

Abstract
An NF1 microdeletion is the single most commonly reported mutation in individuals with neurofibromatosis type 1 (NF1). Individuals with an NF1 microdeletion have, as a group, more neurofibromas at a younger age than the group of all individuals with NF1. We report that NF1 microdeletion individuals additionally have a substantially higher lifetime risk for the development of malignant peripheral nerve sheath tumors than individuals with NF1 who do not have an NF1 microdeletion. This should be taken into account in the medical follow-up of individuals with an NF1 microdeletion.
Neurofibromatosis type 1 (NF1 [MIM 162200]) is an autosomal dominant disorder with a prevalence of 1/4,000 (Huson et al. 1989). NF1 is a tumor-suppressor gene located on chromosome band 17q11.2. The main features of NF1 are benign neurofibromas, café-au-lait spots, axillary freckling, Lisch nodules, and learning disabilities. Most of the patients have a small truncating mutation in the NF1 gene (point mutation, splice mutation, small deletion, insertion, or duplication). However, 5%–10% of individuals with NF1 have a microdeletion (Clementi et al. 1996; Cnossen et al. 1997; Rasmussen et al. 1998). Most of these have a 1.5-Mb deletion incorporating the entire NF1 region (López-Correa et al. 2000; Dorschner et al. 2000). In rare cases, both bigger and smaller microdeletions have been observed (López-Correa et al. 1999; Dorschner et al. 2000; personal observation). Genome database analysis indicates that the common NF1 microdeletion region contains at least 15 genes/transcripts, including NF1 and the three embedded genes. Most of these genes have unknown functions.
The NF1 microdeletion patients tend to have a different phenotype as compared with the general NF1 group. NF1 microdeletion patients frequently have more neurofibromas at an earlier age, a lower mean IQ, and nonfamilial facial features (Kayes et al. 1994; Wu et al. 1995; Leppig et al. 1997; Tonsgard et al. 1997; Wu et al. 1997). Although, as a group, the NF1 microdeletion patients have distinct features, it remains difficult at the individual level to distinguish between deletion and nondeletion patients solely on the basis of clinical observations, and, at this time, there is a lack of genotype-phenotype correlation for individuals with NF1. Because of the enormous clinical variability in phenotypic expression, a medical surveillance program for all individuals with NF1 is necessary to detect a large range of possible complications. Each complication occurs in only a relatively small percentage of individuals with NF1 (Huson and Hughes 1994). The lifetime risk for malignant peripheral nerve sheath tumor (MPNST) in individuals with NF1 may be 8%–13%, as reported in a recent study (Evans et al. 2002), and it would be important to identify a subgroup of individuals with NF1 who have a higher risk of MPNST.
Elsewhere, we reported germline mutations in seven patients with NF1 and MPNSTs (Wu et al. 1999). Three of these had an NF1 microdeletion, suggesting a possibly increased incidence of MPNSTs in NF1 microdeletion patients. Dorschner et al. also reported two patients with MPNSTs in a group of 17 individuals with an NF1 microdeletion and suggested a predisposition to malignancies in NF1 microdeletion cases (Dorschner et al. 2000). We wanted to test the hypothesis that individuals with an NF1 microdeletion represent a specific subgroup of individuals with NF1 and a higher risk of MPNST.
In a multicenter study (Wu et al. 1999; Birindelli et al. 2001; Leroy et al. 2001; unpublished cases), we tested the frequency of NF1 microdeletions on a large number (38) of patients with NF1 and MPNST (all diagnosed as having NF1 according to the National Institutes of Health criteria [Stumpf et al. 1987]). This study on archival material was approved by the institutional review boards of the different institutes. On histology, all tumors corresponded to sarcomatous cellular proliferations, consisting of highly cellular, intersecting bundles of spindle cells, with unequivocal nuclear atypia, prominent typical and/or atypical mitotic figures, and variable areas of necrosis. Tumors were graded as low grade (I), intermediate grade (II), or high grade (III), on the basis of differentiation, mitotic rate, and amount of tumor necrosis (table 1). Of the 38 MPNST cases, 24 were high grade, 2 low grade, 9 intermediate grade, and 2 of unknown grades. Only two MPNSTs showed features enabling classification as either triton or epitheloid tumors.
