

Abstract
Non-alcoholic fatty liver disease (NAFLD) is a multifactorial disease that encompasses a spectrum of pathological conditions, ranging from simple steatosis (NAFL), nonalcoholic steatohepatitis (NASH), fibrosis/cirrhosis which can further progress to hepatocellular carcinoma and liver failure. The progression of NAFL to NASH and liver fibrosis is closely associated with a series of liver injury resulting from lipotoxicity, oxidative stress, redox imbalance (excessive nitric oxide), ER stress, inflammation and apoptosis that occur sequentially in different liver cells which ultimately leads to the activation of liver regeneration and fibrogenesis, augmenting collagen and extracellular matrix deposition and promoting liver fibrosis and cirrhosis. Type 2 diabetes is a significant risk factor in NAFLD development by accelerating liver damage. Here, we overview recent findings from human study and animal models on the pathophysiological communication among hepatocytes (HCs), Kupffer cells (KCs), hepatic stellate cells (HSCs) and liver sinusoidal endothelial cells (LSECs) during the disease development. The mechanisms of crucial signaling pathways, including Toll-like receptor, TGFβ and hedgehog mediated hepatic injury are also discussed. We further highlight the potentials of precisely targeting hepatic individual cell-type using nanotechnology as therapeutic strategy for the treatment of NASH and liver fibrosis.
Keywords: NAFLD, NASH, Liver fibrosis, Lipotoxicity, Nitric oxide, Oxidative stress, ER stress, Metabolic inflammation, TLRs, Hedgehog and TGFβ
1. Introduction
Non-alcoholic fatty liver disease (NAFLD) is a multifactorial disease that is considered as the most common cause of chronic liver disease [1,2]. NAFLD encompasses a spectrum of pathological conditions, ranging from simple steatosis (NAFL), nonalcoholic steatohepatitis (NASH), fibrosis/cirrhosis which can further progress to hepatocellular carcinoma and liver failure [3]. Initially, a “two-hit” hypothesis was established to describe the mechanistic development of NAFLD [4]. The ‘first hit’ usually is the accumulation of lipids, including triglycerides, free fatty acids (FFAs), and small amount of cholesterol in hepatocytes, which induce NAFL and increase the vulnerability of liver to injury caused by the ‘second hits’. Biopsy diagnosis reveals that more than 5% of lipid content by weight is presented in hepatocytes (macrovesicular) in NAFL. The progression from NAFL to liver fibrosis is closely associated with a series of injuries resulting from lipotoxicity, mitochondrial dysfunction, cellular stress and inflammation in the liver [5] that constitutes the “second hit” thought to be required to progress NAFL to NASH. This ‘two-hit’ hypothesis has been further evolved into “three-hit” and “multiple-hit” hypothesis due to the novel findings from recent research. The defective hepatocyte regeneration that is crucial for the replenishment of dead cells has been proposed to be the “third hit” in NAFLD development [6], and the adverse factors, including insulin resistance, inflammatory mediators derived from adipose tissues, dietary factors, lipopolysaccharide (LPS) generated by gut microbiota and genetic and epigenetic factors are the proposed multiple hits that drive the progression of NAFLD to fibrosis [7]. Given the wide range of pathological conditions involved in NAFLD, an international consensus has renamed NAFLD to Metabolic Associated Liver Disease (MAFLD) [8–10]. These liver injuries ultimately leads to the activation of hepatic stellate cells (HSCs), augmenting collagen and extracellular matrix (ECM) production/deposition, thus, promoting liver fibrosis and cirrhosis [11]. A more in-depth histological report in addition to the grading system within each stage of progression may be found in Brunt et al. [12]. Currently, there is no Food and Drug Administration (FDA)-or European Medicines Agency (EMA)-approved antifibrotic treatments available which posts an urgent need for developing anti-NASH and anti-hepatic fibrosis therapeutics [13,14].
Type 2 diabetes (T2D) is a significant risk factor in NAFLD development by driving the progression of liver injury from simple steatosis (NAFL) to NASH and the subsequent liver fibrosis [15]. Clinically, a recent meta-analysis estimated that the global prevalence of NAFLD among patients with T2D is 55.5%, and the prevalence of NASH among patients with T2D is 37.3% [15]. In patients with asymptomatic T2D, the estimated prevalence of advanced fibrosis is around 5%–7%, which leads to high complications of liver cirrhosis and hepatocellular carcinoma [16,17]. Growing bodies of evidence in both human and animal study have contributed to the establishment of the mechanistic link between hepatic steatosis and insulin resistance. Insulin resistance in hepatocytes (HCs) leads to accumulation of lipids (lipotoxicity) which is associated with dysregulated intermediary metabolism, such as enhancing hepatic de novo lipogenesis and augmenting the flow of fatty acids (FAs) from adipocytes to the liver [18], leading to excessive lipid uptake by the liver which favors progression of NAFL to NASH and cirrhosis. Insulin resistance further impairs mitochondrial fatty acid oxidation and promotes fat accumulation, leading to mitochondrial oxidative stress [19]. Activation of inflammatory kinase JNK by ER stress, in turn, aggravates insulin resistance in HCs through blunting the insulin signaling cascade [20]. In addition, glucotoxicity in T2D patients, which refers to metabolic alterations caused by chronically hyperglycemia and impaired branched chain amino acids metabolism, promotes NASH progression via mTOR signalling [21]. In contrast, study on nondiabetic patients with NAFLD demonstrated that increased peripheral insulin resistance, but not the steatosis or obesity, is the critical link with liver fibrosis [22].
The goal of this review is to provide a synopsis on the current literature investigating cellular and molecular mechanisms regulating liver injury at the different disease stages, from NAFL to NASH and liver fibrosis. Pathophysiological communications among HCs, Kupffer cells (KCs), hepatic stellate cells (HSCs) and liver sinusoidal endothelial cells (LSECs) via mediation of Toll-like receptor, hedgehog and TGFβ signaling pathways are further discussed to highlight the potentials of precision medicine via cell-type specific therapeutic strategy for NASH and liver fibrosis.
2. Biocommunication among liver cells
There are two major cell types in the liver, parenchymal cells and non-parenchymal cells. Parenchymal HCs are the primary component of the liver, occupying 60% of the total cell number and 80% of the total liver volume. They are radially located between the liver capillaries and sinusoids in the lobule (Fig. 1). HCs are the predominant functional units of liver that play pivotal role in energy metabolism, detoxification and protein synthesis owing to the large number of mitochondria, rough endoplasmic reticulum (ER), Golgi apparatus and free ribosomes. HCs possess high regeneration capabilities and play a major role in proliferation after liver injury [23,24]. Sinusoidal cells are non-parenchymal cells, constituting 35% of the total cell number and 17% of the total volume of the liver, which include liver sinusoidal endothelial cells (LSECs), Kupffer cells (KCs), hepatic stellate cells (HSCs) [25,26]. LSECs are the second most abundant cell type of the liver after HCs, forming the sinusoidal wall and represent the interface between the blood and KCs on the one side and HCs and HSCs on the other side. They are not only physical barriers, but also contribute to numerous physiological functions, including metabolite transportation, inflammation, and angiogenesis [27]. HSCs are pericytes resided in the perisinusoidal space (space of Disse) and are surrounded by HCs and LSECs and secrete laminin, proteoglycans, and type IV collagen to form basement membrane [28]. HSCs are predominant cell type primarily responsible for liver fibrosis. Hepatocytes loss, inflammation and metabolic alterations stimulate quiescent HSCs trans-differentiation into proliferative, migratory, and contractile myofibroblast-like cells (activated HSCs) [29]. KCs, the specialized macrophages of the liver, also residing in sinusoids, are vital to hepatic and systematic response to pathogens. KCs have emerged as an essential regulator in liver injury and wound healing (Fig. 1) [30].
Fig. 1.
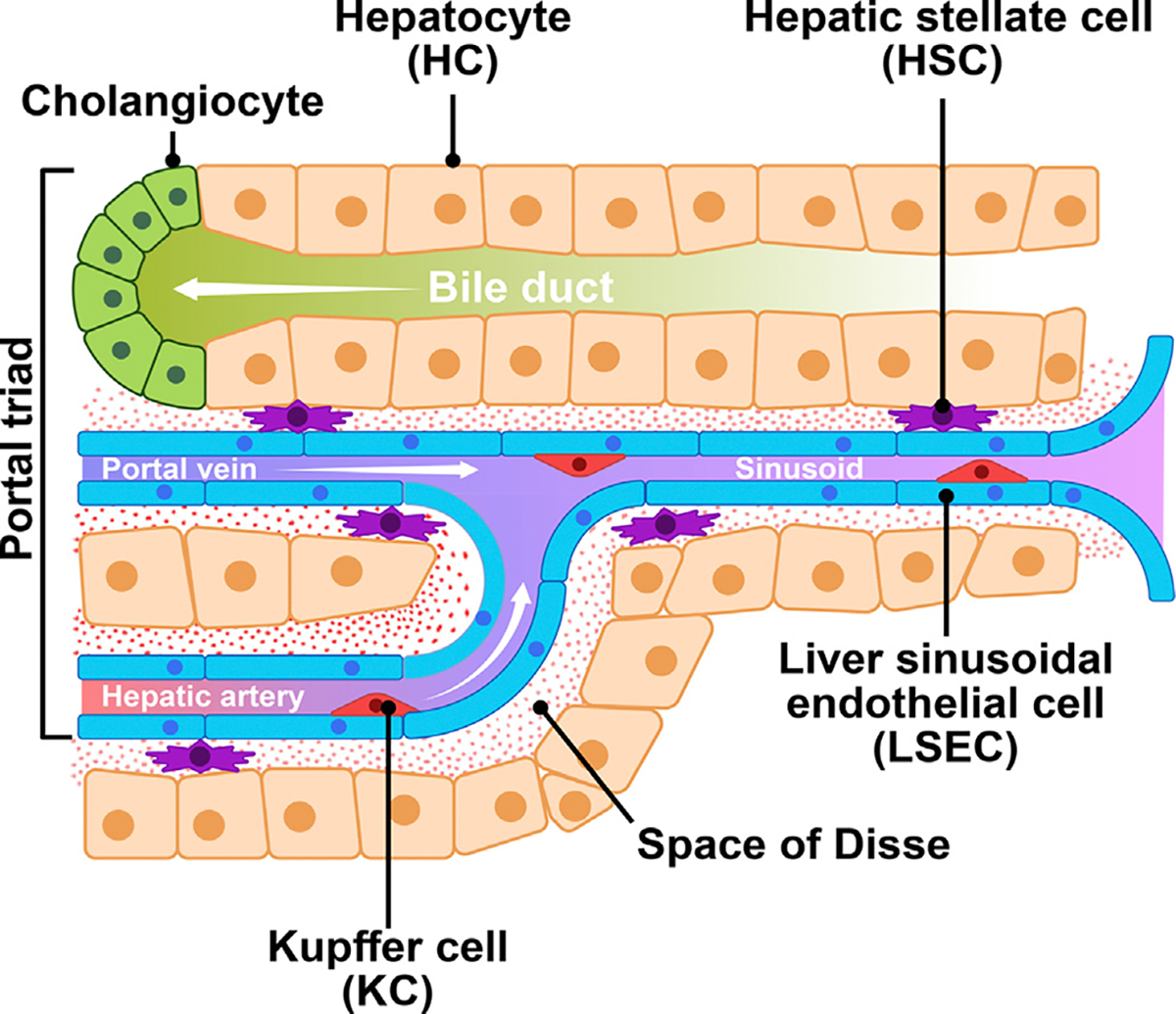
Physiological interaction of hepatocytes (HCs), hepatic stellate cells (HSCs), Kupffer cells (KCs) and liver sinusoidal endothelial cells (LSECs) in the liver.
In the liver, the space of Disse is filled with permeable connective tissue which favours biological exchange between portal blood flow from gastrointestinal tract and HCs. Here, bio-communication takes place among different neighboring cell types, including HCs, biliary epithelial cells, hepatic progenitor cells (HPSc), KCs, bone marrow-derived macrophages, HSCs, LSECs, infiltrating immune cells, and nerve cells via soluble mediators and cytokines [31]. The lipotoxicity induced by the excessive accumulation of lipids in HCs upon metabolic dysregulation, promotes the occurrence of oxidative and ER stress, metabolic inflammation, hepatocyte ballooning, apoptosis and cell death. These events may, ultimately, be followed by the occurrence of fibrogenic activation leading to the initiation and progression of fibrosis through complex processes involving nearby cells, mainly including LSECs, HSCs and KCs (Fig. 2), as it will be presented in this review.
Fig. 2.

