

Abstract
Background
Bevacizumab (Avastin®) is a monoclonal antibody targeting the vascular endothelial growth factor (VEGF). Used alone or in combination with chemotherapy and/or immunotherapy, Avastin® has shown promising efficacy in many cancers. This study compared the efficacy and safety of TAB008 with Avastin® sourced from the EU (bevacizumab-EU), in patients with non-squamous non-small cell lung cancer (nsNSCLC).
Method
In this randomized, double-blind, multicenter, phase III similarity study, treatment naïve for metastatic lung cancer., EGFR wild-type, locally advanced, metastatic, or recurrent non-squamous, non-small cell, lung cancer (nsNSCLC) patients were enrolled and randomized (1:1) into TAB008 or Avastin® groups. Patients received TAB008 or Avastin® 15 mg/kg intravenously plus paclitaxel/carboplatin for 4–6 cycles followed by TAB008 or Avastin® 7.5 mg/kg until disease progression, unacceptable toxicity or death. The primary endpoint compared the objective response rate (ORR) within 6 cycles as read by an independent radiological review committee (IRRC). Secondary endpoints compared disease control rate (DCR) Within 6 cycles, duration of response (DoR), progression-free survival (PFS), a year overall survival rate (OSR), overall survival (OS), safety, immunogenicity, and steady-state pharmacokinetics.
Results
A total of 549 nsNSCLC patients were enrolled (277 in TAB008 group and 272 in Avastin® group). In the full analysis set, ORRs were 55.957% for TAB008 and 55.720% for Avastin®, and the ORR ratio was 1 (90% CI 0.89–1.14), well within the predefined equivalence margin of 0.75–1.33. No significant differences were found in DCR within 6 cycles (95.703% vs 95.367%, p = 0.8536), DoR (8.17 vs 7.3 months, p = 0.3526), PFS (9.10 vs. 7.97 months, p = 0.9457), 1 year overall survival rate (66.2% vs 68%, p = 0.6793), or OS (20.4 vs 17.6 months, p = 0.6549). Serious adverse events (SAEs) occurred in 37.55% (104/277) of patients in the TAB008 group and 34.32% (93/271) in the Avastin® group. Anti-drug antibodies were reported in 3 of 277 (1.08%) TAB008 patients, and 5 of 271 (1.85%) Avastin® patients, neutralizing antibody (Nab) was positive in 1 patient on Avastin®, which became negative upon follow-up. The steady-state trough concentrations (Cssmin) were 106.13 μg/mL in TAB008 group and 96.03 μg/mL in Avastin® groups, with the treatment group ratio of LS geometric means fully contained within the bioequivalence limits of 80.00–125.00% (90% CI was 101.74–120.05%).
Conclusions
TAB008 is similar to Avastin® in terms of efficacy, safety, and pharmacokinetic parameters, with comparable immunogenicity.
Trial registration
ClinicalTrials.gov number; NCT05427305.
Keywords: Avastin® biosimilar, TAB008, Non squamous non-small cell lung cancer (nsNSCLC), EGFR wild-type
Introduction
Bevacizumab (Avastin®) has played a prodigious role in human cancer control (Garcia et al. 2020), with a prolific range of indications for a targeting agent, alone and in combination with chemotherapy, interferon, and checkpoint inhibitors. Avastin® has been approved in colorectal cancer (2004), lung cancer (2006), breast cancer (2008, revoked in 2011 in the United States, sustained in the European Union), glioblastoma and renal cell carcinoma (2009), and cervical and ovarian cancer (2014). In 2018, checkpoint inhibitor potentiation in the IMpower150 study (Socinski et al. 2018) led to approval in frontline treatment for non-squamous, non-small cell lung cancer (nsNSCLC), followed by landmark approval together with atezolizumab as frontline therapy in hepatocellular carcinoma in the IMbrave150 superiority study surpassing survival endpoints achieved by sorafenib (Finn et al. 2020). In China, Avastin® has been approved for colorectal cancer in 2010, NSCLC in 2015, and glioblastoma in 2020.
