

Abstract
2-methyl-3-hydroxybutyryl-CoA dehydrogenase (MHBD) deficiency is a novel inborn error of isoleucine degradation. In this article, we report the elucidation of the molecular basis of MHBD deficiency. To this end, we purified the enzyme from bovine liver. MALDI-TOF mass spectrometry analysis revealed that the purified protein was identical to bovine 3-hydroxyacyl-CoA dehydrogenase type II. The human homolog of this bovine enzyme is a short-chain 3-hydroxyacyl-CoA dehydrogenase, also known as the “endoplasmic reticulum–associated amyloid-β binding protein” (ERAB). This led to the identification of the X-chromosomal gene involved, which previously had been denoted “HADH2.” Sequence analysis of the HADH2 gene from patients with MHBD deficiency revealed the presence of two missense mutations (R130C and L122V). Heterologous expression of the mutant cDNAs in Escherichia coli showed that both mutations almost completely abolish enzyme activity. This confirms that MHBD deficiency is caused by mutations in the HADH2 gene.
Degradation of the branched-chain amino acid isoleucine in humans takes place in mitochondria via the concerted action of a series of enzymes, during which isoleucine first undergoes transamination to 2-keto-3-methylbutyrate, followed by oxidative decarboxylation to 2-methylbutyryl-CoA. Like any 2-methyl branched-chain fatty acid, 2-methylbutyryl-CoA undergoes further breakdown by β-oxidation via a four-step pathway involving dehydrogenation to tiglyl-CoA, hydration to 2-methyl-3-hydroxybutyryl-CoA, dehydrogenation to 2-methylacetoacetyl-CoA, and finally, thiolytic cleavage to produce acetyl-CoA plus propionyl-CoA.
Numerous patients with a defect in isoleucine metabolism have been described in literature. In most of these patients, the defect is at the level of the last enzyme of the degradation pathway; that is, 2-methylacetoacetyl-CoA thiolase (β-ketothiolase) (MIM 203750). The enzymatic and molecular basis of this defect has been studied in detail (Mitchell and Fukao 2001). Patients with 2-methylacetoacetyl-CoA thiolase deficiency typically suffer from intermittent, severe ketoacidosis with vomiting and hematemesis. Patients usually develop normally, although a minority of patients show severe clinical abnormalities that may progress to coma and death. Patients with β-ketothiolase deficiency usually excrete tiglylglycine, 2-methyl-3-hydroxybutyrate, and 2-methylacetoacetate in excess amounts.
Recently, two novel defects in the isoleucine breakdown pathway have been described in single patients, including 2-methylbutyryl-CoA dehydrogenase deficiency (MIM 600301) (Gibson et al. 2000) and 2-methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency (MHBD deficiency [MIM 300256]) (Zschocke et al. 2000). The first patient with MHBD deficiency (patient 1) (table 1) was born at term and recovered well from an episode of metabolic decompensation and lactic acidosis (Zschocke et al. 2000). Psychomotor development was only moderately delayed at age 1 year, but the patient subsequently showed a gradual loss of mental and motor skills, which progressed with profound developmental regression, choreoathetosis, near blindness, and epilepsy. Brain MRI showed a slight frontotemporal atrophy. These clinical symptoms differ markedly from those observed in patients with β-ketothiolase deficiency. Subsequently, additional patients suffering from MHBD deficiency have been identified.
Table 1.