The progression of NAFL to NASH and liver fibrosis is associated with a series of liver injury resulting from lipotoxicity, oxidative stress, nitric oxide, ER stress, inflammation and apoptosis that occur sequentially in different liver cells which ultimately leads to the activation of liver regeneration and fibrogenesis, augmenting collagen and extracellular matrix deposition and promoting liver fibrosis and cirrhosis. Toll-like receptor, TGFβ and hedgehog signalling are key pathways mediated hepatic injury.
3. Lipotoxicity and oxidative stress take a Toll on liver cells in the progression of NAFL to NASH (Fig. 2)
3.1. HCs: The primary target of lipotoxicity and oxidative stress in NAFLD
3.1.1. Lipotoxic lipid species
Hepatic steatosis is initiated by increased influx of lipids to the liver or decreased lipid disposal. The main sources of FA are plasma free fatty acid (FFA) from high adipose tissue lipolysis, increased de novo lipogenesis [32], dietary FAs, reduced fatty acid oxidation as well as decreased secretion of very-low-density lipoproteins (VLDL). HCs cope with the excessive lipids by enhancing synthesis of triglycerides and storage as lipid droplets, resulting in lipid accumulation and steatosis [5,33]. The ability of triglyceride synthesis to compensate for increased hepatic FA exposure appears to determine whether or not lipotoxicity results [34]. For example, studies of mouse models with NASH showed that despite reducing steatosis, the inhibition of liver triglyceride synthesis increased hepatic FFA accumulation and the severity of liver injury and fibrosis. The extent of triglyceride accumulation has been the basis for grading the severity of steatosis in NAFLD. However, triglycerides per se are not hepatotoxic [34]. It is the accumulation of some other types of lipids, including FAs, diacylglycerol, oxysterols, cholesterol, and phospholipids that cause HC injury [35]. The demonstration of lipotoxicity induced by lipids other than triglycerides led to the development of therapeutic strategies aiming to block the hepatic accumulation of lipotoxic lipids [36]. Lipotoxicity, therefore, initiates NASH development and is a new therapeutic target.
3.1.2. Mechanisms of hepatic lipotoxicity in HCs
Abnormal accumulation of lipotoxic lipids cause liver damages via the following mechanisms: (i) increased FFAs overwhelms the capacity of mitochondrial and peroxisomal fatty acid oxidation, resulting in overproduction of reactive oxygen species (ROS) that may be immediately toxic or deplete antioxidants, rendering HCs more vulnerable to other adverse factors that generate oxidative stress [37]. (ii) Accumulation of FAs within mitochondria further compromises mitochondrial respiration [35] that makes mitochondria more vulnerable to other insults, e.g., inflammatory cytokines, leading to the release of mitochondrial factors that promote apoptosis [35]. (iii) Accumulation of FAs can exacerbate insulin resistance and hyperinsulinemia [38], resulting in further hepatic lipid accumulation and promoting inflammatory [39] and fibrogenic responses. (iv) Lipotoxicity can disturb a variety of cellular signaling. For example, FAs interact with or modify other molecules, such as transcription factors (hepatocyte nuclear factor-4 alpha), Toll-like receptors (TLRs), nuclear factor-κB (NF-κB) and JNK pathways [35], leading to global changes of metabolic signaling in hepatocytes [40]. (v) In addition to FAs, other types of bioactive lipid molecules, such as oxysterols, diacylglycerol, cholesterol, and phospholipids also module metabolic signaling pathways and compromise cell viability [41]. Prolonged lipotoxicity may induce oxidative and ER stress [42], impair autophagy [43] and promote metabolic inflammation that potentiates liver cell injury and cell death.
The progression of NAFL towards NASH is characterized by increased hepatic steatosis, inflammation, and accompanied with specific histological signatures, such as hepatocellular ballooning observed in NASH and liver fibrosis development (Fig. 2) [44]. In patients with NASH, HCs become more susceptible to damage and incapable of resisting injury due to increased accumulation of lipid toxic metabolites, triglycerides, and ROS [20,44]. At the initial progression of NAFL towards NASH, HCs typically undergo cell cycle arrest and express apoptosis markers like caspases and Fas receptors [45,46]. However, ballooned HCs are resistant to apoptosis due to the activation of Hedgehog signaling that inhibits expression of caspases, but accompanied with the activation of pro-inflammatory and pro-fibrogenetic factors [45,47,48]. Thus, necrosis (uncontrolled cell death) overtakes the HCs as disease progresses.
3.1.3. Mitochondrial dysfunction and oxidative stress in HCs
Healthy hepatocytes contain remarkably high number of mitochondria. Generally, mitochondria convert ~2% of consumed O2 into ROS and are the major site of cellular ROS formation [49,50]. With increased levels of lipids and ROS in HCs during early stages of NAFLD, mitochondria initially undergo adaptive remodeling as a protective mechanism [51,52], characterized by morphological changes through mitochondrial fission and fusion processes along with variations in energy expenditure and gene expression. However, as NAFLD progresses toward NASH, damaged HCs display maladaptive remodeling characterized by reduced ATP production, increased oxidative stress [53], enlarged mitochondria (3–10 μm in diameter) with reduced number of cristae and presence of crystalline bodies [54,55]. In patients with NAFLD, aberrant lipid metabolism enhances the damage of mitochondrial DNA and mitochondrial respiratory chain (MRC) proteins [56] as well as activates JNK bound to MRC complexes, leading to excessive ROS production [52,57]. Consistently, progression of NASH was evident by increased ROS level and reduced catalase activity which compromised detoxification and enhanced mitochondrial damage in HCs [58]. Moreover, accumulation of cholesterol further leads to a loss of reduced glutathione, an anti-oxidant defense mechanism, leading to inhibition of FA β-oxidation and exacerbation of lipotoxicity [59]. The damaged HCs further induce release of mitochondria-derived damage associated molecular patterns (DAMPs) through necrosis which, in turn, activates the NLRP3 inflammasome pathway [60] and induces metabolic inflammation in the liver.
3.1.4. Nitric oxide in HCs
Nitric Oxide (NO), the most investigated member of the reactive nitrogen species (RNS) family, mediates diverse functions in liver pathophysiology [61]. NO is synthetized enzymatically from L-arginine by three NO synthase isoforms, endothelial (eNOS), inducible (iNOS), and neuronal NOS (nNOS) [62]. iNOS is primarily expressed in HCs, but also in LSECs, HSCs and cholangiocytes [63]. NO can either play a protective role during insulin resistance-induced HCs death and liver generation [64] or partially promotes liver steatosis and NASH progression [40], indicating the importance of fine-tuning of NO metabolism. Constant excessive production of NO creates nitrosative stress inside HCs, resulting in hepatic mass damage mostly via nitrosylation of thiol residues. Proinflammatory cytokines IL-1β and TNFα induce iNOS mediated NO production in HCs has been observed during NAFLD progression [65]. Clinically, both iNOS and nitrotyrosine levels are significantly higher in NASH patients, and a major role for NO0 during NASH-associated fibrogenesis has been suggested. In addition, RNS is synergistic with ROS to produce more damage to HCs either through generation of 0OH or 0NO2 radicals or peroxynitrite (ONOO−) radical. Each chemical reaction involves excessive generation of and oNO radical and contributes to the pathology seen in NAFLD and fibrosis [61].
3.2. Redox imbalance activates HSCs and contribute to fibrosis
Redox imbalance due to excessive reactive oxygen and nitrogen species (ROS and RNS, respectively) produced either as byproduct of metabolic process or from other enzymatic sources also play a major role in HSCs activation. Besides HSCs, damaged HCs, KCs, or infiltrating lymphocytes also produces ROS/RNS to stimulate HSCs activation and proliferation as well as increased production of ECM [66]. Recent studies demonstrate a strong link between immune reaction and ROS imbalance that leads to advanced fibrosis in NAFLD patients. In a mouse model of NASH [67], augmented NO metabolites in serum correlated with the severity of autoimmune hepatitis and fibrosis [68].
NO is involved in nitrosative stress in liver fibrosis which are associated with HSCs activation, including contractility, proliferation and ECM deposition. Endothelins (ET) and NO are well known modulators of HSC contractility and relaxation. NO inhibitors, N-nitro-L-arginine methyl ester (L-NAME) and endothelin-1, were shown to attenuate HSCs contractility [69]. A study in CCl4 induce-liver fibrosis in rats further showed that induction of NO synthesis by L-arginine partially protected the liver from ECM collagen deposition, reduced several markers of liver injury and adverse hepatic remodeling. In contrast, administration of NO inhibitors L-NAME or aminoguanidine increased collagen deposition and liver damage [70], suggesting that endogenous NO protects the liver from injury, probably by decreasing the toxicity of free radicals and/or by attenuating the activity of HSCs. Moreover, administration of NO antagonists induces neutrophil infiltration in the hepatic parenchyma and exacerbates liver injury due to the increased ECM collagen accumulation, whereas increased NO protects HCs from injury by reducing the adhesion of neutrophils [69]. NO was also reported to prevent ROS mediated HSC proliferation. Together, this evidence suggests that upregulation of NO protects liver from collagen deposition and proliferation, thereby, liver fibrosis. The most likely mechanism(s) of action involves scavenging reactive free radicals and decreasing HSC activity.
3.3. Excessive NO in LSECs induced fibrosis associated capillarization
The healthy liver contains LSECs that are very fenestrated in which the holes that go through the cells are organized in sieve plates and allow solutes in the blood plasma, including chylomicron remnants and lipoproteins, to perfuse through to the space of Disse. A key feature of LSECs is that they are responsible for approximately 45% of all the endocytic activity of the liver [71,72], due to the rich profile of scavenger receptors expressed on the cell surface, primarily MR/CD206/SR-E3, SR-H1/2 (Stabilins), and FcγRIIb/CD32b as well as other minor receptors such as SR-A and SR-B [73].
Healthy LSECs secrete a constant and constitutive level of NO via eNOS in response to normal stimuli such as shear stress by the blood and vascular endothelial growth factor (VEGF) secretion. NO produced by eNOS maintains liver homeostasis and, importantly, maintains the quiescence of HSCs and KCs [74]. During early events of simple steatosis, the LSECs remain largely untouched. This is the period in which the HCs are accumulating lipid droplets and begin to suffer from lipotoxicity. LSECs do not store lipids, although they may suffer from the effects of lipotoxicity. The induction of iNOS generates an increase of NO which interacts with ROS to form , HNO2, NO2 radicals, and ONOO− [75]. The first response to these radicals is an alteration of phenotype in which the fenestrae begin to disappear in a process called defenestration or capillarization presumably due to the nitrosylation of cellular components [75]. Although the precise stimuli for the induction of capillarization is not known and may be the result of multiple inputs, the onset of capillarization decreases LSEC permeability which affects lipoprotein secretion from HCs and the de novo synthesis of lipids within the HCs [76,77]. Recent investigations into how resilient LSECs are to the induction of capillarization are beginning to suggest that the pro-inflammatory phenotype of LSECs occurs while the fenestrae are preserved. Kus et al reported that, in C57BL/6 mice fed a high-fat diet (60% Kcal of fat) for 2 to 20 weeks, the sieve plates were preserved throughout the time course showing that both fenestrae diameter and porosity (measured by electron and atomic force microscopy) did not decrease relative to the mice fed on control diets [78]. These studies need to be reinforced and repeated in a larger scope to ensure that these observations are not within the narrow confines of the animal model or diet. It is also important to investigate whether this phenotype occurs in patients with NAFLD.
The progression of NAFLD to steatohepatitis is promoted by NADPH oxidase, NOX1. In human patients and in mice fed a high-fat/high cholesterol diet, NOX1 expression is increased and with concomitant higher levels of alanine aminotransferase (ALT) and cleaved caspase-3, markers of liver damage, which were attenuated in NOX1 knockout mice fed on the same diet. NOX1 upregulation also increased the levels of nitrotyrosine adducts within the sinusoids, together with ballooning HCs and contracting HSCs, resulting in the reduction of blood flow through the sinusoids which is associated with liver fibrosis development [79].
4. ER stress and insulin resistance augments HC injury
The unfolded protein response (UPR) is a signalling system emanating from the ER in response to disruption of ER homeostasis [80]. Under physiological conditions, the UPR is critical for the development and function of highly active secretory cells, including hepatocytes and cells in exocrine and endocrine glands. The endoplasmic reticulum (ER) plays a major role in proteins synthesis. An organized quality control system consisting of molecular chaperones and folding proteins ensures that the ER lumen is an optimal environment for proper protein folding and development [80]. In states of physiological stress, such as an increased demand for protein synthesis or the disruption of protein chaperones in the ER lumen (e.g., owing to pharmacological treatment), protein misfolding can occur. In mammalian cells, the UPR is governed by signaling cascades initiated by three ER-localized protein sensors: the dsRNA-dependent protein kinase (PKR)-like ER kinase (PERK); inositol-requiring enzyme 1α (IRE1α), and activating transcription factor 6 (ATF6) [81]. Diverse functions have been ascribed to the mammalian UPR, including a role in the hepatic lipogenic pathway [80].