Table 1.
Overview of Patients with NF1 in This Report
| Sample | Dela | Ageb | Gradec | Familiald | Teste | Reference |
| NF 00-4 | + | 15 | III | − | F | Unpublished |
| NF 00-3 | + | 22 | III | + | F | Unpublished |
| 755 | + | 23 | II | + | BP | Unpublished |
| 14 L | + | 29 | II | − | BP | Leroy et al. 2001 |
| NF 90-8 | + | 17 | IIIe | U | F | Wu et al. 1999 |
| NF 96-1 | + | 27 | III | − | F | Wu et al. 1999 |
| C12 | + | 58f | U | − | F | Wu et al. 1999 |
| 2 L | + | 18 | I | − | BP | Leroy et al. 2001 |
| 10 L | + | 20 | III | − | SQ, M | Leroy et al. 2001 |
| 1 B | − | 16 | III | + | M | Birindelli et al. 2001 |
| 2 B | − | 14 | II | − | M | Birindelli et al. 2001 |
| 3 B | − | 27 | III | − | M | Birindelli et al. 2001 |
| 4 B | − | 39 | III | − | M | Birindelli et al. 2001 |
| 6 B | − | 33 | III | + | M | Birindelli et al. 2001 |
| 7 B | − | 30 | III | − | M | Birindelli et al. 2001 |
| 8 B | − | 20 | III | − | M | Birindelli et al. 2001 |
| 12 B | − | 32 | III | U | M | Birindelli et al. 2001 |
| NF 89-3 | − | 15 | U | U | F | Wu et al. 1999 |
| NF 90-1 | − | 23 | III | − | F | Wu et al. 1999 |
| NF 88-3 | − | 33 | IIIt | U | F | Wu et al. 1999 |
| NF 88-18 | − | 60 | II | − | F | Wu et al. 1999 |
| 1 L | − | 20 | II | + | M | Leroy et al. 2001 |
| 6 L | − | 31 | I | − | M | Leroy et al. 2001 |
| NFR117 | − | 25 | II | + | M | Unpublished |
| 17 L | − | 35 | III | − | M | Leroy et al. 2001 |
| 11 L | − | 18 | III | − | M | Leroy et al. 2001 |
| 3 L | − | 56 | II | + | M | Leroy et al. 2001 |
| 9 L | − | 49 | III | − | M | Leroy et al. 2001 |
| NFR525 | − | 31 | III | + | M | Unpublished |
| 560 | − | 22 | II | + | M | Unpublished |
| 632 | − | 30 | II | + | M | Unpublished |
| 790 | − | 19 | III | + | M | Unpublished |
| 5 L | − | 55 | III | + | M | Leroy et al. 2001 |
| 8 L | − | 42 | III | − | M | Leroy et al. 2001 |
| 12 L | − | 18 | III | + | M | Leroy et al. 2001 |
| NF 93-2 | − | 28 | III | + | M | Unpublished |
| NF 93-1 | − | 41 | III | − | M | Unpublished |
| NF 96-4 | − | ? | U | U | M | Unpublished |
Presence (+) or absence (−) of deletion.
Age in years at diagnosis of the MPNST.
Grade of MPNST: e = epithelioid, t = triton.
Familial (+) or sporadic (−) occurrence of NF1; U = unknown.
The test performed to confirm the presence or absence of a deletion is indicated: M = marker analysis, BP = specific break point PCR, F = FISH analysis, SQ = semi-quantitated.
Outlier in age at diagnosis distribution.