MHBD Activity in Cultured Skin Fibroblasts, Mutation Analysis of HADH2, and the Effect of the Mutations on MHBD Protein[Note]
|
Mutation Analysis |
||||||
| Subject | Sex | MHBD Activity(nmol/min−1.mg−1) | cDNA | gDNA | Protein | Mutation in Exon |
| Patient: | ||||||
| 1 | M | .6 | 388C→T | 388C→T | R130C | 4 |
| 2 | F | 1.0 | 388C→T | 388C→C/T | R130C | 4 |
| 3 | M | .6 | 388C→T | 388C→T | R130C | 4 |
| 4 | M | .7 | NDa | 388C→T | R130C | 4 |
| 5 | M | 1.8 | 364C→G | 364C→G | L122V | 4 |
| Controls (n=15) | 7.1 ± 0.8 | |||||
Note.— MHBD activity in cultured skin fibroblasts and mutation analysis of the coding gene from patients with MHBD deficiency. MHBD activity in skin fibroblasts was measured spectrophotometrically in the reverse direction by following the decrease in absorbance at 340 nm at 37°C. The standard reaction medium with a total volume of 250 μl contained the following components: 50 mM MES/100 mM potassium phosphate buffer pH 6.5, 0.1% (wt/vol) Triton X-100, 0.1 mM NADH, and 0.2 mg/ml fibroblast homogenate protein. Reactions were started by the addition of 2-methyl-acetoacetyl-CoA at a final concentration of 0.05 mM. Activity is given as a mean of two independent measurements. Results from controls is given as mean ± SD (number of cell lines studied).
ND = not done.
The patients studied in the present report have all been described before: patient 1 in Zschocke et al. (2000), patients 2 and 3 in Ensenauer et al. (2002), and patients 4 and 5 in Poll-The et al. (2001) and Sass and Sperl (2001), respectively. Virtually all patients identified so far show neurological abnormalities, including psychomotor retardation and loss of mental and motor skills, with the exception of patient 5, who had psychomotor retardation but no progressive loss of mental and motor skills. Patients with MHBD deficiency excrete excess amounts of tiglylglycine and 2-methyl-3-hydroxybutyrate, although 2-methylacetoacetate is absent in urine.
To elucidate the molecular basis of MHBD deficiency, we purified 2-methyl-3-hydroxyacyl-CoA dehydrogenase from bovine liver, elaborating on earlier work by Schulz and coworkers, who were the first to show that the conversion of 2-methyl-3-hydroxybutyryl-CoA to 2-methylacetoacetyl-CoA is brought about by a distinct 3-hydroxyacyl-CoA dehydrogenase (Luo et al. 1995; Mao et al. 1995). The final purification protocol consists of a series of four standard liquid chromatography steps; results are summarized in table 2. The purified protein was analyzed using SDS-PAGE (Laemmli 1970), followed by silver staining (Rabilloud et al. 1988). The result, shown in figure 1, revealed that bovine liver MHBD is a protein with an apparent molecular weight of 28 kDa. Attempts to directly sequence the protein by Edman degradation failed, probably because of a blocked N-terminus. To resolve this problem, a different strategy was chosen, and the 28-kDa protein band was digested in gel with trypsin after SDS-PAGE and subjected to MALDI-TOF mass spectrometry analysis. For this purpose, ∼5 μg of purified bovine liver MHBD was loaded on a 12% SDS-PAGE gel. After electrophoresis and CBB R250 staining, protein-containing gel slices were S-alkylated, digested with trypsin (Roche Molecular Biochemicals, sequencing grade), and extracted as described elsewhere (Shevchenko et al. 1996). Extracted peptides were purified and concentrated using Zip-Tips (Millipore). Peptides were eluted from the Zip-Tips with 10 μl of 1% formic acid, 60% acetonitrile. The peptide solution was mixed with an equal volume of 10 mg/ml α-cyano-4-hydroxycinnamic acid (Sigma Chemical) solution in acetonitrile/ethanol (1:1, vol/vol). Aliquots of 0.5 μl were spotted on the target and allowed to dry at room temperature. MALDI-TOF MS spectra were acquired on a Micromass TofSpec 2EC (Micromass, Wythenshawe), equipped with a 2-GHz digitizer. The resulting peptide spectra were used to search a nonredundant protein sequence database (Swiss-Prot/TrEMBL) using the Proteinprobe program. Using this method, we were able to identify MHBD as bovine 3-hydroxyacyl-CoA dehydrogenase type II (HADH2) (Swiss-Prot: locus HCD2_BOVIN, accession number O02691), with 12 of the 14 peptides matching and a coverage percentage of 62% (data not shown). This protein sequence was subsequently used as query to screen the human EST database, which led to the identification of its presumed human homolog, which has been described before as a short-chain 3-hydroxyacyl-CoA dehydrogenase, also known as the “endoplasmatic reticulum–associated amyloid β-binding” protein (ERAB [GenBank accession numbers U96132 and AF037438]). The gene involved is located on the X-chromosome and contains 6 exons (GenBank accession number NM_004493 [Xp11.2, gi:4758503]).