Insulin resistance, the hallmark of type 2 diabetes, with its associated hyperlipidaemia and metabolic inflammation is a risk factor for NAFLD and the subsequent fibrosis. Ample evidence from animal models and human patients with insulin resistance (either with or without Type 2 diabetes) displays similar lipid abnormalities which include: hypertriglyceridemia, low plasma concentrations of HDL, and the appearance of small, dense LDL [82]. A key underlying pathological mechanism of this profile appears to be hepatic VLDL overproduction. The link between insulin resistance and increased VLDL secretion has been thought to be derived from increased delivery of FAs to the liver secondary to increased lipolysis within adipose tissue, increased hepatic lipogenesis, increased levels and activity of microsomal triglyceride transfer protein, and loss of insulin’s ability to direct apolipoprotein B (ApoB) toward degradation [82]. It is now clear that, in response to increased lipid delivery to hepatocytes, significant amount of ApoB is synthesized and accumulated in the ER which coupled with the accumulated lipids in HCs induces chronic metabolic imbalance and disturbs the metabolic homeostasis of ER and compromises its functionality [83]. Overproduction of ApoB in the ER of the injured HCs leads to increased production of misfolded proteins in the ER lumen, and induces ER stress. Although the hepatic UPR may be triggered to alleviate ER protein load and reduce HC damage [81]. However, this response can be overwhelmed in the long term, leading to HC cell death.
In addition, the induced ER stress can further activate inflammatory kinases JNK and NF-κB, leading to the activation of metabolic inflammation and hepatic insulin resistance [81,84]. In the insulin resistance state, hepatic de novo lipogenesis and VLDL assembly are further augmented due to the activation of SREBP-1c, SREBP-2 and their downstream genes involved in lipid synthesis. Thus, the lipid overload-induced ER stress, lipotoxicity and insulin resistance enter a vicious cycle, which eventually induce HC apoptosis and cell death [85]. Clinically, augmented ER stress is clearly observed in biopsies of patients with different stages of liver fibrosis as well as in animal models of fibrosis induced by CCl4 or high-fat diet [86].
5. Crosstalk among different liver cell types via toll-like receptor signaling drives NAFL progression to NASH and fibrosis
5.1. Damaged HCs activates inflammation via KCs and Toll-like receptors (TLRs)
Inflammation is critically involved in the transition from NAFL to NASH in the liver. Generally, the optimal triglyceride deposition as lipid droplets in HCs is hepatoprotective as it attenuates lipotoxicity by reducing extracellular FFAs [47]. Otherwise, incorrect lipid droplet deposition or mobilization and protein expression can enhance HC lipotoxicity, leading to NAFLD progression. HCs express membrane and cytoplasmic pattern recognition receptors (PRRs), like TLR-2, -4, -5, and -9 and nucleotide-binding oligomerization domain (NOD) -1 and -2, etc., which easily sense their respective pathogens and ligands/metabolites, leading to increased local inflammation and the development of injury and insulin resistance [87]. Role of TLRs in HCs is diverse. Deficiency of TLR-2, TLR-4, or TLR-9 receptors in HCs attenuated hepatic inflammation, insulin resistance, oxidative stress and hepatic steatosis [88,89]. Conversely, deficiency of TLR-5 receptors strongly impaired bacterial clearance by the liver and aggravated NAFLD development from steatosis to NASH and fibrosis upon HFD or MCD diet challenge [90].
NASH-driven hepatic inflammation and fibrosis in HCs involve various forms of lytic death, including apoptosis, necrosis, necroptosis, pyroptosis, and ferroptosis which participated in the release of DAMPs and strong inflammatory responses [47,91]. Apoptotic HCs can directly initiate inflammation through KCs/macrophage activation after engulfment of apoptotic bodies which is further evidenced by increased levels of caspase generated keratin-8 fragments, a circulating surrogate biomarker of HC apoptosis, with NASH and fibrosis resolution [92]. Clinically, increased accumulation of senescent HCs are observed in fibrosis stage and T2D [93].
5.2. TLR signaling regulates transition KCs between M2 and M1 during NASH
5.2.1. Stimuli regulate switch of KCs from M2 to M1 state
KCs are the macrophages in the liver and were first identified by Carl Wilhelm von Kupffer in 1876 [94]. Gut-derived antigens drain through the portal vein and arrive in the liver with KCs as the first line of defense and tolerance of foreign molecules. KCs have several pattern recognition receptors such as the mannose receptor, Toll-like receptors, scavenger receptors, and NOD-like receptors that recognize DAMPs and pathogen-associated molecular patters (PAMPs).
Under normal conditions, macrophages are tolerant and trend in the M2 state which suppressess inflammation by IL-4, IL-10, and IL-13 cytokines [30,95]. In contrast, macrophages in the M1 state are often key players in chronic inflammatory diseases such as inflammatory bowel disease (IBD) [96] and atherosclerosis [97]. During the development of NAFLD, multiple factors participate in driving the transition of KCs from M2 to M1 state. These include (i) the decreased blood flow and partial occlusion of the sinusoids places a strain on the LSECs, sending them into a pro-inflammatory phenotype; (ii) HSC activation also secretes a number of pro-inflammatory factors that stimulate the activation of KCs; (iii) Intestinal leakage during NASH and a co-morbidity of obesity increases the levels of bacterial-derived lipopolysaccharides (LPS) in the blood. LPS interacts with TLR-4 resulting in increased secretion of TNFα, IL-1β and IL-6 which promote inflammation [95]; (iv) Higher circulating FFAs from adipose tissue and absorbed from dietary intake activate inflammasomes to further increase the chronic inflammatory condition of NAFLD [98]. After these stimuli increase inflammation, KCs begin to trend to the M1 phenotype from the M2 state established in NAFL which may be usher in the next phase of chronic inflammation-NASH. The balance of M1/M2 states may be a key determinant for the progression of chronic liver inflammation into fibrosis. This has been tested in an experimental setting in which blocking IL-10 with antibodies promoted the M1 phenotype. The promotion of the M2 phenotype under these conditions destroyed M1 KCs, thus ameliorating NAFLD [99].
5.2.2. Activation of TLR-9 signaling keeps KCs in M1 state
TLR-9 is a significant DAMP that is involved with many chronic liver diseases. During HC ballooning and necrosis, mitochondria and other internal organelles are exposed to KCs and other immune cells which can be released into blood circulation. Plasma from mice and patients with NASH contains high levels of mitochondrial DNA (mtDNA) and intact mitochondria which can activate TLR9 [89]. Elevated oxidized mtDNA was also detected in liver biopsies from patients with NASH [51]. Development of NASH in mice fed a high fat diet (HFD) required TLR9 on lysozyme-expressing cells which can be blocked or ameliorated by a clinically applicable TLR-9 antagonist (IRS954) when administered to mice during or before NASH development [89]. Specifically, IRS954 was administered to mice after 8 weeks of HFD administration and efficacy was assessed at week 12. The results showed a significant reduction in steatosis, inflammation, HC ballooning, ALT and the expression of IL1β, IL-6 and TNFα inflammatory cytokines. In addition, knocking out expression of TLR-9 in mouse models diminishes the NASH phenotype. Without TLR-9, KCs trended strongly to the M2 phenotype and were very resistant to M1 activation [100]. These and other lines of evidence strongly suggest that TLR-9 is a major factor in determining the M1 phenotype in KCs to perpetuate NASH going forward into fibrosis.
5.3. TLRs mediate activation of HSCs and contribute to liver fibrosis
Altered lipid metabolism in HCs results in accumulation of free cholesterol and the associated lipotoxicity which is thought to activate resident KCs and trigger secretion of inflammatory cytokines, e.g., TNFα and IL-6, to induce HSC activation [101]. It is also suggested that accumulated free cholesterol in HSCs may directly activate inflammation in the cells in a TLR-4 dependent mechanism, and promote liver fibrosis. Activated human HSCs abundantly express TLR-4 and CD14 which respond to gut bacterial endotoxin LPS and promote release of pro-inflammatory cytokines [102]. Similarly, activated mouse HSCs are enriched in TLR-2, TLR-4, and TLR-9 expression and promote expression of inflammatory cytokines and adhesion molecules, including NF-κB, JNK, TGFβ, IL-6, IL-8, CCL2 (MCP-1), MIP-2, intercellular cell adhesion molecule 1 (ICAM1), vascular cell adhesion molecule 1 (VCAM1) and E-selectin, thus, stimulate pro-fibotic gene expression in HSCs [103]. Although TLR-4 is expressed ubiquitously in liver, studies have demonstrated that TLR-4 expressed on HSCs is crucial for liver fibrosis [88]. For instance, the TLR-4 specific ligand, gut bacterial endotoxin LPS, can activate TLR-4 signaling and the subsequent release of various chemokines and adhesion molecules which attract macrophage migration into the liver and directly activates HSCs as well as the development of liver fibrosis. Resident KCs in liver can also produce TGFβ and activate TLR4 signaling in HSCs that may also promote liver fibrosis through chronic inflammation [102]. In the CCl4 or bile duct ligation induced liver fibrosis animal models, release of DAMP molecules from damaged HCs interacted with TLR-9 and participated in HSCs activation and liver fibrosis while knockdown of TLR-9 reduced fibrosis resolution prominently [102].
In case of unresolved chronic inflammation, HSCs acquire a senescent phenotype over time characterized by permanent cell cycle arrest. Compared to the young and dynamic stage, senescent HSCs produce more proinflammatory cytokines (e.g., IL-6, IL-8, IL-11), less ECM components and more matrix metalloproteinases (MMPs) proteins, thus favoring their immune clearance and attenuating fibrosis progression [104]. The senescent HSCs can encode cytokines or ligands/molecules that potentiate natural killer (NK) cell function. For instance, senescent HSCs express NK cell receptor ligands, MICA, ULBP2 and CD58, on the cell surface which facilitate their removal by hepatic resident NK cells through immunosurveillance mechanism [104].
6. TGFβ signaling promotes liver fibrosis
6.1. TGFβ1 secreted by HCs and HSCs induces liver fibrosis
TGFβ1 belongs to the TGFβ superfamily of related proteins including growth differentiation factors (GDFs), bone morphogenetic proteins (BMPs) and activins [105]. It is a pleotropic cytokine and potent inducer of fibrosis in several organs like heart, lung, liver etc. In liver, TGFβ1 broadly regulates liver pathophysiology sequentially, encompassing an early induction of HCs apoptosis, differentiation of activated HSCs to myofibroblasts during liver fibrosis to cirrhosis and finally to hepatocellular carcinoma (HCC) [106]. Initially, the profibrogenic effect of TGFβ was recognized in HSCs activation and collagen deposition; later it was also identified in HCs and hepatic progenitor cells of liver (Fig. 2) [107].
TGFβ1 is a ligand for the TGFβ1 receptor (TGFβ1R) on the cell surface and propagates signaling through the SMAD family of proteins. The TGFβ1R is phosphorylated on serine and threonine residues and promote the activation of SMAD2 and SMAD3 kinase activity which may further bind to SMAD4 to form a SMAD2/3/4 complex [108]. The complex can then translocate to the nucleus and activate genes/pathways involved in liver fibrosis, including MAPK, PI3K, NF-κB, NOX4 and connective tissue growth factor (CTGF) pathways [109]. TGFβ1 is kept in check by BMP-7, a molecule that interacts with other receptors and signals through SMAD1/5/8 mediation. Both SMAD1/5/8 and SMAD2/3 complexes compete for SMAD4 to reach a balance in the cells. Continual insult in the liver may favor SMAD2/3 formation with SMAD4, leading to the increase of SMAD2/3/4 complex and the associated fibrogenesis. SMAD6 and SMAD7 are inhibitory in nature and can counteract SMAD2/3 activity. In fact, a recent report suggests that SMAD3 is the primary driver for the promotion of fibrosis as the knock-out of SMAD2 enhanced SMAD3 activity even though both are complexed together in the competition of SMAD4 binding [110]. SMADs are regulated by multiple mechanisms including SUMOlyation (ubiquitination), phosphorylation, and poly-ADP ribosylation which may be complex in the setting of the multi-factorial inputs of NAFLD that lead up to liver fibrosis [111].
