluorouracil toxicity in yeast. Nucleic Acids Res 34:140–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Shane B, Stokstad EL. 1985. Vitamin B12–folate interrelationships. Annu. Rev. Nutr 5:115–41 [DOI] [PubMed] [Google Scholar]
- 125.Sheppard NG, Jarl L, Mahadessian D, Strittmatter L, Schmidt A, et al. 2015. The folate-coupled enzyme MTHFD2 is a nuclear protein and promotes cell proliferation. Sci. Rep 5:15029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Shin YS, Chan C, Vidal AJ, Brody T, Stokstad EL. 1976. Subcellular localization of γ-glutamyl carboxypeptidase and of folates. Biochim. Biophys. Acta 444:794–801 [DOI] [PubMed] [Google Scholar]
- 127.Smithells RW, Sheppard S, Schorah CJ. 1976. Vitamin deficiencies and neural tube defects. Arch. Dis. Child 51:944–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sommer H, Santi DV. 1974. Purification and amino acid analysis of an active site peptide from thymidylate synthetase containing covalently bound 5-fluoro-2′-deoxyuridylate and methylenetetrahydrofolate. Biochem. Biophys. Res. Commun 57:689–95 [DOI] [PubMed] [Google Scholar]
- 129.Steegers-Theunissen RP, Boers GH, Trijbels FJ, Eskes TK. 1991. Neural-tube defects and derangement of homocysteine metabolism. N. Engl. J. Med 324:199–200 [DOI] [PubMed] [Google Scholar]
- 130.Stover PJ. 2004. Physiology of folate and vitamin B12 in health and disease. Nutr. Rev 62:S3–12 [DOI] [PubMed] [Google Scholar]
- 131.Strong WB, Schirch V. 1989. In vitro conversion of formate to serine: effect of tetrahydropteroylpolyglutamates and serine hydroxymethyltransferase on the rate of 10-formyltetrahydrofolate synthetase. Biochemistry 28:9430–39 [DOI] [PubMed] [Google Scholar]
- 132.Strong WB, Tendler SJ, Seither RL, Goldman ID, Schirch V. 1990. Purification and properties of serine hydroxymethyltransferase and C1-tetrahydrofolate synthase from L1210 cells. J. Biol. Chem 265:12149–55 [PubMed] [Google Scholar]
- 133.Studzinski GP, Shankavaram UT, Moore DC, Reddy PV. 1991. Association of c-myc protein with enzymes of DNA replication in high molecular weight fractions from mammalian cells. J. Cell. Physiol 147:412–19 [DOI] [PubMed] [Google Scholar]
- 134.Suarez L, Hendricks K, Felkner M, Gunter E. 2009. Maternal serum B12 levels and risk for neural tube defects in a Texas-Mexico border population. Birth Defects Res. A Clin. Mol. Teratol 85:574–81 [DOI] [PubMed] [Google Scholar]
- 135.Subramanyam C, Honn SC, Reed WC, Reddy GP. 1990. Nuclear localization of 68 kDa calmodulin-binding protein is associated with the onset of DNA replication. J. Cell. Physiol 144:423–28 [DOI] [PubMed] [Google Scholar]
- 136.Suh JR, Herbig AK, Stover PJ. 2001. New perspectives on folate catabolism. Annu. Rev. Nutr 21:255–82 [DOI] [PubMed] [Google Scholar]
- 137.Tibbetts AS, Appling DR. 2010. Compartmentalization of mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr 30:57–81 [DOI] [PubMed] [Google Scholar]
- 138.Touzot F, Hacein-Bey-Abina S, Fischer A, Cavazzana M. 2014. Gene therapy for inherited immunodeficiency. Expert Opin. Biol. Ther 14:789–98 [DOI] [PubMed] [Google Scholar]
- 139.Tsang BL, Devine OJ, Cordero AM, Marchetta CM, Mulinare J, et al. 2015. Assessing the association between the methylenetetrahydrofolate reductase (MTHFR) 677C>T polymorphism and blood folate concentrations: a systematic review and meta-analysis of trials and observational studies. Am. J. Clin. Nutr 101:1286–94 [DOI] [PubMed] [Google Scholar]
- 140.US Preventive Services Task Force, Bibbins-Domingo K, Grossman DC, Curry SJ, Davidson KW, et al. 2017. Folic acid supplementation for the prevention of neural tube defects: US Preventive Services Task Force Recommendation Statement. JAMA 317:183–89 [DOI] [PubMed] [Google Scholar]
- 141.van den Donk M, Pellis L, Crott JW, van Engeland M, Friederich P, et al. 2007. Folic acid and vitamin B-12 supplementation does not favorably influence uracil incorporation and promoter methylation in rectal mucosa DNA of subjects with previous colorectal adenomas. J. Nutr 137:2114–20 [DOI] [PubMed] [Google Scholar]
- 142.van der Wilt CL, Backus HH, Smid K, Comijn L, Veerman G, et al. 2001. Modulation of both endogenous folates and thymidine enhance the therapeutic efficacy of thymidylate synthase inhibitors. Cancer Res 61:3675–81 [PubMed] [Google Scholar]
- 143.Veer Reddy GP, Pardee AB. 1983. Inhibitor evidence for allosteric interaction in the replitase multienzyme complex. Nature 304:86–88 [DOI] [PubMed] [Google Scholar]