Avastin® was initially approved in nsNSCLC based on superiority data emanating from the phase III E4599 study (Sandler et al. 2006) in the US, a similar design was replicated in China, the BEYOND study (Zhou et al. 2014). Patients were randomized to carboplatin (AUC = 6 mg/ml/min) plus paclitaxel (200 mg/m2 in the US, 175 mg/m2 in China) with or without Avastin® (15 mg/kg). The overall response rates (ORR), progression-free survival (PFS) and overall survival (OS) were 35%, 6.2 months (HR = 0.66), and 12.3 months (HR = 0.79) for E4599 study, and 54%, 9.2 months (HR = 0.4), and 24.3 months (HR = 0.68) for the BEYOND study. Superior efficacy emanated from the 26 to 27% EGFR mutant tumors, whereby PFS and OS were 12.4 and 24.3 months, respectively, but even in EGFR wild-type tumors, patient PFS and OS of 8.3 and 20.3 months with hazard ratios of 0.33 and 0.57 were still higher than in Western studies.
Based on consistent confirmation of Avastin® treatment effect on ORR risk ratio in meta-analysis of multiple large randomized studies in frontline NSCLC patients, justified selection of this population as adequately sensitive to demonstrate clinical equivalence, with ORR as the primary equivalence endpoint (He et al. 2016). FDA’s meta-analysis of 4 clinical studies yielded a risk ratio (RR) of 0.53, considering an ORR of 38% in the Avastin® plus chemotherapy doublet, a 10% drop out rate, at 80% power, with 70% confidence interval, minimal sample size would be just over 600 patients. However, a much higher ORR has been achieved in EGFR wild-type Chinese populations; sample size would be justifiably smaller.
TAB008 is a biosimilar candidate of Avastin®, non-clinical and preclinical studies confirming the similarity. The phase I study (Wang et al. 2019) in healthy Chinese male volunteers demonstrated the pharmacokinetic, safety, and immunogenicity similarity of TAB008 to Avastin®.
This phase III study aims to demonstrate the efficacy, safety, immunogenicity, and pharmacokinetic profiles of TAB008 and Avastin® in Patients with advanced or recurrent nsNSCLC.
Method
Study design
This randomized, double-blind, equivalence study was conducted in 53 medical centers in China. Treatment naïve for metastatic lung cancer, EGFR wild-type (by PCR or NGS) nsNSCLC patients were enrolled. Patients had to be between 18 and 75 years of age; stage IIIB to IV pathology confirmed nsNSCLC; ECOG PS 0–1; have adequate organ function; no uncontrollable infectious or serious illnesses; and most importantly, measurable lesion according to Response Evaluation Criteria in Solid Tumor (RECIST) version 1.1. Major exclusion criteria included tumors invading major blood vessels, previous major cardiovascular accidents (stroke, heart attack, uncontrollable hypertension), bleeding diathesis, proteinuria; prior history of malignancy other than NSCLC. Patients underwent tumor assessment using contrast-enhanced CT scans every two cycles for the first six cycles, then every four cycles thereafter until disease progression. The concentration of TAB008 in serum was quantitatively detected by ELISA, and the immunogenicity assessment methodology of anti-TAB008 and Avastin monoclonal antibodies in serum were determined by electrochemiluminescence (ECL). The study was approved by the National Medical Products Administration (NMPA) in China, and the ethics committees of each center, all patients provided written informed consent. The study was conducted in compliance with the International Council for Harmonization Good Clinical Practice guidelines and the Helsinki Declaration.
Treatment
Eligible patients were randomized 1:1 to receive TAB008 or Avastin® 15 mg/kg every 3 weeks for 6 cycles, then 7.5 mg/kg until disease progression, intolerable toxicity, withdrawal of consent, lost to follow-up, or death. All patients received carboplatin (area under the curve (AUC) = 5.0 mg/ml/min) and paclitaxel (175 mg/m2) every 3 weeks for between 4 and 6 cycles. The dose adjustment for TAB008, Avastin®, and chemotherapy were chosen to be consistent with the product labeling or standard of care in china.