Several years ago, with regard to the general functions of TGFβ1, Boris Hinz developed a hypothesis that describes the mechanism of the extracellular matrix and the pro-fibrotic TGFβ1 cooperating to regulate the remodeling activities of stromal cells. Matrix straining and stiffening decrease the threshold for TGFβ1 activation in the ECM by increasing the mechanical resistance to cell pulling [112]. In the liver, activation of TGFβ1 by KCs overcompensates in the injury process (e.g., HC ballooning and lipotoxicity) and scar development begins with HSC activation and the deposition of collagen in the Space of Disse [113]. Activated HSCs or myofibroblasts continually secrete TGFβ1 along with TNFα to recruit more macrophages (M1) and leukocytes to the liver in a positive feedback loop that further increases inflammation and stimulates hasty mechanisms for healing, leading to augmented collagen deposition [114,115]. Knockdown of transient receptor potential melastatin7 (TRPM7) attenuates TGFβ1-induced expression of myofibroblast markers, increases the ratio of MMPs/tissue inhibitor of metalloproteinases (TIMPs), and decrease SMAD2/SMAD3-associated collagen production [116]. Migration and proliferation of activated HSCs are hallmark of liver fibrosis. TGFβ induces HSCs migration through guanosine triphosphate (Rho GTPase) signaling activation [117] and proliferation by inducing platelet-derived growth factor receptor-beta expression and oxidative and activating Ca2+/calmodulindependent protein kinase II (CaMKII) [116]. Therapeutic strategies, e.g., blocking circulating TGFβ1, antagonizing its receptors, or blocking its activation at the cell surface, have been challenging due to adverse effects of systemic TGFβ antagonism, including induction of inflammation and cancer [116,118].
6.2. BMP-9 mediates signaling between HSCs and LSECs in liver regeneration
BMP-9 is a member of the TGFβ1 family that is closely involved with liver regeneration and fibrosis. This paracrine hormone is constitutively secreted by HSCs which is upregulated when HSCs are activated during liver injury or disease [119]. BMP-9 binds to activin receptor-like kinase 1 (ALK1) receptors on the surface of LSECs and transduce their signals through the SMAD system to active gene expression [120]. BMP-9 has been described as a profibrotic factor since its increased secretion during HSC activation promotes fibrosis. It has been reported that using antibodies against BMP-9 neutralizes the upgraded BMP-9 in a mouse model and attenuate liver fibrosis, suggesting that this molecule may serve as a therapeutic target of liver fibrosis[121].
Emerging evidence have demonstrated that function of BMP-9 in LSECs is associated with genetic background. For example, the most prominent strain of mice used in liver injury models is the C57BL/6 due to its propensity to overeat and develop NAFLD on HFD diets. Knocking out of BMP-9 in BALB/c mice did not result in a significant phenotype. In contrast, mouse in the 129/Ola background is more prone to vascular alterations and its LSECs are more likely to be impacted than other hepatic cell types [122]. This is due to the defectiveness of LSECs in the 129/Ola strain, especially in the male mice. LSECs from the 129/Ola BMP-9 KO mice were capillarized and had lower overall expression of terminally differentiated markers such as the Stabilin receptors, mannose receptor, Ehd3 endocytic receptor and Gata4 that regulates fenestration [123,124]. Addition of BMP-9 partially restored some but not all of these markers in LSECs isolated from 129/Ola BMP-9 KO livers [124]. Collectively, these data implicate that BMP-9 is a regulator of liver homeostasis and is required for liver regeneration. Low constitutive levels of BMP-9 are optimal for adequate response to mild insults and may prevent liver injury to some degree due to the liver’s unique regenerative properties [125,126]. However, BMP-9 is detrimental at high levels, a situation found in a setting of inflammation or steatohepatitis, and adversely affects LSEC morphology and function as observed in the 129/Ola mouse model studies [124,127].
7. Hedgehog signaling in NASH and liver fibrosis
Hedgehog (Hh) is a classic morphogen secreted by ligand-producing cells and diffuses into the extracellular space to regulate Hh-responsive target cells[128]. Mammals have three different Hh ligands: Sonic (Shh), Indian (Ihh) and Desert (Dhh) hedgehog. The Hh signalling pathway is a highly complex signalling cascade with many players and intricate regulation. However, it can be simplified into four major elements: i) the ligand Hh; ii) the receptor Patched (Patch [PTCH1]); iii) the signal transducer G protein-coupled receptor Smoothened (Smo); and iv) the effector transcription factor, Gli (GLI1) [129].
Of the 3 hedgehog ligands, Shh has the greatest impact in fatty liver development and progression. Shh regulates a plethora of biological processes in cells expressing Shh receptors (patched 1), including proliferation, differentiation and viability as well as adult liver regeneration [130]. Shh in healthy adult livers is not detectable. However, damaged HCs quickly induces expression of Shh. For example, ER stress inducers stimulate Shh expression in HCs [131]. More importantly, liver injury induced by FFA-associated lipotoxicity also enhances Shh expression [131]. In contrast, HCs with deleted caspase-2 are protected from apoptosis and do not induce Shh expression when challenged with FFA, palmitic acid [132]. Activation of Shh has been linked to the outcomes of patients with NASH [48]. Ballooning HCs produce Shh [133] which can be released either directly into the ECM or incorporated into exosomes, the small membrane vesicles that transduce stress signal to distant cells. Increased secretion of exosomes from apoptotic HCs regulate other HCs, as well as KCs, HSCs and LSECs [134]. Non-lipid particle-associated Shh binds to matrix proteins, i.e., lypican 3, to increase its concentration near the Shh producing cells in livers of patients with NASH. Shh also associates with lipoproteins which allows the ligand to initiate signalling in distant cells [135]. Activation of Shh stimulates Shh-responsive cells to produce other factors that regulate wound healing. For example, Shh signalling induces expression of TGFβ, CTGF, amphiregulin, jagged, and Wnt ligands by HSCs [136]. Shh stimulates LSECs to produce VEGF [137] and induces ductal cells to express osteopontin and chemokines that recruit various types of immune cells to the injured liver [138]. These immune cells further secret diverse cytokines, e.g., IL-1β, IL-6, TNFα and INFγ to exacerbate hepatic inflammation and liver injury. Shh also regulates macrophage polarization, thereby modulating the local balance of inflammatory, anti-inflammatory, and fibrogenic cytokines [138]. Through this process, apoptotic HCs induce a liver regenerative response to replace the dying cells [134]. Inhibition in hedgehog signalling compromises liver regeneration and replacement for HC loss [139]. However, activation of hedgehog signalling also promotes development of NASH-associated cirrhosis [140], HCC [141]. Therefore, hedgehog signalling must be tightly regulated to assure the appropriate programming for hepatic wound healing and regeneration.
Similar to Shh, Ihh is upregulated in animal models of NAFLD consuming a HFD, primarily through the inflammation component of this disease [142]. Increased Ihh secretion in the fatty liver promotes fibrosis through transdifferentiation of HSCs mediated by Myc and TGF-β2 signaling. This results in the release of Wnt5a from HSCs which may result in HCC. Once HCC develops, it is poorly differentiated and also secretes Wnt5a to act in an autocrine manner to further tumorigenesis [143]. Dhh is restricted to the nervous system and to testis and plays no known role in NAFLD development or progression [144].
8. Perspective therapeutic strategy of precision medicine for NASH and liver fibrosis treatment
The current therapeutic strategy for patients diagnosed with mild forms of NALFD is diet and life style modification, such as body weight control and adequate physical activity. With more severe forms of NASH, there are also other disease modalities to tackle. In diabetes associated NAFLD, insulin sensitizers, including metformin, thiazolidinediones, glucagon-like peptide-1 receptor agonists, and the dipeptidyl peptidase IV inhibitors, have been used in NASH management alongside with dietary control and behavior changes which alleviate the progression of NASH in most cases [145].
For several decades, physicians and researchers have been developing drug carriers that will increase the drug delivery to liver for targeting NASH and liver fibrosis and decrease the side effects of drug metabolism. Nanoparticle-based drug delivery approach has been shown to be the promising remedies for this purpose. Of all the classes of nanoparticles available to treat fatty liver and other hepatic diseases, liposome (LNP) based formulations are the most successful due to their high stability, low toxicity and scale-up production methods [146]. The advantage of liposomes is that they have the ability to encapsulate both hydrophobic and hydrophilic drugs and the hydrophilic membrane shell may be modified by chemical moieties for aiming the liposome to target a specific liver cell-type (Fig. 3) [147]. The lipid composition of the LNP is of particular importance as it determines the half-life in circulation, including how the LNP interacts with targeted cells and how the LNP escapes the endosomal pathway to deliver the cargo into cellular cytoplasm [148,149]. For example, SB431542 is a TGFβ inhibitor with low water solubility and poor bioavailability as a stand-alone drug. When encapsulated by an oleate-based liposome, it had relatively good uptake and efficacy in liver with few side effects in other organs when used in an experimental CCl4-induced liver fibrosis mouse model [150]. Specific HC targeting may be accomplished through the asialoglycoprotein receptor (ASGPR) which is only expressed on HCs that recognize galactose or N-acetylgalactosamine ligands with high affinity [151,152]. LSECs may be specifically targeted with hyaluronic acid (HA)-based liposomes as the HARE/Stabilin-2 receptor specifically recognizes HA [153,154]. HSCs may be targeted using liposomes with PDGFR-β-targeting antibodies for specific binding to HSC cell surfaces [155]. KCs and other macrophages may be targeted with liposomes containing phosphatidylserine (PS) lipids (Fig. 3). Since both KCs and LSECs express Stabilin-1 which will bind with PS, and even Stabilin-2 expressed by LSECs will bind with PS [156], PS may not be a very good molecule to exclusively target KCs. However, if the liposome is greater than 200 nm in diameter, it will favor KC uptake over LSEC internalization through phagocytosis [157]. More importantly, even though liposomes of many different types tend to accumulate in the liver, targeting specific liver cell type will increase efficacy of the pharmaceutical cargos and decrease off-target effects, and therefore reduce side effects.
Fig. 3.

Liposomes have the ability to encapsulate hydrophobic and hydrophilic drugs. The membrane shell may be modified by chemical moieties for aiming the liposome to target a specific liver cell-type. HC targeting may be accomplished through the asialoglycoprotein receptor HSCs may be targeted using liposomes with PDGFR-b-targeting antibodies for specific binding to HSC cell surfaces. LSECs may be specifically targeted with hyaluronic acid-based liposomes and KCs and other macrophages may be targeted with liposomes containing phosphatidylserine lipids.
Currently, no nanoparticle or liposomal formulation has been approved for the treatment of NALFD and liver fibrosis. However, there are some nanoparticle formulations in clinical trials. For example, a natural mutation in patatin-like phospholipase domain-containing 3 (PNPLA3) protein at I148M reduces it’s enzymatically activity in remodeling lipid droplets during normal lipid droplet catabolism [158]. An antisense oligonucleotide (ASO) targeting the mRNA of PNPLA3-I148M improved overall NASH outcomes in a mouse model and this ASO is currently in phase I human trials in people with this same mutation in PNPLA3 (Ionis & AstraZeneca) [159]. Similarly, targeting the 17-β hydroxysteroid dehydrogenase 13 (HSD17B13) using RNAi silencing (Alnylam and Regeneron) has being developed for the treatment of NASH. Although HSD17B13 is not a very well-characterized enzyme, it is known to be localized on lipid droplets similar to PNPLA3 and a number of polymorphisms have been linked to human NASH [160,161]. Both ASO and RNAi-based strategies use liposomal encapsulation as well as naked nucleic acid strategies targeting galactose to the ASGP receptor for efficient uptake in HCs are also under development [162,163].
9. Concluding remarks and future perspectives
The fundamental metabolic perturbations that trigger NAFL and its progression to NASH and liver fibrosis in diabetic subjects are insulin resistance which is centrally involved in the pathogenesis of lipotoxic induced tissue injury, a pathological condition in which accumulation of harmful lipids (e.g, FFAs) in HCs perturbs cellular organelle function, causing a series of liver damage by oxidative stress, excessive nitric oxide, ER stress, inflammatory cytokines and apoptosis that occur sequentially in the neighbouring parenchymal cells (HCs) and non-parenchymal cells (e.g., HSCs, KCs and LSECs), which induce a hepatic regeneration and wound-healing response to replace dead HCs. The repair process is intricate and involves activation of multiple signalling pathways, including redox signalling, oxidative stress, neurohumoral signaling, pro-inflammatory cascades, growth factors, senescence and programming cell death, which are deleterious when deregulated.
Deregulated wound-healing responses worsen clinical outcomes by promoting development of fibrosis, cirrhosis and liver cancer.