The presence of microdeletions was assessed on nontumor tissue by different techniques, depending on the available material for analysis: a specific breakpoint PCR (López-Correa et al. 2001), FISH, semiquantitated PCR, and/or marker analysis (see table 1). The FISH experiments were performed with PAC clone RPCI5-926B9 located within NF1 (López-Correa et al. 1999). In several cases, only DNA was available, and FISH could not be performed. In these cases, a specific breakpoint PCR was performed to detect the common breakpoint junction fragment. If a specific deletion junction fragment was not detected, marker analysis was performed by assessing heterozygosity for five markers located in the NF1 microdeletion region (D17S1800: 5′NF1-1, 3′NF1-1, 3′NF1-2, 3′NF1-3, and IVS/38-GT; see also López-Correa et al. [1999]). We excluded an NF1 microdeletion if heterozygosity for markers located in the microdeletion region was observed in nontumor DNA. We realize that we might have missed cases with a smaller deletion confined to the NF1 gene region. Only cases showing no heterozygosity for any of the five dinucleotide repeats were further analyzed with semiquantitative PCR for a potential microdeletion.
For the semiquantitated PCR analysis, exon 48 of NF1 was amplified, together with exon 24 of CFTR as a control fragment (25 cycles). This semiquantitated PCR analysis was optimized using known NF1 microdeletion and nonmicrodeletion individuals. The relative intensity of the NF1 fragment was scored after electrophoresis on an ABI PRISM 310 Genetic Analyzer. All primer sequences and PCR conditions are available from the authors on request.
Nine of 38 patients with NF1 and MPNSTs (23.7%) showed an NF1 microdeletion in the nontumor tissue (table 1). This microdeletion frequency is significantly higher (P<.005; binomial distribution) than expected in the general NF1 population (5%–10%) (Clementi et al. 1996; Cnossen et al. 1997; Rasmussen et al. 1998). To estimate the lifetime risk of MPNST in NF1 microdeletion patients (Bayes’s Theorem), we used conservative estimates of the different probabilities. We used 20% (this study 23.7%) for the frequency of an NF1 microdeletion in individuals with NF1 and MPNST (P[Del/MPNST, NF1]) and 10% (probably an overestimation) for the frequency that a patient with NF1 is a carrier of a microdeletion (P[Del/NF1]). P(MPNST/NF1) is an estimate of the lifetime risk for the development of MPNST in the general NF1 population.
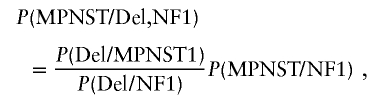 |
and
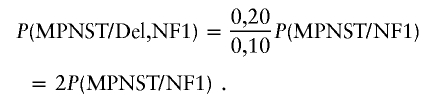 |
Using these parameters, we estimate that the lifetime risk for MPNST in NF1 microdeletion patients is twice as high as in the general NF1 population.
Previous cross-sectional studies showed that only 1%–2% of individuals with NF1 have MPNST (Huson et al. 1989). Specialist series found higher risks of MPNST in NF1, owing to a bias toward patients with important complications that require specialist treatment in a hospital setting (Greager et al. 1992). The lifetime risk of MPNST in patients with NF1 was recently estimated to be 8%–13%, in a population-based study with near-total ascertainment for NF1-related MPNST, using a dual approach for ascertainment (NF1 register and Cancer registry) (Evans et al. 2002). Evans et al. (2002) also found that the majority of patients with NF1 and MPNST clinically present with their malignancy in the 3rd or 4th decade of life (median 26 years). This is at a much younger age than individuals who have sporadic MPNST but are not afflicted with NF1 (median 62 years).
In the NF1-microdeletion patient group reported here, the age at diagnosis does not have a normal distribution curve because of an outlier (patient C12, 58 years). We observed a substantial difference in the median age at diagnosis between deletion and nondeletion cases: 22 years (microdeletion) versus 30 years (nonmicrodeletion). This difference of 8 years is not significant (P=.15; Mann-Whitney U test). After exclusion of the outlier (C12), we observe a significant difference (P=.04; Mann-Whitney U test) of 9 years in the median age of onset (21 years vs. 30 years). Therefore, it might be interesting to assess the significance of this difference in age at diagnosis in a larger study.