Randomization and masking
Eligible subjects were randomized 1:1 by the Interactive Web Response System (IWRS) into the TAB008 group or Avastin® group. Investigators, patients were blinded until database lock to ensure the objectivity of efficacy and safety evaluation. Randomization was stratified by Eastern Cooperative Oncology Group (ECOG) performance status (0 vs. 1), disease stage (III vs. IV), and presence or absence of brain metastasis.
Endpoints
The primary endpoint would be overall response rate (ORR) within the first 6 cycles of treatment determined by the independent radiology review committee (IRRC). Secondary endpoints included disease control rate (DCR = CR + PR + SD) within 6 cycles, duration of response (DoR), progression-free survival (PFS), overall survival rate (OSR) at 1 year, overall survival (OS), safety, immunogenicity, and pharmacokinetic bioequivalence of steady-state trough concentration. Tumor response and progression were assessed according to RECIST v 1.1. Safety was evaluated by the type, incidence, severity, and relationship of adverse events (AEs) to the study drug. AEs were graded according to National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) version 4.03. Pharmacokinetic analysis was performed to ensure at least 140 patients had data from the full sampling set of 4 dose points, blood sampling was done before dosing on day 1 of Cycles 1, 5, 6, and 7, and immunogenicity samples were collected pre-dose in cycle 1, 2, 3, 5, 7, and 28 ± 7 days after the last dose in all patients.
Statistical analysis
Patients with EGFR mutations were excluded from the study, so the lower limit of the 90% confidence interval of ORR of 46% from the BEYOND study (Sandler et al. 2006) was chosen as the target ORR for this study. Sample size was calculated to achieve > 80% power to demonstrate equivalence between TAB008 and Avastin® on the primary efficacy endpoint (risk ratio of ORR) with the 90% confidence interval of the pre-specified “equivalence” margin of 0.75–1.33. Considering a 10% dropout rate, the total sample size would be 546 patients, 273 patients per arm.
The primary and secondary efficacy analyses were based on the full analysis set (FAS), based on the intention to treat principle, and consisted of all randomized patients that received at least one study dose. The per-protocol set (PPS) consisted of all FAS patients including all randomized patients who received at least one drug treatment and at least one tumor evaluation without any major protocol deviation. Safety set (SS) was analyzed for all patients who received at least one dose of the study drug. The pharmacokinetic analysis set (PKS) consisted of subjects who completed sample collection at cycle 5, 6, and 7, and the PKS was used for PK analysis.
The Cox proportional hazards model was used to calculate the HR for the primary analysis. Survival curves were estimated using the Kaplan–Meier method. The 90% confidence interval (CI) of the ratio of the best objective response rate between the two groups was calculated using the Miettinen and Nurminen method. Subgroup analyses were carried out to assess treatment effects by pre-specified demographic factors. The equivalence analysis refers to the bioequivalence evaluation method. If the geometric mean ratio and 90% confidence interval of the mean steady-state trough concentration of TAB008 monoclonal antibody and Avastin® fall within the range of 80.00–125.00%, then the steady-state trough concentration of the two drugs under the same treatment scheme is judged to be bioequivalent. All statistical analyses were carried out using SAS version 9.2. All reported AEs were coded according to the MedDRA v20.0, and the severity of AE was graded by the investigator according to the CTCAE v4.03.
Results
Patient disposition
From October 30, 2017 to November 8, 2019, 904 patients were screened, 549 patients were randomized to TAB008 (n = 277) or Avastin® (n = 272). Of the randomized patients, 277 (100%) and 271 (99.6%) patients in TAB008 and Avastin® groups received at least 1 dose of study drug and were included in the FAS and SS; 256 (92.4%) and 259 (95.2%) subjects in TAB008 and Avastin® group had at least 1 tumor assessment after receiving study drug and consist of PPS (Fig. 1). In TAB008 group, there were 17 (6.14%) subjects who received chemotherapy and 15 (5.42%) subjects who received radiotherapy. In Avastin® group, there were 11 (4.06%) subjects who received chemotherapy and 9 (3.32%) subjects who received radiotherapy. Most of the screen failures could be attributed to having the EGFR activating mutation. The primary analysis was conducted based on the data base lock on March 24, 2020.