Currently, there is no approved drugs for NASH and liver fibrosis treatment. NASH is a heterogeneous disease with multiple adverse factors contributing to the disseise development. Ideal drugs for both NASH and fibrosis should be able to improve the liver microenvironment and tissue histology as well as its complications such as cardiovascular disease. Reversal of NASH without worsening of fibrosis has been a primary focus of the novel therapeutics in clinical trials. Unfortunately, the outcomes from clinical trials showed no treatment was able to significantly improve histological status of NASH and the resolution of fibrosis. Nevertheless, novel therapeutic strategy for NASH and fibrosis has been under developing which takes into consideration of insulin sensitization, anti-inflammatory, antifibrotic and drug delivery approaches. Future research should focus on combination therapies should shed light on the prevention and treatment of NASH and liver fibrosis.
Acknowledgments
This work was supported by a grant from British Heart Foundation (UK) (PG/19/86134788) and a grant from Northern Ireland Chest Heart & Stroke (2019_08) to Q. Su; National Institutes of Health grant HL130864 to E. Harris.
Abbreviations:
- ALK1
Activin receptor-like kinase 1
- ALT
Alanine aminotransferase
- ApoB
apolipoprotein B
- ASGPR
Asialoglycoprotein receptor
- ASO
Antisense oligonucleotide
- ATP
Adenosine triphosphate
- BMPs
Bone morphogenetic proteins
- CaMKII
Ca2+/calmodulindependent protein kinase I
- CCl4
Carbon tetrachloride
- CTGF
Connective tissue growth factor
- DAMPs
Damage associated molecular patterns
- ECM
extracellular matrix
- EMA
European Medicines Agency
- ER
Endoplasmic Reticulum
- ET-1
Endothelin-1
- FDA
Food and Drug Administration
- FA
Fatty acid
- FFAs
Free fatty acids
- GDFs
Growth differentiation factors
- HA
Hyaluronic acid
- HCC
Hepatocellular carcinoma
- HCs
Hepatocytes
- HFD
High fat diet
- Hh
Hedgehog
- HNF-4α
Hepatocyte nuclear factor-4 alpha
- HPSc
Hepatic progenitor cells
- HSCs
Hepatic stellate cells
- HSD17B13
17-β hydroxysteroid dehydrogenase 13
- HIF-1α
Hypoxia-inducible factor-1alpha
- ICAM1
Intercellular cell adhesion molecule 1
- IL-1β
Interleukin-1beta
- IL-6
Interleukins-6
- INFγ
Interferon gamma
- JNK
c-Jun N-terminal kinase
- KCs
Kupffer cells
- L-NAME
N-nitro-L-arginine methyl ester
- LNP
Liposome based nanoparticles
- LSECs
Liver sinusoidal endothelial cells
- MAFLD
Metabolic associated fatty liver disease
- MAPK
Mitogen activated protein kinase
- MCD
Methionine-choline-deficient
- MCP-1
Monocyte chemoattractant protein-1
- MICA
Major Histocompatibility Complex (MHC) Class I Chain-Related gene A
- MIP-2
macrophage inflammatory protein-2
- MMPs
Matrix metalloproteinases
- MRC
mitochondrial respiratory chain
- NAFLD
Non-alcoholic fatty liver disease
- NASH
Non-alcoholic steatohepatitis
- NF-κB
Nuclear factor-kappaB
- NK
Natural killer
- NLRP3
NACHT, LRR and PYD domains-containing protein 3
- NO
Nitric oxide
- NOD
Nucleotide-binding oligomerization domain
- NOS
Nitric oxide synthase
- NOX1
NADPH oxidase1
- PAMPs
Pathogen-associated molecular patterns
- PDGFR-β
Platelet derived growth factor receptor-beta
- PI3K
Phosphatidyl inositol-3 kinase
- PNPLA3
Patatin-like phospholipase domain-containing 3
- PS
Phosphatidylserine
- RNS
Reactive nitrogen species
- ROS
Reactive oxygen species
- Shh
Sonic hedgehog
- Smo
Smoothened
- SREBP-1c
Sterol regulatory element binding protein-1c
- T2D
Type 2 diabetes
- TGF-β
Transforming growth factor-beta
- TIMPs
Tissue inhibitor of metalloproteinases
- TLRs
Toll-like receptors
- TNFα
Tumor necrosis factor – alpha
- TRPM7
Transient receptor potential melastatin7
- ULBP2
UL16 binding protein 2
- UPR
Unfolded protein response
- VCAM1
Vascular cell adhesion molecule 1
- VEGF
Vascular endothelial growth factor
- VLDL
Very low density lipoprotein
- Wnt/β-catenin/Wnt signalling
Wingless and Int-1 signalling
Footnotes
This review is part of the Advanced Drug Delivery Reviews theme issue on “Fibrosis & Drug Delivery”.
References
- [1].Targher G, Byrne CD, Clinical Review: Nonalcoholic fatty liver disease: a novel cardiometabolic risk factor for type 2 diabetes and its complications, J. Clin. Endocrinol. Metab. 98 (2013) 483–495. [DOI] [PubMed] [Google Scholar]
- [2].Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, George J, Bugianesi E, Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention, Nat. Rev. Gastroenterol. Hepatol. 15 (2018) 11–20. [DOI] [PubMed] [Google Scholar]
- [3].Hazlehurst JM, Woods C, Marjot T, Cobbold JF, Tomlinson JW, Non-alcoholic fatty liver disease and diabetes, Metabolism 65 (2016) 1096–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Day CP, From fat to inflammation, Gastroenterology 130 (2006) 207–210. [DOI] [PubMed] [Google Scholar]
- [5].Su Q, Kumar V, Sud N, Mahato RI, MicroRNAs in the pathogenesis and treatment of progressive liver injury in NAFLD and liver fibrosis, Adv. Drug Deliv. Rev. 129 (2018) 54–63. [DOI] [PubMed] [Google Scholar]
- [6].Jou J, Choi S, Diehl A, Mechanisms of disease progression in nonalcoholic fatty liver disease, Semin. Liver Dis. 28 (2008) 370–379. [DOI] [PubMed] [Google Scholar]
- [7].Buzzetti E, Pinzani M, Tsochatzis EA, The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD), Metabolism 65 (2016) 1038–1048. [DOI] [PubMed] [Google Scholar]
- [8].Eslam M, Sanyal AJ, George J, International Consensus P MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease, Gastroenterology 158 (1999–2014) (2020) e1. [DOI] [PubMed] [Google Scholar]
- [9].Eslam M, Newsome PN, Sarin SK, Anstee QM, Targher G, Romero-Gomez M, Zelber-Sagi S, Wai-Sun Wong V, Dufour JF, Schattenberg JM, Kawaguchi T, Arrese M, Valenti L, Shiha G, Tiribelli C, Yki-Jarvinen H, Fan JG, Gronbaek H, Yilmaz Y, Cortez-Pinto H, Oliveira CP, Bedossa P, Adams LA, Zheng MH, Fouad Y, Chan WK, Mendez-Sanchez N, Ahn SH, Castera L, Bugianesi E, Ratziu V, George J, A new definition for metabolic dysfunction-associated fatty liver disease: an international expert consensus statement, J. Hepatol. 73 (2020) 202–209. [DOI] [PubMed] [Google Scholar]
- [10].Fouad Y, Waked I, Bollipo S, Gomaa A, Ajlouni Y, Attia D, What’s in a name? Renaming ’NAFLD’ to ’MAFLD’, Liver Int. 40 (2020) 1254–1261. [DOI] [PubMed] [Google Scholar]
- [11].Bertolani C, Marra F, The role of adipokines in liver fibrosis, Pathophysiology 15 (2008) 91–101. [DOI] [PubMed] [Google Scholar]
- [12].Brunt EM, Kleiner DE, Carpenter DH, Rinella M, Harrison SA, Loomba R, Younossi Z, Neuschwander-Tetri BA, Sanyal AJ, American Association for the Study of Liver Diseases NTF, NAFLD: reporting histologic findings in clinical practice, Hepatology 73 (2021) 2028–2038. [DOI] [PubMed] [Google Scholar]
- [13].Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ, Mechanisms of NAFLD development and therapeutic strategies, Nat. Med. 24 (2018) 908–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tapper EB and Parikh ND. Mortality due to cirrhosis and liver cancer in the United States, 1999–2016: observational study. Bmj. 2018:k2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Younossi ZM, Golabi P, de Avila L, Paik JM, Srishord M, Fukui N, Qiu Y, Burns L, Afendy A, Nader F, The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: a systematic review and meta-analysis, J. Hepatol. 71 (2019) 793–801. [DOI] [PubMed] [Google Scholar]
- [16].Tilg H, Moschen AR, Roden M, NAFLD and diabetes mellitus, Nat. Rev. Gastroenterol. Hepatol. 14 (2017) 32–42. [DOI] [PubMed] [Google Scholar]
- [17].Trombetta M, Spiazzi G, Zoppini G, Muggeo M, Review article: type 2 diabetes and chronic liver disease in the Verona diabetes study, Aliment. Pharmacol. Ther. 22 (Suppl. 2) (2005) 24–27. [DOI] [PubMed] [Google Scholar]
- [18].Jelenik T, Kaul K, Séquaris G, Flögel U, Phielix E, Kotzka J, Knebel B, Fahlbusch P, Hörbelt T, Lehr S, Reinbeck AL, Müller-Wieland D, Esposito I, Shulman GI, Szendroedi J, Roden M, Mechanisms of insulin resistance in primary and secondary nonalcoholic fatty liver, Diabetes 66 (2017) 2241–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Satapati S, Kucejova B, Duarte JAG, Fletcher JA, Reynolds L, Sunny NE, He T, Nair LA, Livingston KA, Fu X, Merritt ME, Sherry AD, Malloy CR, Shelton JM, Lambert J, Parks EJ, Corbin I, Magnuson MA, Browning JD, Burgess SC, Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver, J. Clin. Investig. 126 (2016) 1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Musso G, Cassader M, Paschetta E, Gambino R, Bioactive lipid species and metabolic pathways in progression and resolution of nonalcoholic steatohepatitis, Gastroenterology 155 (2018) 282–302.e8. [DOI] [PubMed] [Google Scholar]
- [21].Gaggini M, Carli F, Rosso C, Buzzigoli E, Marietti M, Della Latta V, Ciociaro D, Abate ML, Gambino R, Cassader M, Bugianesi E, Gastaldelli A, Altered amino acid concentrations in NAFLD: Impact of obesity and insulin resistance, Hepatology (Baltimore, MD) 67 (2018) 145–158. [DOI] [PubMed] [Google Scholar]
- [22].Rosso C, Mezzabotta L, Gaggini M, Salomone F, Gambino R, Marengo A, Saba F, Vanni E, Younes R, Saponaro C, Buzzigoli E, Caviglia GP, Abate ML, Smedile A, Rizzetto M, Cassader M, Gastaldelli A, Bugianesi E, Peripheral insulin resistance predicts liver damage in nondiabetic subjects with nonalcoholic fatty liver disease, Hepatology (Baltimore, MD) 63 (2016) 107–116. [DOI] [PubMed] [Google Scholar]
- [23].Ang CH, Hsu SH, Guo F, Tan CT, Yu VC, Visvader JE, Chow PKH, Fu NY, Lgr5+ pericentral hepatocytes are self-maintained in normal liver regeneration and susceptible to hepatocarcinogenesis, PNAS 116 (2019) 19530–19540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chen F, Jimenez RJ, Sharma K, Luu HY, Hsu BY, Ravindranathan A, Stohr BA, Willenbring H, Broad distribution of hepatocyte proliferation in liver homeostasis and regeneration, Cell Stem Cell 26 (2020) 27–33.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Knook DL, Sleyster EC, Isolated parenchymal, Kupffer and endothelial rat liver cells characterized by their lysosomal enzyme content, Biochem. Biophys. Res. Commun. 96 (1980) 250–257. [DOI] [PubMed] [Google Scholar]
- [26].Blouin A, Bolender RP, Weibel ER, Distribution of organelles and membranes between hepatocytes and nonhepatocytes in the rat liver parenchyma. A stereological study, J. Cell Biol. 72 (1977) 441–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Poisson J, Lemoinne S, Boulanger C, Durand F, Moreau R, Valla D, Rautou PE, Liver sinusoidal endothelial cells: Physiology and role in liver diseases, J. Hepatol. 66 (2017) 212–227. [DOI] [PubMed] [Google Scholar]
- [28].Marrone G, Shah VH, Gracia-Sancho J, Sinusoidal communication in liver fibrosis and regeneration, J. Hepatol. 65 (2016) 608–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Khomich O, Ivanov AV, Bartosch B, Metabolic hallmarks of hepatic stellate cells in liver fibrosis, Cells 9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Dixon LJ, Barnes M, Tang H, Pritchard MT, Nagy LE, Kupffer cells in the liver, Compr Physiol 3 (2013) 785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Heymann F, Tacke F, Immunology in the liver–from homeostasis to disease, Nat. Rev. Gastroenterol. Hepatol. 13 (2016) 88–110. [DOI] [PubMed] [Google Scholar]
