

Abstract
The cyclooctyne‐functionalized alcohol (1R,8S,9S)‐bicyclo‐[6.1.0]non‐4‐yn‐9‐ylmethanol (BCN‐OH) is applied as initiator for the organo‐catalyzed ring‐opening polymerization (ROP) of morpholine‐2,5‐diones based on the l‐amino acids valine, isoleucine, and phenylalanine. The ROP is catalyzed by a binary system of 1,8‐diazabicyclo[5.4.0]undec‐7‐ene (DBU) and 1‐(3,5‐bis(trifluoromethyl)phenyl)‐3‐cyclohexylthiourea (TU) applying a feed ratio of [M]/[I]/[DBU]/[TU] of 100/1/1/10. Kinetic studies reveal that BCN‐OH is capable to initiate the polymerization of morpholine‐2,5‐diones, which proceed in a controlled manner until monomer conversions of 80%. Characterization by means of 1H NMR spectroscopy, size exclusion chromatography (SEC), and matrix‐assisted laser desorption/ionization‐time of flight‐mass spectrometry confirm the covalent attachment of the cyclooctyne moiety as α‐end group of the poly(morpholine‐2,5‐dione)s with maximum dispersities of 1.25. As a proof of concept, a vitamin A end‐functionalized poly(2‐ethyl‐2‐oxazoline) is coupled to a poly(ester amide) by strain‐promoted azide‐alkyne cycloaddition. Characterization of the block copolymer by SEC and DOSY NMR spectroscopy confirm the successful attachment of the two building blocks. The versatile cyclooctyne moiety shall facilitate a metal‐free attachment of other polymer blocks, targeting ligands or dyes at the α‐end group of well‐defined poly(morpholine‐2,5‐dione)s. In consequence, the approach provides access to a new generation of functionalized poly(ester amide)s, which can be customized for specific needs.
Keywords: click chemistry; morpholine‐2,5‐dione; poly(ester amide); polydepsipeptide; ring‐opening polymerization; strain‐promoted azide‐alkyne cycloaddition (SPAAC)
This is the first report of a successful initiation of the ring‐opening polymerization of morpholine‐2,5‐diones based on l‐valine, l‐isoleucine, and l‐phenylalanine using a cyclooctyne derivative as an initiator. The post‐polymerization modification via strain‐promoted azide‐alkyne cycloaddition is demonstrated by coupling a poly(ester amide) with a hydrophilic and vitamin A end‐functionalized “stealth” polymer.
1. Introduction
Poly(ester amide)s are in the focus of research as alternatives to polyesters for biomedical applications.[ 1 , 2 , 3 ] In addition to the ester moieties, these biodegradable and biocompatible polymers also feature more stable amide bonds in the backbone. This provides more parameters to tailor the polymers to specific applications.[ 2 , 3 ] For example, an advanced adjustment of the degradation of the corresponding drug delivery systems, which may occur under acidic, basic or enzymatic conditions, is possible.[ 1 , 4 ] Furthermore, the hydrophobicity and, thus, the interactions between polymer and drug can be influenced by the composition of the ester and amide moieties, but also by the choice of suitable monomer substituents.[ 3 , 4 ] By incorporating aromatic substituents, an additional attractive interaction with drugs, which also often contain conjugated systems, can be introduced via π–π stacking.[ 5 ]
The ring‐opening polymerization (ROP) of morpholine‐2,5‐diones represents one option to obtain poly(ester amide)s.[ 1 , 6 , 7 ] As these monomers are not commercially available, the synthesis of the respective compounds, for example, from naturally occurring l‐amino acids, is necessary.[ 1 , 5 , 8 , 9 , 10 ] The homopolymerization of a morpholine‐2,5‐dione by ROP was first described by Helder et al.[ 6 ] in 1985. Here, 6‐methylmorpholine‐2,5‐dione was polymerized in bulk in the presence of tin(II)‐ethylhexanoate as catalyst. Since then, there have been many developments in this field.[ 1 , 11 , 12 , 13 ] In addition to metal‐based ROP, a polymerization using enzyme‐catalyzed approaches has been mainly described by Feng et al.[ 12 , 14 , 15 , 16 , 17 ]
Organo‐based ROP of morpholine‐2,5‐diones was pioneered by Hedrick and coworkers in 2005, describing the use of N‐heterocyclic carbenes and thiourea for catalysis.[ 18 , 19 ] Since then, 1,5,7‐triazabicyclo[4.4.0]dec‐5‐ene (TBD) and 1,8‐diazabicyclo[5.4.0]undec‐7‐ene (DBU) have come into focus as catalysts.[ 20 , 21 ] Moreover, narrower dispersities and higher monomer conversions have been achieved in combination with thiourea derivatives as cocatalysts.[ 1 , 5 , 10 , 22 , 23 ] The possibility to perform the polymerization at room temperature using such catalyst systems might enable the use of temperature‐sensitive reagents during the reaction.