Table 2.
Summary of the Purification of MHBD from Bovine Liver[Note]
| Protein (mg) | Specific Activity (μmol/min.mg) | Total Activity (μmol/min) | Purification Factor | Recovery (%) | |
| Supernatant | 347.8 | .28 | 98.3 | 1 | 100 |
| S-Sepharose FF CL-6B | 52.4 | .90 | 47.4 | 3.2 | 48.2 |
| Hydroxylapatite CHT-II | 4.3 | 3.92 | 17.1 | 14 | 17.4 |
| Blue-Sepharose CL-6B | 1.1 | 51.8 | 5.7 | 185 | 5.8 |
| HiTrap butyl FF | .6 | 53.0 | 3.2 | 189 | 3.3 |
Note.— All steps were performed at 4°C. Approximately 10 g of bovine liver was minced and homogenized in a buffer containing 10 mM MES pH 6.0, 10% (vol/vol) glycerol, 2 mM DTT, and 100 mM KCl, using a Potter-Elvehjem homogenizer. After sonication, the homogenate was centrifuged at 20,000×gav for 1 h at 4°C, and the supernatant was diluted with a buffer containing 10 mM MES pH 6.0, 10% (vol/vol) glycerol and 2 mM DTT. This was loaded on an S-Sepharose FF CL-6B column (1.6×10.5 cm; Pharmacia Biotech), and bound proteins were eluted from the column using a linear gradient of KCl in the same buffer up to a final concentration of 400 mM. Fractions were collected and assayed for MHBD activity, as described elsewhere (Zschocke et al. 2000). Protein concentrations were determined, as described elsewhere, using bovine serum albumin as standard (Bradford 1976). Fractions with MHBD activity were pooled, diluted in a 20-mM potassium phosphate buffer (pH 7.4) containing 10% (vol/vol) glycerol and 5 mM DTT, and loaded on a 5-ml hydroxylapatite CHT-II column (Biorad). Bound proteins were eluted with a linear gradient of potassium phosphate up to a final concentration of 300 mM. Fractions with highest MHBD activity were pooled, diluted in a 20-mM potassium phosphate buffer pH 7.4 containing 10% (vol/vol) glycerol and 5 mM DTT, and loaded on a Blue-Sepharose CL-6B column (Pharmacia Biotech). The column was developed with a linear gradient of KCl up to a final concentration of 1 M. Fractions with highest MHBD activity were pooled, and an equal volume of a 20-mM potassium phosphate pH 7.4 buffer containing 10% (vol/vol) glycerol, 5 mM DTT, and 3 M ammonium sulfate was added slowly. After 1 h on ice, aggregates were removed by centrifugation at 10,000×gav for 10 min. The supernatant was loaded on a 1-ml HiTrap butyl FF column (Pharmacia Biotech) equilibrated with a solution containing 20 mM potassium phosphate pH 7.4 buffer containing 10% (vol/vol) glycerol, 5 mM DTT, and 1.5 M ammonium sulfate. After sample application, the column was washed and developed with linear gradient of ammonium sulfate to a final concentration of 0 M. Fractions were collected, assayed for MHBD activity, and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), as described elsewhere (Laemmli 1970), followed by silver staining (Rabilloud et al. 1988).