- [32].Ye L, Cao Z, Lai X, Shi Y, Zhou N, Niacin Ameliorates Hepatic Steatosis by Inhibiting De Novo Lipogenesis Via a GPR109A-Mediated PKC-ERK1/2-AMPK Signaling Pathway in C57BL/6 Mice Fed a High-Fat Diet, J. Nutr. 150 (2020) 672–684. [DOI] [PubMed] [Google Scholar]
- [33].Su Q, Baker C, Christian P, Naples M, Tong X, Zhang K, Santha M, Adeli K, Hepatic mitochondrial and ER stress induced by defective PPARalpha signaling in the pathogenesis of hepatic steatosis, Am. J. Physiol. Endocrinol. Metab. 306 (2014) E1264–E1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yamaguchi K, Yang L, McCall S, Huang J, Yu XX, Pandey SK, Bhanot S, Monia BP, Li Y-X, Diehl AM, Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis, Hepatology 45 (2007) 1366–1374. [DOI] [PubMed] [Google Scholar]
- [35].Machado MV, Diehl AM, Pathogenesis of Nonalcoholic Steatohepatitis, Gastroenterology 150 (2016) 1769–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Filozof C, Goldstein BJ, Williams RN, Sanyal A, Non-alcoholic steatohepatitis: limited available treatment options but promising drugs in development and recent progress towards a regulatory approval pathway, Drugs 75 (2015) 1373–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Begriche K, Massart J, Robin M-A, Bonnet F, Fromenty B, Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease, Hepatology 58 (2013) 1497–1507. [DOI] [PubMed] [Google Scholar]
- [38].Gruben N, Shiri-Sverdlov R, Koonen DPY, Hofker MH, Nonalcoholic fatty liver disease: A main driver of insulin resistance or a dangerous liaison?, Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1842 (2014) 2329–2343 [DOI] [PubMed] [Google Scholar]
- [39].Miao H, Zhang Y, Lu Z, Liu Q, Gan L, FOXO1 involvement in insulin resistance-related pro-inflammatory cytokine production in hepatocytes, Inflamm. Res. 61 (2012) 349–358. [DOI] [PubMed] [Google Scholar]
- [40].Liu Q, Rehman H, Krishnasamy Y, Ramshesh VK, Theruvath TP, Chavin KD, Schnellmann RG, Lemasters JJ, Zhong Z, Role of inducible nitric oxide synthase in mitochondrial depolarization and graft injury after transplantation of fatty livers, Free Radical Biol. Med. 53 (2012) 250–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Cusi K, Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: pathophysiology and clinical implications, Gastroenterology 142 (2012) 711–725.e6. [DOI] [PubMed] [Google Scholar]
- [42].Ashraf NU, Sheikh TA, Endoplasmic reticulum stress and Oxidative stress in the pathogenesis of Non-alcoholic fatty liver disease, Free Radical Res. 49 (2015) 1405–1418. [DOI] [PubMed] [Google Scholar]
- [43].Amir M, Czaja MJ, Autophagy in nonalcoholic steatohepatitis, Expert Rev. Gastroenterol. Hepatol. 5 (2014) 159–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Canivet CM, Bonnafous S, Rousseau D, Leclere PS, Lacas-Gervais S, Patouraux S, Sans A, Luci C, Bailly-Maitre B, Iannelli A, Tran A, Anty R, Gual P, Hepatic FNDC5 is a potential local protective factor against Non-Alcoholic Fatty Liver, Biochim. Biophys. Acta, Mol. Basis Dis. 1866 (2020) 165705. [DOI] [PubMed] [Google Scholar]
- [45].Ferreira DMS, Castro RE, Machado MV, Evangelista T, Silvestre A, Costa A, Coutinho J, Carepa F, Cortez-Pinto H, Rodrigues CMP, Apoptosis and insulin resistance in liver and peripheral tissues of morbidly obese patients is associated with different stages of non-alcoholic fatty liver disease, Diabetologia 54 (2011) 1788–1798. [DOI] [PubMed] [Google Scholar]
- [46].Zhang P, Wang P-X, Zhao L-P, Zhang X, Ji Y-X, Zhang X-J, Fang C, Lu Y-X, Yang X, Gao M-M, Zhang Y, Tian S, Zhu X-Y, Gong J, Ma X-L, Li F, Wang Z, Huang Z, She Z-G, Li H, The deubiquitinating enzyme TNFAIP3 mediates inactivation of hepatic ASK1 and ameliorates nonalcoholic steatohepatitis, Nat. Med. 24 (2018) 84–94. [DOI] [PubMed] [Google Scholar]
- [47].Ibrahim SH, Hirsova P, Gores GJ, Non-alcoholic steatohepatitis pathogenesis: sublethal hepatocyte injury as a driver of liver inflammation, Gut 67 (2018) 963–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Machado M, Angulo P, Diehl A, Fibrosis in nonalcoholic fatty liver disease: mechanisms and clinical implications, Semin. Liver Dis. 35 (2015) 132–145. [DOI] [PubMed] [Google Scholar]
- [49].Holmström KM, Finkel T, Cellular mechanisms and physiological consequences of redox-dependent signalling, Nat. Rev. Mol. Cell Biol. 15 (2014) 411–421. [DOI] [PubMed] [Google Scholar]
- [50].Kotiadis VN, Duchen MR, Osellame LD, Mitochondrial quality control and communications with the nucleus are important in maintaining mitochondrial function and cell health, Biochim. Biophys. Acta, Lipids Lipid Metab. 1840 (2014) 1254–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, Jankowiak F, Herder C, Carstensen M, Krausch M, Knoefel WT, Schlensak M, Roden M, Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis, Cell Metab. 21 (2015) 739–746. [DOI] [PubMed] [Google Scholar]
- [52].Mansouri A, Gattolliat C-H, Asselah T, Mitochondrial dysfunction and signaling in chronic liver diseases, Gastroenterology 155 (2018) 629–647. [DOI] [PubMed] [Google Scholar]
- [53].Boland ML, Oldham S, Boland BB, Will S, Lapointe J-M, Guionaud S, Rhodes CJ, Trevaskis JL, Nonalcoholic steatohepatitis severity is defined by a failure in compensatory antioxidant capacity in the setting of mitochondrial dysfunction, World J. Gastroenterol. 24 (2018) 1748–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Lotowska JM, Sobaniec-Lotowska ME, Bockowska SB, Lebensztejn DM, Pediatric non-alcoholic steatohepatitis: the first report on the ultrastructure of hepatocyte mitochondria, World J. Gastroenterol. 20 (2014) 4335–4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kleiner DE, Makhlouf HR, Histology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis in adults and children, Clin. Liver Dis. 20 (2016) 293–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Yamada T, Murata D, Adachi Y, Itoh K, Kameoka S, Igarashi A, Kato T, Araki Y, Huganir RL, Dawson TM, Yanagawa T, Okamoto K, Iijima M, Sesaki H, Mitochondrial stasis reveals p62-mediated ubiquitination in parkin-independent mitophagy and mitigates nonalcoholic fatty liver disease, Cell Metab. 28 (2018) 588–604.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Win S, Than TA, Le BHA, García-Ruiz C, Fernandez-Checa JC, Kaplowitz N, Sab (Sh3bp5) dependence of JNK mediated inhibition of mitochondrial respiration in palmitic acid induced hepatocyte lipotoxicity, J. Hepatol. 62 (2015) 1367–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Baker PR, Friedman JE, Mitochondrial role in the neonatal predisposition to developing nonalcoholic fatty liver disease, J. Clin. Investig. 128 (2018) 3692–3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Schuster S, Cabrera D, Arrese M, Feldstein AE, Triggering and resolution of inflammation in NASH, Nat. Rev. Gastroenterol. Hepatol. 15 (2018) 349–364. [DOI] [PubMed] [Google Scholar]
- [60].Marques PE, Oliveira AG, Pereira RV, David BA, Gomides LF, Saraiva AM, Pires DA, Novaes JT, Patricio DO, Cisalpino D, Menezes-Garcia Z, Leevy WM, Chapman SE, Mahecha G, Marques RE, Guabiraba R, Martins VP, Souza DG, Mansur DS, Teixeira MM, Leite MF, Menezes GB, Hepatic DNA deposition drives drug-induced liver injury and inflammation in mice, Hepatology (Baltimore, MD) 61 (2015) 348–360. [DOI] [PubMed] [Google Scholar]
- [61].Ramos-Tovar E, Muriel P, Free radicals, antioxidants, nuclear factor-E2-related factor-2 and liver damage, J. Appl. Toxicol. 40 (2020) 151–168. [DOI] [PubMed] [Google Scholar]
- [62].Rochette L, Lorin J, Zeller M, Guilland J-C, Lorgis L, Cottin Y, Vergely C, Nitric oxide synthase inhibition and oxidative stress in cardiovascular diseases: Possible therapeutic targets?, Pharmacol Ther. 140 (2013) 239–257. [DOI] [PubMed] [Google Scholar]
- [63].Yang H, Zhang WJ, Wu LQ, Gu F, Ye LY, Li J, Xu SQ, Xu YP and Lou JN, Protection of liver sinusoidal endothelial cells from hypoxia-reoxygenation induced apoptosis by alpha-1 antitrypsin in vitro, Zhonghua Yi Xue Za Zhi 85 (2005) 106–110. [PubMed] [Google Scholar]
- [64].Carnovale CE, Ronco MT, Role of nitric oxide in liver regeneration, Ann. Hepatol. 11 (2012) 636–647. [PubMed] [Google Scholar]
- [65].Guo Z, Shao L, Zheng L, Du Q, Li P, John B, Geller DA, miRNA-939 regulates human inducible nitric oxide synthase posttranscriptional gene expression in human hepatocytes, PNAS 109 (2012) 5826–5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Tsuchida T, Friedman SL, Mechanisms of hepatic stellate cell activation, Nat. Rev. Gastroenterol. Hepatol. 14 (2017) 397–411. [DOI] [PubMed] [Google Scholar]
- [67].Sutti S, Jindal A, Locatelli I, Vacchiano M, Gigliotti L, Bozzola C, Albano E, Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH, Hepatology (Baltimore, MD) 59 (2014) 886–897. [DOI] [PubMed] [Google Scholar]
- [68].Beyazit Y, Efe C, Tanoglu A, Purnak T, Sayilir A, Taskıran I, Kekilli M, Turhan T, Ozaslan E, Wahlin S, Nitric oxide is a potential mediator of hepatic inflammation and fibrogenesis in autoimmune hepatitis, Scand. J. Gastroenterol. 50 (2015) 204–210. [DOI] [PubMed] [Google Scholar]
- [69].Ramos-Tovar E, Muriel P, Molecular mechanisms that link oxidative stress, inflammation, and fibrosis in the liver, Antioxidants (Basel) 9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Torok NJ, Dysregulation of redox pathways in liver fibrosis, Am. J. Physiol. Gastrointest. Liver Physiol. 311 (2016) G667–G674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Simon-Santamaria J, Malovic I, Warren A, Oteiza A, Le Couteur D, Smedsrod B, McCourt P, Sorensen KK, Age-related changes in scavenger receptor-mediated endocytosis in rat liver sinusoidal endothelial cells, J. Gerontol. A Biol. Sci. Med. Sci. 65 (2010) 951–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Blouin A, Bolender RP, Weibel ER, Distribution of organelles and membranes between hepatocytes and nonhepatocytes in the rat liver parenchyma. A stereological study, J. Cell Biol. 72 (1977) 441–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Pandey E, Nour AS, Harris EN, Prominent Receptors of Liver Sinusoidal Endothelial Cells in Liver Homeostasis and Disease, Front. Physiol. 11 (2020) 873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Xie G, Wang X, Wang L, Atkinson RD, Kanel GC, Gaarde WA, Deleve LD, Role of differentiation of liver sinusoidal endothelial cells in progression and regression of hepatic fibrosis in rats, Gastroenterology 142 (918–927) (2012) e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Miyao M, Kotani H, Ishida T, Kawai C, Manabe S, Abiru H, Tamaki K, Pivotal role of liver sinusoidal endothelial cells in NAFLD/NASH progression, Lab. Invest. 95 (2015) 1130–1144. [DOI] [PubMed] [Google Scholar]