Fig. 1.

Patient disposition
Baseline characteristics and demographics of patients were balanced between TAB008 group and Avastin® group (Table 1). In the Avastin® group, a subject not treated with investigational drug was eliminated.
Table 1.
Patient demographics and baseline characteristics (FAS)
| TAB008 + CP (n = 277) | Avastin® + CP (n = 271) | |
|---|---|---|
| Age mean (SD) | 58.5 (8.89) | 58.1 (8.02) |
| Weight, mean (SD) | 62.0 (10.75) | 63.1 (10.58) |
| Gender | ||
| Male (n, %) | 198 (71.48) | 199 (73.43) |
| Female (n, %) | 79 (28.52) | 72 (26.57) |
| Nationality | ||
| Han (n, %) | 267 (96.39) | 263 (97.05) |
| Others (n, %) | 10 (3.61) | 8 (2.95) |
| Disease stage | ||
| III | 33 (11.91) | 33 (12.18) |
| IV | 244 (88.09) | 238 (87.82) |
| ECOG | ||
| 0 | 69 (24.91) | 67 (24.72) |
| 1 | 208 (75.09) | 204 (75.28) |
| Histology | ||
| Adenocarcinoma (n, %) | 274 (98.92) | 264 (97.42) |
| Adenosquamous carcinoma (mainly adenocarcinoma) (n, %) | 0 (0.00) | 2 (0.74) |
| Large-cell carcinoma (n, %) | 0 (0.00) | 1 (0.37) |
| Others | 3 (1.08) | 4 (1.48) |
SD standard deviation, ECOG Eastern Cooperative Oncology Group
Efficacy
The primary efficacy endpoint of ORR within 6 cycles evaluated by IRRC was 55.957% for the TAB008 arm, and 55.720% for Avastin® arm in FAS (N = 548), calculated risk ratio of 1.00 (90% CI 0.89–1.14), well within the predefined equivalence margin of 0.75–1.33 (Table 2), all were partial responses.
Table 2.
Summary of response evaluated by IRRC (FAS)
| TAB008 (n = 277) | Avastin® (n = 271) | Ratio (90% CI) | |
|---|---|---|---|
| Best overall response (%) | 155 (55.96) | 151 (55.72) | 1 (0.89, 1.14) |
| Complete response | 0 | 0 | |
| Partial response | 155 (55.96) | 151 (55.72) | |
| Stable disease | 90 (32.49) | 96 (35.42) | |
| Disease control rate (DCR) | 245 (88.45) | 247 (91.14) | |
| Disease progression (PD) | 8 | 12 | |
| Unable to evaluate (NE)a | 24 | 12 |
IRRC independent radiological review committee, FAS full analysis set, CI confidential interval
a21 subjects withdrew early from the study for various reasons and 3 cases were considered unable to be evaluated in the TAB008 group. 12 subjects in the Avastin® group withdrew early from the study for various reasons.
TAB008 and Avastin® were similar in the Kaplan–Meier curve for PFS (Fig. 2), and the median PFS are 8.97 months and 7.85 months respectively, with hazard ratio of 0.99(95CI: 0.80,1.22). There were no statistical differences in all other secondary efficacy endpoints including DCR within 6 cycles (95.703% vs 95.367%), DoR (8.05 months vs 7.20 months, p = 0.9457), OSR at 1 year (66.21% vs 68.00%, p = 0.6793), and OS (20.07 months vs 19.09 months, p = 0.6549).
Fig. 2.