Figure 1 .
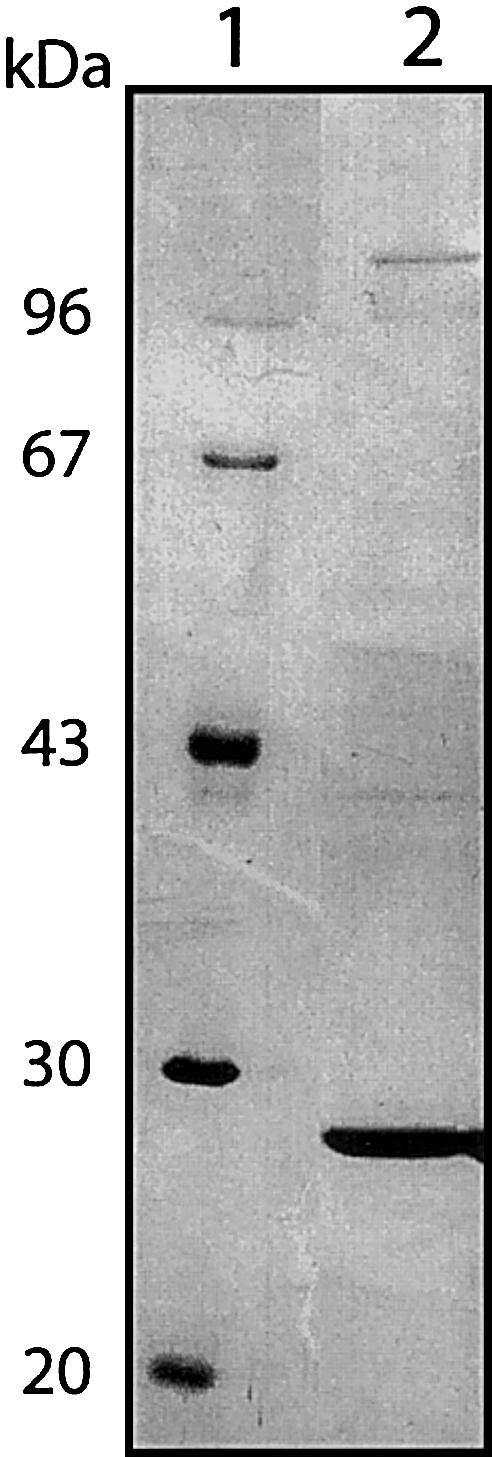
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis of bovine liver MHBD. MHBD was purified from bovine liver and analyzed using SDS-PAGE (Laemmli 1970) followed by silver staining (Rabilloud et al. 1988). Lane 1, molecular weight markers; Lane 2, purified bovine liver MHBD.