- [76].Tanaka M, Iwakiri Y, The hepatic lymphatic vascular system: structure, function, markers, and lymphangiogenesis, Cell Mol. Gastroenterol. Hepatol. 2 (2016) 733–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Herrnberger L, Hennig R, Kremer W, Hellerbrand C, Goepferich A, Kalbitzer HR, Tamm ER, Formation of fenestrae in murine liver sinusoids depends on plasmalemma vesicle-associated protein and is required for lipoprotein passage, PLoS ONE 9 (2014) e115005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Kus E, Kaczara P, Czyzynska-Cichon I, Szafranska K, Zapotoczny B, Kij A, Sowinska A, Kotlinowski J, Mateuszuk L, Czarnowska E, Szymonski M, Chlopicki S, LSEC fenestrae are preserved despite pro-inflammatory phenotype of liver sinusoidal endothelial cells in mice on high fat diet, Front. Physiol. 10 (2019) 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Matsumoto M, Zhang J, Zhang X, Liu J, Jiang JX, Yamaguchi K, Taruno A, Katsuyama M, Iwata K, Ibi M, Cui W, Matsuno K, Marunaka Y, Itoh Y, Torok NJ, Yabe-Nishimura C, The NOX1 isoform of NADPH oxidase is involved in dysfunction of liver sinusoids in nonalcoholic fatty liver disease, Free Radic. Biol. Med. 115 (2018) 412–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Rutkowski DT, Wu J, Back S-H, Callaghan MU, Ferris SP, Iqbal J, Clark R, Miao H, Hassler JR, Fornek J, Katze MG, Hussain MM, Song B, Swathirajan J, Wang J, Yau GDY, Kaufman RJ, UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators, Dev. Cell 15 (2008) 829–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Su Q, Tsai J, Xu E, Qiu W, Bereczki E, Santha M, Adeli K, Apolipoprotein B100 acts as a molecular link between lipid-induced endoplasmic reticulum stress and hepatic insulin resistance, Hepatology 50 (2009) 77–84. [DOI] [PubMed] [Google Scholar]
- [82].Su Q, Rutledge AC, Dekker M, Adeli K, Apolipoprotein B: not just a biomarker but a causal factor in hepatic endoplasmic reticulum stress and insulin resistance, Clinical Lipidology. 5 (2010) 267–276. [Google Scholar]
- [83].Halbleib K, Pesek K, Covino R, Hofbauer HF, Wunnicke D, Hänelt I, Hummer G, Ernst R, Activation of the unfolded protein response by lipid bilayer stress, Mol. Cell 67 (2017) 673–684.e8. [DOI] [PubMed] [Google Scholar]
- [84].Sepulveda D, Rojas-Rivera D, Rodríguez DA, Groenendyk J, Köhler A, Lebeaupin C, Ito S, Urra H, Carreras-Sureda A, Hazari Y, Vasseur-Cognet M, Ali MMU, Chevet E, Campos G, Godoy P, Vaisar T, Bailly-Maitre B, Nagata K, Michalak M, Sierralta J, Hetz C, Interactome screening identifies the ER luminal chaperone Hsp47 as a regulator of the unfolded protein response transducer IRE1α, Mol. Cell 69 (2018) 238–252.e7. [DOI] [PubMed] [Google Scholar]
- [85].Zeng X, Zhu M, Liu X, Chen X, Yuan Y, Li L, Liu J, Lu Y, Cheng J, Chen Y, Oleic acid ameliorates palmitic acid induced hepatocellular lipotoxicity by inhibition of ER stress and pyroptosis, Nutr. Metab. (Lond). 17 (2020) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Koo JH, Lee HJ, Kim W, Kim SG, Endoplasmic reticulum stress in hepatic stellate cells promotes liver fibrosis via PERK-mediated degradation of HNRNPA1 and up-regulation of SMAD2, Gastroenterology 150 (2016) 181–193.e8. [DOI] [PubMed] [Google Scholar]
- [87].Cai J, Zhang X-J, Li H, The role of innate immune cells in nonalcoholic steatohepatitis, Hepatology (Baltimore, MD) 70 (2019) 1026–1037. [DOI] [PubMed] [Google Scholar]
- [88].Zhao G-N, Zhang P, Gong J, Zhang X-J, Wang P-X, Yin M, Jiang Z, Shen L-J, Ji Y-X, Tong J, Wang Y, Wei Q-F, Wang Y, Zhu X-Y, Zhang X, Fang J, Xie Q, She Z-G, Wang Z, Huang Z, Li H, Tmbim1 is a multivesicular body regulator that protects against non-alcoholic fatty liver disease in mice and monkeys by targeting the lysosomal degradation of Tlr4, Nat. Med. 23 (2017) 742–752. [DOI] [PubMed] [Google Scholar]
- [89].Garcia-Martinez I, Santoro N, Chen Y, Hoque R, Ouyang X, Caprio S, Shlomchik MJ, Coffman RL, Candia A, Mehal WZ, Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9, J. Clin. Invest. 126 (2016) 859–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Etienne-Mesmin L, Vijay-Kumar M, Gewirtz AT, Chassaing B, Hepatocyte toll-like receptor 5 promotes bacterial clearance and protects mice against high-fat diet-induced liver disease, Cell. Molecular Gastroenterol. Hepatol. 2 (2016) 584–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Gautheron J, Gores GJ, Rodrigues CMP, Lytic cell death in metabolic liver disease, J. Hepatol. 73 (2020) 394–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Eguchi A, Wree A, Feldstein AE, Biomarkers of liver cell death, J. Hepatol. 60 (2014) 1063–1074. [DOI] [PubMed] [Google Scholar]
- [93].Aravinthan A, Scarpini C, Tachtatzis P, Verma S, Penrhyn-Lowe S, Harvey R, Davies SE, Allison M, Coleman N, Alexander G, Hepatocyte senescence predicts progression in non-alcohol-related fatty liver disease, J. Hepatol. 58 (2013) 549–556. [DOI] [PubMed] [Google Scholar]
- [94].Haubrich WS, Kupffer of Kupffer cells, Gastroenterology 127 (2004) 16. [DOI] [PubMed] [Google Scholar]
- [95].Hritz I, Mandrekar P, Velayudham A, Catalano D, Dolganiuc A, Kodys K, Kurt-Jones E, Szabo G, The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88, Hepatology 48 (2008) 1224–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Hunter MM, Wang A, Parhar KS, Johnston MJ, Van Rooijen N, Beck PL, McKay DM, In vitro-derived alternatively activated macrophages reduce colonic inflammation in mice, Gastroenterology 138 (2010) 1395–1405. [DOI] [PubMed] [Google Scholar]
- [97].Moore KJ, Tabas I, Macrophages in the pathogenesis of atherosclerosis, Cell 145 (2011) 341–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, Szabo G, Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells, Hepatology 54 (2011) 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Wan J, Benkdane M, Teixeira-Clerc F, Bonnafous S, Louvet A, Lafdil F, Pecker F, Tran A, Gual P, Mallat A, Lotersztajn S, Pavoine C, M2 Kupffer cells promote M1 Kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease, Hepatology 59 (2014) 130–142. [DOI] [PubMed] [Google Scholar]
- [100].Mridha AR, Haczeyni F, Yeh MM, Haigh WG, Ioannou GN, Barn V, Ajamieh H, Adams L, Hamdorf JM, Teoh NC, Farrell GC, TLR9 is up-regulated in human and murine NASH: pivotal role in inflammatory recruitment and cell survival, Clin. Sci. (Lond). 131 (2017) 2145–2159. [DOI] [PubMed] [Google Scholar]
- [101].Ioannou GN, The Role of Cholesterol in the Pathogenesis of NASH, Trends Endocrinol. Metab.: TEM 27 (2016) 84–95. [DOI] [PubMed] [Google Scholar]
- [102].Nakamoto N, Kanai T, Role of toll-like receptors in immune activation and tolerance in the liver, Front. Immunol. 5 (2014) 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Kiziltas S, Toll-like receptors in pathophysiology of liver diseases, World J. Hepatol. 8 (2016) 1354–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Papatheodoridi A-M, Chrysavgis L, Koutsilieris M, Chatzigeorgiou A, The role of senescence in the development of nonalcoholic fatty liver disease and progression to nonalcoholic steatohepatitis, Hepatology (Baltimore, MD) 71 (2020) 363–374. [DOI] [PubMed] [Google Scholar]
- [105].Massague J, Wotton D, Transcriptional control by the TGF-beta/Smad signaling system, EMBO J. 19 (2000) 1745–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Fabregat I, Moreno-Càceres J, Sánchez A, Dooley S, Dewidar B, Giannelli G, Ten Dijke P, Consortium IL, TGF-β signalling and liver disease, The FEBS journal. 283 (2016) 2219–2232. [DOI] [PubMed] [Google Scholar]
- [107].Dewidar B, Meyer C, Dooley S, Meindl B, Nadja, TGF-β in hepatic stellate cell activation and liver fibrogenesis-updated 2019, Cells 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Ungefroren H, Sebens S, Groth S, Gieseler F, Fandrich F, The Src family kinase inhibitors PP2 and PP1 block TGF-beta1-mediated cellular responses by direct and differential inhibition of type I and type II TGF-beta receptors, Curr. Cancer Drug Targets 11 (2011) 524–535. [DOI] [PubMed] [Google Scholar]
- [109].Derynck R, Zhang YE, Smad-dependent and Smad-independent pathways in TGF-beta family signalling, Nature 425 (2003) 577–584. [DOI] [PubMed] [Google Scholar]
- [110].Zhang L, Liu C, Meng XM, Huang C, Xu F, Li J, Smad2 protects against TGF-beta1/Smad3-mediated collagen synthesis in human hepatic stellate cells during hepatic fibrosis, Mol. Cell. Biochem. 400 (2015) 17–28. [DOI] [PubMed] [Google Scholar]
- [111].Dahl M, Maturi V, Lonn P, Papoutsoglou P, Zieba A, Vanlandewijck M, van der Heide LP, Watanabe Y, Soderberg O, Hottiger MO, Heldin CH, Moustakas A, Fine-tuning of Smad protein function by poly(ADP-ribose) polymerases and poly(ADP-ribose) glycohydrolase during transforming growth factor beta signaling, PLoS ONE 9 (2014) e103651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Hinz B, The extracellular matrix and transforming growth factor-beta1: Tale of a strained relationship, Matrix Biol. 47 (2015) 54–65. [DOI] [PubMed] [Google Scholar]
- [113].Rivera CA, Bradford BU, Hunt KJ, Adachi Y, Schrum LW, Koop DR, Burchardt ER, Rippe RA, Thurman RG, Attenuation of CCl(4)-induced hepatic fibrosis by GdCl(3) treatment or dietary glycine, Am. J. Physiol. Gastrointest. Liver Physiol. 281 (2001) G200–G207. [DOI] [PubMed] [Google Scholar]
- [114].Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA, Liver fibrosis and repair: immune regulation of wound healing in a solid organ, Nat. Rev. Immunol. 14 (2014) 181–194. [DOI] [PubMed] [Google Scholar]
- [115].Li Y, Liu R, Wu J, Li X, Self-eating: friend or foe? The emerging role of autophagy in fibrotic diseases, Theranostics. 10 (2020) 7993–8017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Higashi T, Friedman SL, Hoshida Y, Hepatic stellate cells as key target in liver fibrosis, Adv. Drug Deliv. Rev. 121 (2017) 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Li L, Wang J-Y, Yang C-Q, Jiang W, Effect of RhoA on transforming growth factor β1-induced rat hepatic stellate cell migration, Liver International: Official Journal of the International Association for the Study of the Liver. 32 (2012) 1093–1102. [DOI] [PubMed] [Google Scholar]
- [118].Trautwein C, Friedman SL, Schuppan D, Pinzani M, Hepatic fibrosis: concept to treatment, J. Hepatol. 62 (2015) S15–S24. [DOI] [PubMed] [Google Scholar]
- [119].Breitkopf-Heinlein K, Meyer C, Konig C, Gaitantzi H, Addante A, Thomas M, Wiercinska E, Cai C, Li Q, Wan F, Hellerbrand C, Valous NA, Hahnel M, Ehlting C, Bode JG, Muller-Bohl S, Klingmuller U, Altenoder J, Ilkavets I, Goumans MJ, Hawinkels LJ, Lee SJ, Wieland M, Mogler C, Ebert MP, Herrera B, Augustin H, Sanchez A, Dooley S, Ten Dijke P, BMP-9 interferes with liver regeneration and promotes liver fibrosis, Gut 66 (2017) 939–954. [DOI] [PubMed] [Google Scholar]
- [120].Luo K, Signaling cross talk between TGF-beta/Smad and other signaling pathways, Cold Spring Harb Perspect Biol. 9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Li P, Li Y, Zhu L, Yang Z, He J, Wang L, Shang Q, Pan H, Wang H, Ma X, Li B, Fan X, Ge S, Jia R, Zhang H, Targeting secreted cytokine BMP9 gates the attenuation of hepatic fibrosis, Biochim. Biophys. Acta, Mol. Basis Dis. 1864 (2018) 709–720. [DOI] [PubMed] [Google Scholar]