Similar to metal‐catalyzed ROP, primary alcohols such as benzyl alcohol act as initiators in organo‐catalyzed ROP, thereby defining the α‐end group of the resulting poly(ester amide). Although this has been exploited for the formation of block copolymers through utilization of macroinitiators,[ 5 , 20 ] the possibility to attach end groups with added value via this straightforward route has, to the best of our knowledge, not yet been explored.
In particular, cyclooctyne derivatives seem highly promising since they enable the subsequent coupling of various building blocks carrying an azide functionality via strain‐promoted azide‐alkyne cycloaddition (SPAAC). Such a strategy may, for example, be utilized for attachment of labels[ 24 , 25 ] or other polymeric building blocks exhibiting a “stealth” effect.[ 26 , 27 ] In fact, the cyclooctyne derivative (1R,8S,9S)‐bicyclo‐[6.1.0]non‐4‐yn‐9‐ylmethanol (BCN‐OH) has been successfully used for the initiation of the organo‐based ROP of lactide and ε‐caprolactone.[ 28 , 29 ]
Herein, we describe the organo‐catalyzed ROP of morpholine‐2,5‐diones based on the natural α‐amino acids l‐valine (ValG), l‐isoleucine (IleG), and l‐phenylalanine (PheG) initiated by BCN‐OH (Scheme 1 ). Initially, suitable ROP conditions were established using DBU in combination with 1‐(3,5‐bis(trifluoromethyl)phenyl)‐3‐cyclohexylthiourea (TU) as catalytic system at room temperature. The α‐end group fidelity was investigated by mass spectrometry. As a proof of principle, an azide end‐functionalized poly(2‐ethyl‐2‐oxazoline) (PEtOx) additionally carrying retinoic acid amide was conjugated to a cyclooctyne functionalized poly(ester amide) via SPAAC.
Scheme 1.
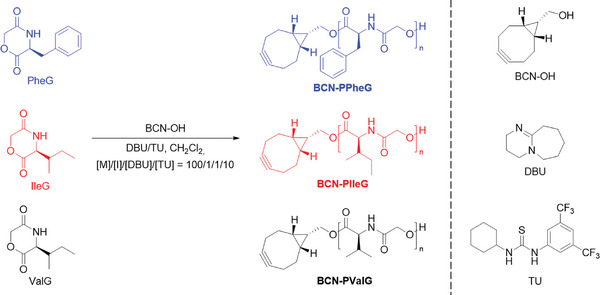
Schematic representation of the synthesis route toward the polymers BCN‐PPheG, BCN‐PIleG, and BCN‐PValG using the corresponding monomers PheG, IleG, and ValG.
2. Results and Discussion
The monomers for the polymerizations described below were synthesized following the commonly used routes.[ 5 , 30 ] Therefore, the corresponding l‐amino acid was reacted with chloroacetyl chloride in tetrahydrofuran in the presence of triethylamine. The obtained precursor subsequently underwent a cyclization reaction at high dilution in N,N‐dimethylformamide using NaHCO3 as base. A detailed description of the procedure is available in the Supporting Information.