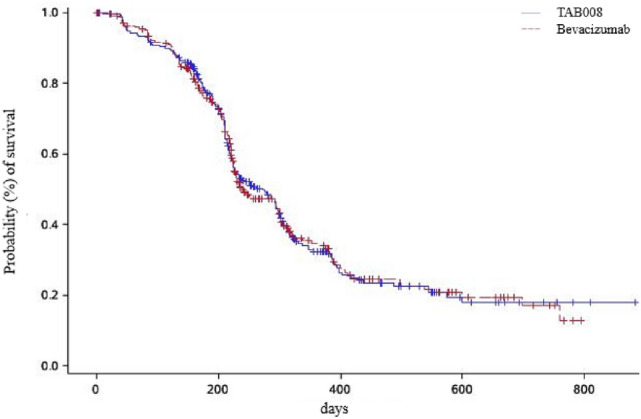
Kaplan–Meier estimate of survival for PFS
Safety
A summary of treatment emergent AEs (TEAEs) is shown in Table 3. There is no significant difference between the TAB008 and Avastin® group in the incidence of TEAEs (99.28% vs 98.89%, p = 0.6830), AEs ≥ grade 3 (78.34% vs 77.86%, p = 0.9180), serious AEs (SAE, 37.55% vs 34.32%, p = 0.4764), fatal AEs (3.61% vs 3.69%, p = 1.0000) (Table 4), and AEs of special interest (AESI, 42.24% vs 40.59%, p = 0.7289).
Table 3.
Summary of AEs
| AE type, n (%) | TAB008 (N = 277) | Avastin® (N = 271) | p value |
|---|---|---|---|
| Any AEs | 275 (99.28%) | 268 (98.89%) | 0.6830 |
| Any AE related to investigational drugsa | 221 (79.78%) | 211 (77.86%) | 0.6022 |
| Any AE related to chemotherapya | 267 (96.44%) | 264 (97.42%) | 0.6239 |
| Any significant AEsb | 58 (20.94%) | 44 (16.24%) | 0.1877 |
| AEs leading to investigational drugs discontinuation | 66 (23.83%) | 56 (20.66%) | 0.4117 |
| AE grade ≥ 3c | 217(78.34%) | 211(77.86%) | 0.9180 |
| SAE | 104 (37.55%) | 93 (34.32%) | 0.4764 |
| Fatal AEsd | 10 (3.61%) | 10 (3.69%) | 1.0000 |
| AESIs | 117 (42.24%) | 110 (40.59%) | 0.7289 |
Only treatment emergent AEs are summarized. For each category, patients are included only once, even if they had multiple events in the same category
aAdverse events related to study medicines are defined as definitely related, likely related, or possible related as assessed by the investigator
bSignificant AE refers to any AE requiring specific intervention, but not yet reaching the severity of an SAE
cGrading according to the Common terminology criteria for adverse events (CTCAE), version 4.03
dAEs with fatal outcomes in more than 1 patient included respiratory failure (3 in the TAB008 group and 2 in the Avastin® group)
Table 4.
Summary of fatal AEs
| AE type | TAB008 (N = 277) | Avastin® (N = 271) |
|---|---|---|
| Pulmonary infection | 2 | |
| Respiratory failure | 3 | 2 |
| Lung malignant tumor | 1 | 3 |
| Cerebral infarction | 1 | |
| Intestinal obstruction | 1 | |
| Acute pancreatitis | 1 | |
| Asphyxia | 1 | |
| Innutrition | 1 | |
| Death | 1 | 3 |
| Total (n, %) | 10 (3.61%) | 10 (3.69%) |
The incidences of all drug-related AEs in the TAB008 and Avastin® group are 79.78% and 77.86%(p = 0.6022), respectively, and the most frequent AEs related to investigational drugs ≥ grade 3 are decreased neutrophil count (31% vs. 29.5%), decreased leucocyte count (14.08% vs. 10.70%), and decreased platelet count (6.13% vs. 3.69%). The incidence of AESI is similar between the two groups, and the most frequent AESIs are proteinuria (28.16% vs. 27.31%) and hypertension (17.69% vs. 17.71%) (Fig. 3).
Fig. 3.

Incidence of treatment emergent adverse events of special interest
Pharmacokinetics
In the PKS (n = 140), the steady-state trough concentrations of the two groups were similar, with geometric mean ratios of 106.13 and 96.03, respectively, and 90% CI of (101.74%, 120.05%), which fell within the range of 80.00% to 125.00%, indicating that the steady-state trough concentrations of TAB008 and Avastin® were bioequivalent.