- [122].Bourdeau A, Faughnan ME, Letarte M, Endoglin-deficient mice, a unique model to study hereditary hemorrhagic telangiectasia, Trends Cardiovasc. Med. 10 (2000) 279–285. [DOI] [PubMed] [Google Scholar]
- [123].Geraud C, Schledzewski K, Demory A, Klein D, Kaus M, Peyre F, Sticht C, Evdokimov K, Lu S, Schmieder A, Goerdt S, Liver sinusoidal endothelium: a microenvironment-dependent differentiation program in rat including the novel junctional protein liver endothelial differentiation-associated protein-1, Hepatology 52 (2010) 313–326. [DOI] [PubMed] [Google Scholar]
- [124].Desroches-Castan A, Tillet E, Ricard N, Ouarne M, Mallet C, Belmudes L, Coute Y, Boillot O, Scoazec JY, Bailly S, Feige JJ, Bone morphogenetic protein 9 is a paracrine factor controlling liver sinusoidal endothelial cell fenestration and protecting against hepatic fibrosis, Hepatology 70 (2019) 1392–1408. [DOI] [PubMed] [Google Scholar]
- [125].Wang Y, Ma C, Sun T, Ren L, Potential roles of bone morphogenetic protein-9 in glucose and lipid homeostasis, J. Physiol. Biochem. (2020). [DOI] [PubMed] [Google Scholar]
- [126].John M, Kim KJ, Bae SDW, Qiao L, George J, Role of BMP-9 in human liver disease, Gut 68 (2019) 2097–2100. [DOI] [PubMed] [Google Scholar]
- [127].Desroches-Castan A, Tillet E, Ricard N, Ouarne M, Mallet C, Feige JJ, Bailly S, Differential consequences of bmp9 deletion on sinusoidal endothelial cell differentiation and liver fibrosis in 129/Ola and C57BL/6 Mice, Cells 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Briscoe J, Thérond PP, The mechanisms of Hedgehog signalling and its roles in development and disease, Nat. Rev. Mol. Cell Bio. 14 (2013) 416–429. [DOI] [PubMed] [Google Scholar]
- [129].Machado MV, Diehl AM, Hedgehog signalling in liver pathophysiology, J. Hepatol. 68 (2018) 550–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Swiderska-Syn M, Syn WK, Xie G, Krüger L, Machado MV, Karaca G, Michelotti GA, Choi SS, Premont RT, Diehl AM, Myofibroblastic cells function as progenitors to regenerate murine livers after partial hepatectomy, Gut 63 (2014) 1333–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Rangwala F, Guy CD, Lu J, Suzuki A, Burchette JL, Abdelmalek MF, Chen W, Diehl AM, Increased production of sonic hedgehog by ballooned hepatocytes, J. Pathol. 224 (2011) 401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Machado MV, Michelotti GA, Pereira Td.A., Boursier J, Kruger L, Swiderska-Syn M, Karaca G, Xie G, Guy CD, Bohinc B, Lindblom KR, Johnson E, Kornbluth S, Diehl AM, Reduced lipoapoptosis, hedgehog pathway activation and fibrosis in caspase-2 deficient mice with non-alcoholic steatohepatitis, Gut 64 (2015) 1148–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Guy CD, Suzuki A, Zdanowicz M, Abdelmalek MF, Burchette J, Unalp A, Diehl AM, Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease, Hepatology 55 (2012) 1711–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Nojima H, Freeman CM, Schuster RM, Japtok L, Kleuser B, Edwards MJ, Gulbins E, Lentsch AB, Hepatocyte exosomes mediate liver repair and regeneration via sphingosine-1-phosphate, J. Hepatol. 64 (2016) 60–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Briscoe J, Palm W, Swierczynska MM, Kumari V, Ehrhart-Bornstein M, Bornstein SR, Eaton S, Secretion and Signaling Activities of Lipoprotein-Associated Hedgehog and Non-Sterol-Modified Hedgehog in Flies and Mammals, PLoS Biol. 11 (2013) e1001505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Xie G, Karaca G, Swiderska-Syn M, Michelotti GA, Krüger L, Chen Y, Premont RT, Choi SS, Diehl AM, Cross-talk between Notch and Hedgehog regulates hepatic stellate cell fate in mice, Hepatology 58 (2013) 1801–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Xie G, Choi SS, Syn W-K, Michelotti GA, Swiderska M, Karaca G, Chan IS, Chen Y, Diehl AM, Hedgehog signalling regulates liver sinusoidal endothelial cell capillarisation, Gut 62 (2013) 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Pereira TA, Xie G, Choi SS, Syn W-K, Voieta I, Lu J, Chan IS, Swiderska M, Amaral KB, Antunes CM, Secor WE, Witek RP, Lambertucci JR, Pereira FL, Diehl AM, Macrophage-derived hedgehog ligands promotes fibrogenic and angiogenic responses in human schistosomiasis mansoni, Liver International. 33 (2013) 149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Machado MV, Michelotti GA, Pereira TA, Xie G, Premont R, Cortez-Pinto H, Diehl AM, Accumulation of duct cells with activated YAP parallels fibrosis progression in non-alcoholic fatty liver disease, J. Hepatol. 63 (2015) 962–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Moylan CA, Pang H, Dellinger A, Suzuki A, Garrett ME, Guy CD, Murphy SK, Ashley-Koch AE, Choi SS, Michelotti GA, Hampton DD, Chen Y, Tillmann HL, Hauser MA, Abdelmalek MF, Diehl AM, Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease, Hepatology 59 (2014) 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Zheng XIN, Zeng WEN, Gai X, Xu Q, Li C, Liang Z, Tuo H, Liu Q, Role of the Hedgehog pathway in hepatocellular carcinoma (Review), Oncol. Rep. 30 (2013) 2020–2026. [DOI] [PubMed] [Google Scholar]
- [142].Sundaram SS, Swiderska-Syn M, Sokol RJ, Halbower AC, Capocelli KE, Pan Z, Robbins K, Graham B, Diehl AM, Nocturnal hypoxia activation of the hedgehog signaling pathway affects pediatric nonalcoholic fatty liver disease severity, Hepatology Communications 3 (2019) 883–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Chong YC, Lim TE, Fu Y, Shin EM, Tergaonkar V, Han W, Indian Hedgehog links obesity to development of hepatocellular carcinoma, Oncogene 38 (2018) 2206–2222. [DOI] [PubMed] [Google Scholar]
- [144].Merchant JL, Saqui-Salces M, Inhibition of Hedgehog signaling in the gastrointestinal tract: targeting the cancer microenvironment, Cancer Treat. Rev. 40 (2014) 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Tuleta I, Frangogiannis NG, Diabetic fibrosis, Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1867 (2021) 166044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Yonezawa S, Koide H, Asai T, Recent advances in siRNA delivery mediated by lipid-based nanoparticles, Adv. Drug Deliv. Rev. (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Allen TM, Cullis PR, Liposomal drug delivery systems: from concept to clinical applications, Adv. Drug Deliv. Rev. 65 (2013) 36–48. [DOI] [PubMed] [Google Scholar]
- [148].Paunovska K, Da Silva Sanchez AJ, Sago CD, Gan Z, Lokugamage MP, Islam FZ, Kalathoor S, Krupczak BR, Dahlman JE, Nanoparticles Containing Oxidized Cholesterol Deliver mRNA to the Liver Microenvironment at Clinically Relevant Doses, Adv. Mater. 31 (2019) e1807748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Kauffman KJ, Dorkin JR, Yang JH, Heartlein MW, DeRosa F, Mir FF, Fenton OS, Anderson DG, Optimization of lipid nanoparticle formulations for mRNA delivery in vivo with fractional factorial and definitive screening designs, Nano Lett. 15 (2015) 7300–7306. [DOI] [PubMed] [Google Scholar]
- [150].Zhang J, Li R, Liu Q, Zhou J, Huang H, Huang Y, Zhang Z, Wu T, Tang Q, Huang C, Zhao Y, Zhang G, Mo L, Li Y, He J, SB431542-loaded liposomes alleviate liver fibrosis by suppressing TGF-beta signaling, Mol. Pharm. 17 (2020) 4152–4162. [DOI] [PubMed] [Google Scholar]
- [151].Liu X, Han M, Xu J, Geng S, Zhang Y, Ye X, Gou J, Yin T, He H, Tang X, Asialoglycoprotein receptor-targeted liposomes loaded with a norcantharimide derivative for hepatocyte-selective targeting, Int. J. Pharm. 520 (2017) 98–110. [DOI] [PubMed] [Google Scholar]
- [152].Mbatha L, Chakravorty S, de Koning CB, van Otterlo WA, Arbuthnot P, Ariatti M, Singh M, Spacer length: a determining factor in the design of galactosyl ligands for hepatoma cell-specific liposomal gene delivery, Curr. Drug Deliv. 13 (2016) 935–945. [DOI] [PubMed] [Google Scholar]
- [153].Toriyabe N, Hayashi Y, Hyodo M, Harashima H, Synthesis and evaluation of stearylated hyaluronic acid for the active delivery of liposomes to liver endothelial cells, Biol. Pharm. Bull. 34 (2011) 1084–1089. [DOI] [PubMed] [Google Scholar]
- [154].Campbell F, Bos FL, Sieber S, Arias-Alpizar G, Koch BE, Huwyler J, Kros A, Bussmann J, Directing nanoparticle biodistribution through evasion and exploitation of Stab2-dependent nanoparticle uptake, ACS Nano 12 (2018) 2138–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Hsu CY, Chen CH, Aljuffali IA, Dai YS, Fang JY, Nanovesicle delivery to the liver via retinol binding protein and platelet-derived growth factor receptors: how targeting ligands affect biodistribution, Nanomedicine (Lond). 12 (2017) 317–331. [DOI] [PubMed] [Google Scholar]
- [156].Park SY, Kim IS, Stabilin receptors: role as phosphatidylserine receptors, Biomolecules. 9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Elvevold K, Smedsrod B, Martinez I, The liver sinusoidal endothelial cell: a cell type of controversial and confusing identity, Am. J. Physiol. Gastrointest. Liver Physiol. 294 (2008) G391–G400. [DOI] [PubMed] [Google Scholar]
- [158].BasuRay S, Wang Y, Smagris E, Cohen JC, Hobbs HH, Accumulation of PNPLA3 on lipid droplets is the basis of associated hepatic steatosis, Proc. Natl. Acad. Sci. USA 116 (2019) 9521–9526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Linden D, Ahnmark A, Pingitore P, Ciociola E, Ahlstedt I, Andreasson AC, Sasidharan K, Madeyski-Bengtson K, Zurek M, Mancina RM, Lindblom A, Bjursell M, Bottcher G, Stahlman M, Bohlooly YM, Haynes WG, Carlsson B, Graham M, Lee R, Murray S, Valenti L, Bhanot S, Akerblad P, Romeo S, Pnpla3 silencing with antisense oligonucleotides ameliorates nonalcoholic steatohepatitis and fibrosis in Pnpla3 I148M knock-in mice, Mol Metab. 22 (2019) 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Ma Y, Belyaeva OV, Brown PM, Fujita K, Valles K, Karki S, de Boer YS, Koh C, Chen Y, Du X, Handelman SK, Chen V, Speliotes EK, Nestlerode C, Thomas E, Kleiner DE, Zmuda JM, Sanyal AJ, Kedishvili NY, Liang TJ, Rotman Y, 17-Beta hydroxysteroid dehydrogenase 13 is a hepatic retinol dehydrogenase associated with histological features of nonalcoholic fatty liver disease, Hepatology 69 (2019) 1504–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Dong XC, A closer look at the mysterious HSD17B13, J. Lipid Res. 61 (2020) 1361–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Tanowitz M, Hettrick L, Revenko A, Kinberger GA, Prakash TP, Seth PP, Asialoglycoprotein receptor 1 mediates productive uptake of N-acetylgalactosamine-conjugated and unconjugated phosphorothioate antisense oligonucleotides into liver hepatocytes, Nucleic Acids Res. 45 (2017) 12388–12400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Bottger J, Arnold K, Thiel C, Rennert C, Aleithe S, Hofmann U, Vlaic S, Sales S, Shevchenko A, Matz-Soja M, RNAi in murine hepatocytes: the agony of choice–a study of the influence of lipid-based transfection reagents on hepatocyte metabolism, Arch. Toxicol. 89 (2015) 1579–1588. [DOI] [PubMed] [Google Scholar]