2.1. Kinetic Studies
Similar to reaction conditions typically used for the DBU/TU‐catalyzed ROP of morpholine‐2,5‐diones,[ 5 , 10 , 22 , 23 , 30 ] the reactions were carried out at room temperature in CH2Cl2 at a [M]/[I]/[DBU]/[TU] ratio of 100/1/1/10.
Kinetic studies were performed to investigate the polymerization of PheG, IleG, and ValG using BCN‐OH as initiator (Scheme 1). Samples taken from the reaction solutions at specific time intervals were analyzed by means of 1H NMR spectroscopy and size exclusion chromatography (SEC) to determine the monomer conversions and the number average of the molar masses (M n).
First, an initial monomer concentration ([M]0) of 0.5 mol L−1 was set in the ROP of PheG, i.e., the same concentration used in the adapted polymerization procedure by Göppert et al.[ 5 ] However, the formation of a high molar mass shoulder of the distribution was observed in the SEC elugrams already at a monomer conversion of 42% (Figure S2, Supporting Information). As evident from the increase of the M n values with the monomer conversions with a flattening slope, molar mass control could not be achieved at these reaction conditions. Thereby, the dispersities increased up to 1.40 during the considered reaction time frame. These data point toward the presence of a significant amount of intermolecular chain transfer reactions. Reducing the monomer concentration or increasing the amount of TU may result in an improvement of reaction control.[ 5 ] This is reasonable as a lower monomer and polymer concentration would decrease the probability of intermolecular transesterifications. At a [M]0 of 0.3 mol L−1, unimodal molar mass distributions were maintained until conversions around 50%, and dispersities remained below 1.3 even at monomer conversions above 80% (Figure S3, Supporting Information). Accordingly, these polymerization conditions were also applied for the ROP of IleG and ValG.
Figure 1 displays the kinetic plots based on the obtained data from 1H NMR spectroscopy and SEC. In particular, the ROP of PheG and ValG followed a linear trend in the semi‐logarithmic kinetic plot. The polymerization of PheG proceeded significantly faster than that of ValG, as evidenced by the increased slope. In contrast, a significant decrease of the slope in the semilogarithmic plot for the ROP of IleG was observed in an early stage of the reaction. Despite that, the molar mass of the BCN‐PIleG increased in a linear fashion with the monomer conversion, which was also the case for BCN‐PValG. In case of BCN‐PPheG, the slope flattened slightly above conversions of around 50%, but molar mass control was significantly improved compared to the ROP conducted at a [M]0 of 0.5 mol L−1.
Figure 1.
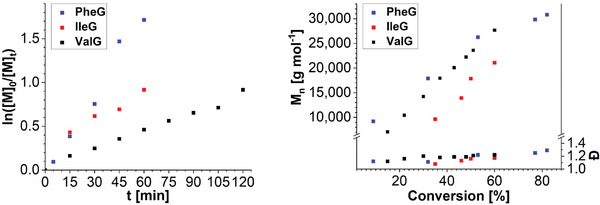
Semilogarithmic kinetic plots (left) and plots of M n and Ð determined by SEC against the monomer conversion (right) for the BCN‐OH‐initiated polymerizations of PheG, IleG, and ValG ([M]/[I]/[DBU]/[TU] = 100/1/1/10, [M]0 = 0.3 mol L−1, room temperature).
The SEC elugrams of the kinetic samples obtained from the polymerization of IleG displayed a slight low molar mass tailing (Figure S3, Supporting Information), which may indicate that chain terminations or intramolecular chain transfers could not be fully avoided, although dispersities remained below 1.16. Unimodal and narrow molar mass distributions with dispersities below 1.21 were found throughout the ROP of ValG. A slight high molar mass shoulder only occurred at conversions above 60%.