Immunogenicity
A total of 8 subjects developed anti-drug antibodies (ADA), including 3 subjects (1.08%) in the TAB008 group and 5 subjects (1.85%) in the Avastin® group. Only one subject (Avastin® group) developed a neutralizing antibody, and this subject tested negative for neutralizing antibodies at the end of the study (see Table 5).
Table 5.
Summary of immunogenicity (SS)
| TAB008 (n = 277) | Avastin® (n = 271) | |
|---|---|---|
| ADA positive | 3 (1.08%) | 5 (1.85%) |
| Nab positive | 0 (0%) | 1 (0.37%) |
Discussion
The Biologics Price Competition and Innovation Act of 2009 (BPCI Act) created an abbreviated licensure pathway for biological products shown to be “biosimilar” to or “interchangeable” with an FDA-licensed biological product (the “reference product”) in the US. Biosimilar development guidelines based on “totality of the Evidence “approach (safety, purity, and potency from a full complement of clinical and non-clinical studies) have been issued by the regulatory authorities of the EMA, FDA, and the NMPA. This phase III equivalence study of TAB008 has confirmed safety (AEs. SAEs, AESIs, immunogenicity) and potency (ORRs, PFS, OS) to the originator, after confirmation of pharmacokinetic similarity in a stringent phase I clinical study (Wang et al. 2019). Even though the design of this phase III Avastin® originator controlled study in NSCLC is similar to that for ABP-215, and PF-06439535, there are added dimensions of ethnicity, natural disease history (even in EGFR wild-type), dosing intensity (lower paclitaxel and carboplatin dosages), and thus sample size (IBI305: 450 EGFR wild-type patients, QL1101: 535 EGFR wild-type and mutant patients) issues that factor into trial design adjustment.
For patients with EGFR activating mutations, EGFR-tyrosine kinase inhibitors (TKIs) can significantly improve ORR and prolong survival compared to chemotherapy (Fukuoka et al. 2011). From the perspective of patient benefit, patients with EGFR mutations were excluded from this study, as well as known ALK and ROS mutations, which make up around 3.37% and 2.56%, respectively, of Chinese NSCLC patients (Li et al. 2013; Zhu et al. 2019). In this study, the maintenance dose of Avastin® after 6 cycles was 7.5 mg/kg, which was also consistent with that in routine clinical use.
The ORR was 55.72 and 55.96% in TAB008 and Avastin® groups, much higher than expected compared to IBI305 (Avastin® biosimilar) at 43–46% in EGFR wild-type patients in China (Yang et al. 2019) yet comparable to the ORR attained in non-EGFR mutation selected Chinese patients in the BEYOND study at 54%, but since evaluation was performed by blinded independent radiologist review, it should be acceptable. The PFS as assessed by IRRC in the TAB008 and Avastin® groups were 8.97 months and 7.85 months, respectively, which was longer than that reported in the predominantly Caucasian E4599 study (6.2 months) (Sandler et al. 2006) quite similar to the PFS of 7.64 and 7.77 months attained in the IBI305 study, and the 8.3 months PFS reported in EGFR wild-type patients in the BEYOND study, but much shorter than the 12.3 month PFS in its EGFR mutant patients (Zhou et al. 2014).
The types, incidence, and severity of AEs, SAEs, and AEs of special interest were similar between the two groups. All the types and severity of AEs in the TAB008 group were known or expected for Avastin®, and no unexpected adverse events occurred. AEs of special interest included proteinuria, hypertension, bleeding, thromboembolic event, and gastric-intestinal perforation. Almost all AESI were similar between the two groups, except bleeding and thromboembolic event, and most of which were grade 1 or 2, and could be easily controlled clinically. Immunogenicity was low and similar between the two groups, and only one Avastin® patient developing neutralizing antibodies which later abated.
This phase III study and the phase I study (Wang et al. 2019) completed in 2018 demonstrated the similarity of TAB008 in terms of efficacy, safety, pharmacokinetic characteristics, and immunogenicity of Avastin® (AVASTIN.FDA approval Package Insert. 2019). However, lack of reimbursement and high out-of-pocket costs limited its clinical practice (Monk et al. 2017). Because of the similarity and relatively low price, TAB008 can be a high-quality and reliable alternative of Avastin®.