It is known that NF1 microdeletion individuals as a group show a more severe phenotype than the general NF1 population. One might argue that mildly affected NF1 individuals with an MPNST have a lower chance of being diagnosed as NF1 and, thus, a higher chance of not being included in this study. Therefore, this study could have a bias toward individuals more severely affected with NF1 with a higher likelihood of harboring an NF1 microdeletion. We do not think that this bias plays a role in this study because the NF1 MPNST cases were diagnosed in centers with multidisciplinary outpatient clinics for neurofibromatosis. These centers have a high suspicion for the diagnosis of neurofibromatosis in cases presenting with MPNST. In addition, we believe that the potential ascertainment bias for diagnosing an individual more severely affected with MPNST as having NF1 applies also for the referral of an individual with an NF1-compatible phenotype to a multidisciplinary clinic. The proportion of NF1 microdeletion individuals in the total NF1 population is estimated on the basis of series from multidisciplinary clinics. These series might also show an ascertainment bias toward more severely affected cases, resulting in a potential overrepresentation of NF1 microdeletion individuals. As a result, both ascertainment biases (NF1 in MPNST population and NF1 deletion in NF1 multidisciplinary clinic population) will eliminate each other, to a great degree, in the equation.
Our study indicates that NF1 microdeletion patients have a substantially higher risk for the development of MPNSTs compared with nonmicrodeletion NF1 individuals. If we assume that the lifetime risk of MPNST in NF1 individuals is 8%–13% (Evans et al. 2002), then NF1 microdeletion patients have a lifetime risk for MPNST of 16%–26%. We therefore propose routine testing of individuals with NF1 for the presence of a microdeletion and follow-up of NF1 microdeletion patients with a higher level of suspicion for MPNSTs. Complaints of pain and/or rapid growth of tumors should be actively investigated using a low clinical threshold.
Further molecular investigation of tumoral cells of neurofibromas and MPNSTs from both NF1 microdeletion and nonmicrodeletion patients might shed some light on the molecular pathogenesis of the increased susceptibility for MPNST development in NF1 microdeletion patients.
Acknowledgments
We thank the individuals with NF1 who collaborated in this study. T.D. is supported by the Vlaams instituut voor de bevordering van Wetenschappelijk-Technologisch onderzoek in de industrie (IWT). E.L. is a part-time clinical researcher of the Fonds voor Wetenschappelijk Onderzoek Vlaanderen (FWO). This work is also supported by the FWO (grant G.0096.02 to E.L.); the Interuniversity Attraction Poles (IAP) grant from the Federal Office for Scientific, Technical and Cultural Affairs, Belgium (2002–2006; P5/25); and the Deutsche Krebshilfe (70-2794-DeI and 70-2635-Ma3).
Electronic-Database Information
The URL for data presented herein is as follows:
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for NF1) [PubMed]
References
- Birindelli S, Perrone F, Oggionni M, Lavarino C, Pasini B, Vergani B, Ranzani GN, Pierotti MA, Pilotti S (2001) Rb and TP53 pathway alterations in sporadic and NF1-related malignant peripheral nerve sheath tumors. Lab Invest 81:833–844 [DOI] [PubMed] [Google Scholar]
- Clementi M, Boni S, Mammi I, Favarato M, Tenconi R (1996) Clinical application of genetic polymorphism in neurofibromatosis type 1. Ann Genet 39:92–96 [PubMed] [Google Scholar]
- Cnossen MH, van der Est MN, Breuning MH, van Asperen CJ, Breslau-Siderius EJ, van der Ploeg AT, Goede-Bolder A, van den Ouweland AM, Halley DJ, Niermeijer MF (1997) Deletions spanning the neurofibromatosis type 1 gene: implications for genotype-phenotype correlations in neurofibromatosis type 1? Hum Mutat 9:458–464 [DOI] [PubMed] [Google Scholar]