In general, chain transfer reactions through transesterification occurred only towards the end of the polymerizations, which suggested that the desired cyclooctyne end group would be present at the majority of polymer chains if the ROP were terminated at appropriate reaction times.
2.2. Synthesis of Cyclooctyne End‐Functionalized Poly(ester amide)s
The syntheses of BCN‐PPheG, BCN‐PIleG, and BCN‐PValG were performed using the reaction conditions described in the previous section. The ROP of ValG and IleG were driven to conversions around 80%, whereas the polymerization of PheG was terminated at a conversion of 29% (Table 1 ). The expected degrees of polymerization (DPtheo) correspond to molar masses between 6 and 14 kg mol−1. The molar masses determined by SEC significantly exceed those values, hinting toward a large hydrodynamic volume of all poly(ester amide)s in the eluent. In line with the kinetic studies, dispersity values around 1.2 were determined. Whereas the molar mass distribution of BCN‐PIleG revealed a high molar mass shoulder, monomodal distributions were detected for BCN‐PPheG and BCN‐PValG (Figure S5, Supporting Information).
Table 1.
Selected characterization data of the poly(ester amide)s and the block copolymer.
|
Conv. a) [%] |
DPtheo b) |
M n,theo b) [g mol−1] |
M n c) [g mol−1] |
Ɖ c) | |
|---|---|---|---|---|---|
| BCN‐PValG | 83 | 83 | 13 200 | 35 800 | 1.19 |
| BCN‐PIleG | 81 | 81 | 14 000 | 25 800 | 1.25 |
| BCN‐PPheG | 29 | 29 | 6 100 | 20 600 | 1.19 |
| Ret‐PEtOx‐b‐PValG | – | 19/83 | 15 500 | 35 800 | 1.23 |
Determined from the 1H NMR spectra of the reaction solutions;
Calculated from [M]0/[I] and conversion;
Determined by SEC (DMAc, 0.21% w/w LiCl; RID; PS calibration).
Due to the rather high DPtheo values and overlapping signals of the repeating units, end group signals derived from the cyclooctyne end group were difficult to detect and to assign in the 1H NMR spectra, which, nevertheless, confirm the identities of the repeating units (compare assignment in Figure S6, Supporting Information).
The α‐end group fidelity was hence investigated by means of matrix‐assisted laser desorption/ionization‐time of flight‐mass spectrometry (MALDI‐TOF MS). Figure 2 depicts the MALDI mass spectrum of BCN‐PValG. The spectra of BCN‐PIleG and BCN‐PPheG are shown in the Supporting Information (Figures S7 and S8, Supporting Information). In all spectra, the main m/z distribution was assigned to the respective polymer with the desired α‐end group, as evident from the overlay of the measured and calculated isotopic patterns. For BCN‐PValG, the sodium adducts represented the only species detected until m/z values of 16 000. The occurrence of molar mass discrimination effects at such high m/z values is not surprising. Nevertheless, non‐fragmented macromolecules could be ionized in a molar mass range that corresponds to M n,theo. Irrespective of the sample, spectra measured with dithranol as a matrix instead of trans‐2‐[3‐(4‐tert‐butylphenyl)‐2‐methyl‐2‐propenylidene]malononitrile (DCTB) revealed more m/z series. In the former, macromolecules with carboxylic acid or carboxylate end groups were detected frequently. Such polymer chains could theoretically be formed by initiation of the ROP with traces of water. However, their high abundance, particularly in the lower m/z regions of the spectra points toward the formation of those species through fragmentation. Cyclic species indicating intramolecular chain transfer reactions occurring during the ROP were only found in the spectra of BCN‐PPheG. In this case, transesterification reactions were anticipated based on the results of the kinetic studies as described above.
Figure 2.