- 144.Visentin M, Diop-Bove N, Zhao R, Goldman ID. 2014. The intestinal absorption of folates. Annu. Rev. Physiol 76:251–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Viswanathan M, Treiman KA, Kish-Doto J, Middleton JC, Coker-Schwimmer EJ, Nicholson WK. 2017. Folic acid supplementation for the prevention of neural tube defects: an updated evidence report and systematic review for the US Preventive Services Task Force. JAMA 317:190–203 [DOI] [PubMed] [Google Scholar]
- 146.Vollset SE, Clarke R, Lewington S, Ebbing M, Halsey J, et al. 2013. Effects of folic acid supplementation on overall and site-specific cancer incidence during the randomised trials: meta-analyses of data on 50,000 individuals. Lancet 381:1029–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wang YC, Lin WL, Lin YJ, Tang FY, Chen YM, Chiang EP. 2014. A novel role of the tumor suppressor GNMT in cellular defense against DNA damage. Int. J. Cancer 134:799–810 [DOI] [PubMed] [Google Scholar]
- 148.Watkins D, Schwartzentruber JA, Ganesh J, Orange JS, Kaplan BS, et al. 2011. Novel inborn error of folate metabolism: identification by exome capture and sequencing of mutations in the MTHFD1 gene in a single proband. J. Med. Genet 48:590–92 [DOI] [PubMed] [Google Scholar]
- 149.Weir DG, Keating S, Molloy A, McPartlin J, Kennedy S, et al. 1988. Methylation deficiency causes vitamin B12-associated neuropathy in the pig. J. Neurochem 51:1949–52 [DOI] [PubMed] [Google Scholar]
- 150.Werbrouck BF, Pauwels WJ, De Bleecker JL. 2008. A case of 5-fluorouracil-induced peripheral neuropathy. Clin. Toxicol 46:264–66 [DOI] [PubMed] [Google Scholar]
- 151.Wilson A, Platt R, Wu Q, Leclerc D, Christensen B, et al. 1999. A common variant in methionine synthase reductase combined with low cobalamin (vitamin B12) increases risk for spina bifida. Mol. Genet. Metab 67:317–23 [DOI] [PubMed] [Google Scholar]
- 152.Woeller CF, Anderson DD, Szebenyi DM, Stover PJ. 2007. Evidence for small ubiquitin-like modifier-dependent nuclear import of the thymidylate biosynthesis pathway. J. Biol. Chem 282:17623–31 [DOI] [PubMed] [Google Scholar]
- 153.Wolff T, Witkop CT, Miller T, Syed SB. 2009. Folic acid supplementation for the prevention of neural tube defects: an update for the U.S. Preventive Services Task Force. Ann. Intern. Med 150:632–39 [DOI] [PubMed] [Google Scholar]
- 154.Wong NA, Brett L, Stewart M, Leitch A, Longley DB, et al. 2001. Nuclear thymidylate synthase expression, p53 expression and 5FU response in colorectal carcinoma. Br. J. Cancer 85:1937–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Yadav U, Kumar P, Yadav SK, Mishra OP, Rai V. 2015. Polymorphisms in folate metabolism genes as maternal risk factor for neural tube defects: an updated meta-analysis. Metab. Brain Dis 30:7–24 [DOI] [PubMed] [Google Scholar]
- 156.Yan L, Zhao L, Long Y, Zou P, Ji G, et al. 2012. Association of the maternal MTHFR C677T polymorphism with susceptibility to neural tube defects in offsprings: evidence from 25 case-control studies. PLOS ONE 7:e41689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Yang Y, Chen J, Wang B, Ding C, Liu H. 2015. Association between MTHFR C677T polymorphism and neural tube defect risks: a comprehensive evaluation in three groups of NTD patients, mothers, and fathers. Birth Defects Res. A Clin. Mol. Teratol 103:488–500 [DOI] [PubMed] [Google Scholar]
- 158.Yarbro JW. 1992. Mechanism of action of hydroxyurea. Semin. Oncol 19:1–10 [PubMed] [Google Scholar]
- 159.Yeo EJ, Wagner C. 1994. Tissue distribution of glycine N-methyltransferase, a major folate-binding protein of liver. PNAS 91:210–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zamierowski M, Wagner C. 1974. High molecular weight complexes of folic acid in mammalian tissues. Biochem. Biophys. Res. Commun 60:81–87 [DOI] [PubMed] [Google Scholar]
- 161.Zhang T, Lou J, Zhong R, Wu J, Zou L, et al. 2013. Genetic variants in the folate pathway and the risk of neural tube defects: a meta-analysis of the published literature. PLOS ONE 8:e59570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Zhang T, Xin R, Gu X, Wang F, Pei L, et al. 2009. Maternal serum vitamin B12, folate and homocysteine and the risk of neural tube defects in the offspring in a high-risk area of China. Public Health Nutr 12:680–86 [DOI] [PubMed] [Google Scholar]
- 163.Zhao R, Diop-Bove N, Visentin M, Goldman ID. 2011. Mechanisms of membrane transport of folates into cells and across epithelia. Annu. Rev. Nutr 31:177–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Zhao R, Min SH, Wang Y, Campanella E, Low PS, Goldman ID. 2009. A role for the proton-coupled folate transporter (PCFT-SLC46A1) in folate receptor-mediated endocytosis. J. Biol. Chem 284:4267–74 [DOI] [PMC free article] [PubMed] [Google Scholar]