In conclusion, this study in patients with advanced non-squamous NSCLC demonstrated similarity between TAB008 and Avastin® in terms of efficacy, safety, pharmacokinetic characteristics, and immunogenicity. These results, together with non-clinical and phase I study result, confirm the similarity between TAB008 and Avastin®. TAB008 has been submitted to NMPA for marketing application and can be used as a high-quality alternative to Avastin® after approval.
Acknowledgements
Authors would like to express our deepest gratitude to the patients, in addition to the investigators and staff at the study sites for their contribution to the study. The present study was sponsored by TOT BIOPHARM International Company. All the Authors contributed to revision of the drafts, approved the final version, and made the decision to submit the manuscript for publication.
Author contributions
SL and SQ designed this protocol of this clinical trial as the leading Principal investigators of the clinical trial, ZZ, JC, KG, PS, YP, GY, KM, JS, YS, LY, PC and AL all participated in the clinical trial as the investigators and wrote the main manuscript text, JH prepared all of the figures and tables as the biostatistician. All authors reviewed the manuscript.
Data Availability
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Declarations
Conflict of interest
The authors declare no potential conflicts of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- AVASTIN.FDA approval Package Insert. 06/2019
- Finn RS, Qin S, Ikeda M, Galle PR, IMbrave150 Investigators et al (2020) Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med 382(20):1894–1905 [DOI] [PubMed] [Google Scholar]
- Fukuoka M, Wu YL, Thongprasert S et al (2011) Biomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer in Asia (IPASS). J Clin Oncol 29:2866–2874 [DOI] [PubMed] [Google Scholar]
- Garcia J, Hurwitz HI, Sandler AB, Miles D, Coleman RL, Deurloo R, Chinot OL (2020) Bevacizumab (Avastin®) in cancer treatment: a review of 15 years of clinical experience and future outlook. Cancer Treat Rev 86:102017 [DOI] [PubMed] [Google Scholar]
- He K, Chen H, Gwise T, Casak S, Lemery S, Keegan P, Pazdur R, Sridhara R (2016) Statistical considerations in evaluating a biosimilar product in an oncology clinical study. Clin Cancer Res 22(21):5167–5170 [DOI] [PubMed] [Google Scholar]
- Li Y, Li Y, Yang T, Wei S, Wang J, Wang M, Wang Y, Zhou Q, Liu H, Chen J (2013) Clinical significance of EML4-ALK fusion gene and association with EGFR and KRAS gene mutations in 208 chinese patients with non-small cell lung cancer. PLoS ONE 8(1):e52093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk BJ, Lammers PE, Cartwright T et al (2017) Barriers to the Access of Bevacizumab in Patients with Solid Tumors and the Potential Impact of Biosimilars: A Physician Survey. Pharmaceuticals (basel) 10:19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler A, Gray R, Perry MC et al (2006) Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med 355(24):2542–2550 [DOI] [PubMed] [Google Scholar]
- Socinski MA, Jotte RM, Cappuzzo F, IMpower150 Study Group et al (2018) Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med 378(24):2288–2301 [DOI] [PubMed] [Google Scholar]
- Wang J, Qi L, Liu L et al (2019) A phase I, randomized, single-dose study evaluating the biosimilarity of TAB008 to bevacizumab in healthy volunteers. Front Pharmacol 10:905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Wu B, Huang L et al (2019) Biosimilar candidate IBI305 plus paclitaxel/carboplatin for the treatment of non-squamous non-small cell lung cancer. Transl Lung Cancer Res 8(6):989–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Wu Y, Chen G et al (2014) Efficacy and biomarker data from BEYOND: A randomized phase 3 study of first line chemotherapy ± bevacizumab in Chinese patients with advanced non-squamous non-small cell lung cancer. Int J Radiat Oncol 90(5):S17 [Google Scholar]
- Zhu YC, Zhang XG, Lin XP, Wang WX, Li XF, Wu LX, Chen HF, Xu CW, Du KQ (2019) Clinicopathological features and clinical efficacy of crizotinib in Chinese patients with ROS1-positive non-small cell lung cancer. Oncol Lett 17:3466–3474 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.