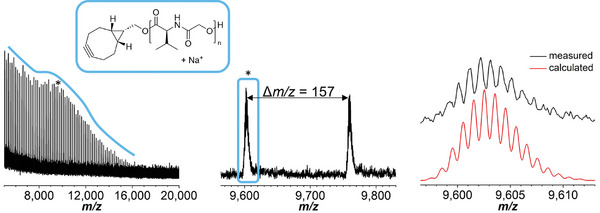
MALDI‐TOF MS analysis of BCN‐PValG. Left: Full mass spectrum (DCTB, NaTFA) of BCN‐PValG with highlighted distribution of the detected species. Middle: Zoom into the spectrum. Right: Overlay of the measured and the calculated isotopic pattern of the detected species.
In summary, the analytical data demonstrate a successful synthesis of BCN‐PPheG, BCN‐PIleG as well as BCN‐PValG as the presence of the desired cyclooctyne moiety was confirmed.
2.3. SPAAC Reaction
To demonstrate the accessibility of the cyclooctyne moiety of the synthesized poly(morpholine‐2,5‐dione)s during a SPAAC, BCN‐PValG was reacted with Ret‐PEtOx‐N3 in CH2Cl2 at room temperature (Scheme 2 ). The azide‐functionalized building block is based on PEtOx as a hydrophilic polymer that exhibits “stealth” properties and vitamin A as a targeting ligand for hepatic stellate cells.[ 27 ]
Scheme 2.
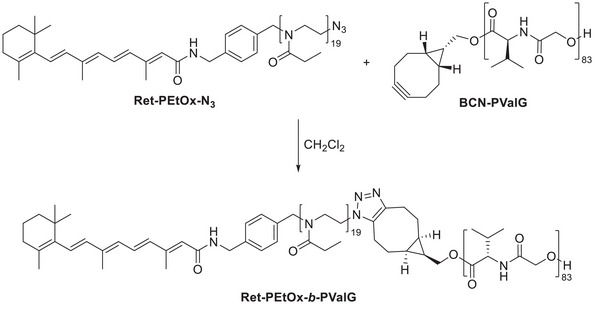
Schematic representation of the SPAAC of BCN‐PValG and Ret‐PEtOx‐N3 to produce an amphiphilic block copolymer functionalized with a biologically active molecule.
Figure S9 (Supporting Information) displays an overlay of the 1H NMR spectra of Ret‐PEtOx‐b‐PValG and the two polymeric educts. Signals assigned to both building blocks were observed in the spectrum of the obtained block copolymer. Those appeared at the same diffusion coefficient in the diffusion‐ordered NMR spectrum (DOSY NMR, Figures S10 and S11, Supporting Information), which indicates a successful coupling of the polymers.
To further confirm the covalent attachment of the two blocks, a SEC measurement was carried out with a refractive index detector (RID) and an additional diode array detector (DAD, Figure 3 ). The block copolymer eluted at slightly lower retention volumes compared to that of BCN‐PValG. This is in line with the large hydrodynamic volume of the poly(ester amide) on the SEC system and the comparably low molar mass of Ret‐PEtOx‐b‐PValG (DP = 19).
Figure 3.
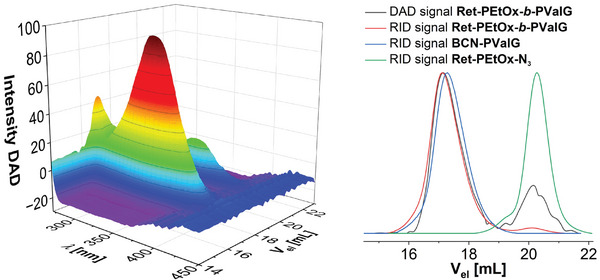
Left: 3D spectrum of the SEC measurement (DMAc, 0.21% w/w LiCl; RID; DAD) of Ret‐PEtOx‐b‐PValG using DAD. Right: Overlay of the elugrams of Ret‐PEtOx‐b‐PValG (red), BCN‐PValG (blue), and Ret‐PEtOx‐N3 (green) recorded with the RID and the elugram of Ret‐PEtOx‐b‐PValG recorded with the DAD (351 nm, gray).