- Dorschner MO, Sybert VP, Weaver M, Pletcher BA, Stephens K (2000) NF1 microdeletion breakpoints are clustered at flanking repetitive sequences. Hum Mol Genet 9:35–46 [DOI] [PubMed] [Google Scholar]
- Evans DG, Baser ME, McGaughran J, Sharif S, Howard E, Moran A (2002) Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet 39:311–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greager JA, Reichard KW, Campana JP, DasGupta TK (1992) Malignant schwannoma of the head and neck. Am J Surg 163:440–442 [DOI] [PubMed] [Google Scholar]
- Huson S, Hughes R (1994) The neurofibromatoses: a pathogenic and clinical overview. Chapman and Hall, London [Google Scholar]
- Huson SM, Compston DA, Clark P, Harper PS (1989) A genetic study of von Recklinghausen neurofibromatosis in south east Wales. I. Prevalence, fitness, mutation rate, and effect of parental transmission on severity. J Med Genet 26:704–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayes LM, Burke W, Riccardi VM, Bennett R, Ehrlich P, Rubenstein A, Stephens K (1994) Deletions spanning the neurofibromatosis 1 gene: identification and phenotype of five patients. Am J Hum Genet 54:424–436 [PMC free article] [PubMed] [Google Scholar]
- Leppig KA, Kaplan P, Viskochil D, Weaver M, Ortenberg J, Stephens K (1997) Familial neurofibromatosis 1 microdeletions: cosegregation with distinct facial phenotype and early onset of cutaneous neurofibromata. Am J Med Genet 73:197–203 [DOI] [PubMed] [Google Scholar]
- Leroy K, Dumas V, Martin-Garcia N, Falzone MC, Voisin MC, Wechsler J, Revuz J, Creange A, Levy E, Lantieri L, Zeller J, Wolkenstein P (2001) Malignant peripheral nerve sheath tumors associated with neurofibromatosis type 1: a clinicopathologic and molecular study of 17 patients. Arch Dermatol 137:908–913 [PubMed] [Google Scholar]
- López-Correa C, Brems H, Lazaro C, Estivill X, Clementi M, Mason S, Rutkowski JL, Marynen P, Legius E (1999) Molecular studies in 20 submicroscopic neurofibromatosis type 1 gene deletions. Hum Mutat 14:387–393 [DOI] [PubMed] [Google Scholar]
- López-Correa C, Brems H, Lazaro C, Marynen P, Legius E (2000) Unequal meiotic crossover: a frequent cause of NF1 microdeletions. Am J Hum Genet 66:1969–1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Correa C, Dorschner M, Brems H, Lazaro C, Clementi M, Upadhyaya M, Dooijes D, Moog U, Kehrer-Sawatzki H, Rutkowski JL, Fryns JP, Marynen P, Stephens K, Legius E (2001) Recombination hotspot in NF1 microdeletion patients. Hum Mol Genet 10:1387–1392 [DOI] [PubMed] [Google Scholar]
- Rasmussen SA, Colman SD, Ho VT, Abernathy CR, Arn PH, Weiss L, Schwartz C, Saul RA, Wallace MR (1998) Constitutional and mosaic large NF1 gene deletions in neurofibromatosis type 1. J Med Genet 35:468–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpf DA, Alksne JF, Annegers JF, Brown SS, Conneally PM, Housman D, Leppert M, Miller JP, Moss ML, Pileggi AJ, Rapin I, Strohman RC, Swanson LW, Zimmerman A (1987) Neurofibromatosis 1988. National Institutes of Health: consensus development conference statement. Arch Neurol 45:575–578 [Google Scholar]
- Tonsgard JH, Yelavarthi KK, Cushner S, Short MP, Lindgren V (1997) Do NF1 gene deletions result in a characteristic phenotype? Am J Med Genet 73:80–86 [DOI] [PubMed] [Google Scholar]
- Wu BL, Austin MA, Schneider GH, Boles RG, Korf BR (1995) Deletion of the entire NF1 gene detected by the FISH: four deletion patients associated with severe manifestations. Am J Med Genet 59:528–535 [DOI] [PubMed] [Google Scholar]
- Wu BL, Schneider GH, Korf BR (1997) Deletion of the entire NF1 gene causing distinct manifestations in a family. Am J Med Genet 69:98–101 [PubMed] [Google Scholar]
- Wu R, López-Correa C, Rutkowski JL, Baumbach LL, Glover TW, Legius E (1999) Germline mutations in NF1 patients with malignancies. Genes Chromosomes Cancer 26:376–380 [PubMed] [Google Scholar]