The DAD confirmed the successful attachment of the two building blocks as the block copolymer was detected at wavelengths corresponding to the absorption of the conjugated system of the retinoic acid amide‐based end group. BCN‐PValG alone does not absorb light between 300 and 450 nm (Figure S12, Supporting Information). A minor amount of Ret‐PEtOx‐N3 was also visible, which may be a result from PEtOx chains lacking the azide functionality.
3. Conclusion
In conclusion, we successfully applied the cyclooctyne‐functionalized alcohol BCN‐OH as initiator for the organo‐catalyzed ROP of the morpholine‐2,5‐diones ValG, IleG, and PheG. Kinetic studies revealed a successful initiation of the polymerization, which was catalyzed by DBU and TU. Characterization of the resulting polymers BCN‐PPheG, BCN‐PIleG, and BCN‐PValG by means of 1H NMR spectroscopy, SEC and, in particular, MALDI‐TOF MS confirmed the preservation and covalent attachment of the cyclooctyne moiety as α‐end group.
As an exemplary hydrophilic polymer featuring a targeting ligand, Ret‐PEtOx‐N3 was successfully coupled to BCN‐PValG as a hydrophobic and degradable building block.
Although the ROP of, in particular, PheG might be further optimized in the future, the versatile cyclooctyne moiety enables a metal‐free attachment of, for example, hydrophilic polymer blocks exhibiting a “stealth” effect, targeting ligands, or dyes at the α‐end group of well‐defined poly(ester amide)s. In consequence, the approach provides access to a new generation of functionalized poly(ester amide)s, which can be customized for the specific needs of various drug delivery systems.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
This project was funded by the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG; Project No. 316213987, SFB 1278 “PolyTarget”, projects A01, A06, Z01). The RapifleX MALDI‐TOF/TOF system was financed by the Thüringer Aufbaubank (funding ID: 2016 IZN 0009). Nicole Fritz is acknowledged of the MALDI‐TOF MS measurements and Dr. Grit Festag for the SEC measurement support.
Open access funding enabled and organized by Projekt DEAL.
Schreiber J., Göppert N. E., Stafast L. M., Weber C., Schubert U. S., Cyclooctyne End‐Functionalized Poly(morpholine‐2,5‐dione)s. Macromol. Rapid Commun. 2025, 46, 2400705. 10.1002/marc.202400705
Contributor Information
Christine Weber, Email: christine.weber@uni-jena.de.
Ulrich S. Schubert, Email: ulrich.schubert@uni-jena.de.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Dirauf M., Muljajew I., Weber C., Schubert U. S., Prog. Polym. Sci. 2022, 129, 101547. [Google Scholar]
- 2. Winnacker M., Rieger B., Polym. Chem. 2016, 7, 7039. [Google Scholar]
- 3. Rodríguez‐Galán A., Franco L., Puiggalí J., in Biodegradable Polymers: Processing, Degradation and Applications (Ed: Felton G. P.), Nova Science Publishers, New York: 2011. [Google Scholar]
- 4. Ghosal K., Latha M. S., Thomas S., Eur. Polym. J. 2014, 60, 58. [Google Scholar]
- 5. Göppert N. E., Dirauf M., Liebing P., Weber C., Schubert U. S., Macromol. Rapid Commun. 2022, 44, e2200651. [DOI] [PubMed] [Google Scholar]
- 6. Helder J., Kohn F. E., Sato S., van den Berg J. W., Feijen J., Makromol. Chem., Rapid Commun. 1985, 6, 9. [Google Scholar]
- 7. P. J. A. in't Veld , Dijkstra P. J., van Lochem J. H., Feijen J., Makromol. Chem. 1990, 191, 1813. [Google Scholar]
- 8. Methoxypolyethylene glycol‐polyester‐amino acid modified disordered copolymer as well as preparation method and application thereof, CN108383999 (A), 2018.
- 9. Ohya Y., Yamamoto H., Nagahama K., Ouchi T., J. Polym. Sci. Part A: Polym. Chem. 2009, 47, 3892. [Google Scholar]
- 10. Lian J., Li M., Wang S., Tao Y., Wang X., Macromolecules 2020, 53, 10830. [Google Scholar]
- 11. Feng Y., Klee D., Höcker H., Macromol. Chem. Phys. 2001, 202, 3120. [Google Scholar]
- 12. Feng Y., Lu J., Behl M., Lendlein A., Macromol. Biosci. 2010, 10, 1008. [DOI] [PubMed] [Google Scholar]
- 13. Feng Y., Klee D., Höcker H., Macromol. Chem. Phys. 1999, 200, 2276. [Google Scholar]
- 14. Feng Y., Knüfermann J., Klee D., Höcker H., Macromol. Chem. Phys. 1999, 200, 1506. [Google Scholar]
- 15. Feng Y., Knüfermann J., Klee D., Höcker H., Macromol. Rapid Commun. 1999, 20, 88. [Google Scholar]
- 16. Feng Y., Klee D., Höcker H., Macromol. Biosci. 2001, 1, 66. [DOI] [PubMed] [Google Scholar]
- 17. Feng Y., Klee D., Keul H., Höcker H., Macromol. Chem. Phys. 2000, 201, 2670. [Google Scholar]
- 18. Pratt R. C., Dove A. P., Lohmeijer B. G. G., Culkin D. A., Waymouth R. M., Hedrick J. L., Polym. Prepr. 2005, 46, 902. [Google Scholar]
- 19. Pratt R. C., Lohmeijer B. G. G., Mason A., Waymouth R. M., Hedrick J. L., Polym. Prepr. 2006, 47, 101. [Google Scholar]
- 20. Dirauf M., Erlebach A., Weber C., Hoeppener S., Buchheim J. R., Sierka M., Schubert U. S., Macromolecules 2020, 53, 3580. [Google Scholar]
- 21. Dirauf M., Bandelli D., Weber C., Görls H., Gottschaldt M., Schubert U. S., Macromol. Rapid Commun. 2018, 39, 1800433. [DOI] [PubMed] [Google Scholar]
- 22. Guo Y.‐T., Shi C., Du T.‐Y., Cheng X.‐Y., Du F.‐S., Li Z.‐C., Macromolecules 2022, 55, 4000. [Google Scholar]
- 23. Shi C.‐X., Guo Y.‐T., Wu Y.‐H., Li Z.‐Y., Wang Y.‐Z., Du F.‐S., Li Z.‐C., Macromolecules 2019, 52, 4260. [Google Scholar]
- 24. Gaitzsch J., Delahaye M., Poma A., Du Prez F., Battaglia G., Polym. Chem. 2016, 7, 3046. [Google Scholar]
- 25. Melnychuk N., Ashokkumar P., Aparin I. O., Klymchenko A. S., ACS Appl. Polym. Mater. 2022, 4, 44. [Google Scholar]
- 26. Gppert N. E., Dirauf M., Weber C., Schubert U. S., Polym. Chem. 2021, 12, 5426. [Google Scholar]
- 27. Stafast L. M., Engel N., Görls H., Weber C., Schubert U. S., Eur. Polym. J. 2023, 184, 111779. [Google Scholar]
- 28. van den Berg S. A., Zuilhof H., Wennekes T., Macromolecules 2016, 49, 2054. [Google Scholar]
- 29. Goeppert N. E., Vollrath A., Stafast L. M., Stumpf S., Schulze B., Hoeppener S., Weber C., Schubert U. S., RSC Appl. Polym. 2024, 2, 184. [Google Scholar]
- 30. Burton T. F., Garisoain Z., Chaix C., Aassine J., Virapin E., Voronova A., Pinaud J., Giani O., ACS Omega 2024, 9, 28583. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.