On the basis of the nucleotide sequence of the human gene, primers were selected and used for the amplification of HADH2 from both cDNA and genomic DNA (table 3). First-strand cDNA synthesis was performed as described (IJlst et al. 1994), using 5–10 μg of total RNA isolated from cultured human skin fibroblasts. MHBD-encoding cDNA was amplified from first-strand cDNA as template in two overlapping fragments. Fragment 1: “−21M13 forward”-tagged −21HADH2 plus “M13 reverse”-tagged +497HADH2; fragment 2: “−21M13 forward”-tagged +352HADH2 plus “M13 reverse”-tagged +837HADH2. PCR conditions for each primer set were 94°C for 2 min; followed by 30 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 1 min; and a final extension step at 72°C for 2 min. Subsequent sequence analysis of these PCR fragments, using both sense and antisense strands, was performed using “−21M13 forward” and “M13 reverse” fluorescent primers on an Applied Biosystems 377A automated sequencer, according to the manufacturer’s protocol. For mutation analysis at the genomic level, DNA was purified from cultured skin fibroblasts, using the Promega Wizard Genomic DNA purification kit, or from blood spots, using Chelex, as described elsewhere (Walsh et al. 1991). The gene was amplified in three fragments using the following primer sets: fragment 1: “−21M13 forward”-tagged Ex1/2frw plus “M13 reverse”-tagged Ex1/2rev; fragment 2: “−21M13 forward”-tagged Ex3/4frw plus “M13 reverse”-tagged Ex3/4rev; fragment 3: “−21M13 forward”-tagged Ex5/6frw plus “M13 reverse”-tagged Ex5/6rev. PCR conditions for each primer set were 2 min at 96°C for 2 min; followed by 30 cycles of 96°C for 30 s, 55°C for 30 s, and 72°C for 2 min; and a final extension step at 72°C for 2 min. Subsequent sequence analysis of these PCR fragments, using both sense and antisense strands, was performed as described above. The results of the sequence analysis of HADH2 from the patients with MHBD deficiency are shown in table 1. In all patients, mutations were identified in their HADH2 cDNA, which could be confirmed by genomic DNA analysis. One missense mutation (C388C→T), resulting in the substitution of the arginine residue at position 130 for a cysteine (R130C), was identified in four of the five patients (patients 1–4) (table 1). The predominance of one missense mutation in a severe X-chromosomal disorder was unexpected. There is no indication that the families of our patients are related. Cytosine at position 388 is part of a CpG dinucleotide that may be methylated and, thus, could represent a mutation hot spot in the HADH2 gene. Family histories of the affected patients were unremarkable. At least one carrier for the 388C→T mutation is asymptomatic. As expected for an X-chromosomal gene, all mutations observed in the cDNA from the male patients (patients 1, 3, 4, and 5) were found to be hemizygous at the genomic level. In the female patient (patient 2), the mutation was found in a heterozygous form at the genomic level, evident from RFLP analysis, as shown in figure 2. In patient 5, a 364C→G mutation was found, which results in the substitution of the leucine residue at position 122 for a valine (L122V). RFLP analysis was used to screen for the 364C→G mutation in the family of patient 5. The results, presented in figure 3, clearly show that the mother is heterozygous for the 364C→G mutation, whereas the mutation is not found in the other members of the family. MHBD is a member of the short-chain dehydrogenase family (conserved domain database at NCBI: pfam00106.6, adh_short), and both missense mutations, R130C and L122V, are within the conserved domain. However, it is not known whether these residues are involved in enzyme activity. Amino acid sequence comparison of human MHBD with members of this family revealed that both residues are not highly conserved.
Table 3.
Primers Used for Amplification and Cloning of Human HADH2[Note]
| Primer | Amplification |
| “−21M13 forward”-tagged −21HADH2 | 5′-tgtaaaacgacggccagtCGTGGAGTGGCCGGCGAC-3′ |
| “M13 reverse”-tagged +497HADH2 | 5′-caggaaacagctatgaccTCCAACCTGACCCTCGAAGG-3′ |
| “−21M13 forward”-tagged +352HADH2 | 5′-tgtaaaacgacggccagtCATACCTTGGAAGACTTCCAG-3′ |
| “M13 reverse”-tagged +837HADH2 | 5′-caggaaacagctatgaccAAAGGAAGGGCAGAGGAGC’-3′ |
| “−21M13 forward”-tagged Ex1/2frw | 5′-tgtaaaacgacggccagtCGTGGAGTGGCCGGCGAC-3′ |
| “M13 reverse”-tagged Ex1/2rev | 5′-caggaaacagctatgaccTGACCTCATGCACACCCTGG-3′ |
| “−21M13 forward”-tagged Ex3/4frw | 5′-tgtaaaacgacggccagtGAGATGAATACCTTCTCCAC-3′ |
| “M13 reverse”-tagged Ex3/4rev | 5′-caggaaacagctatgaccagatctAGAGTAGAAGTCATAGGTGG-3′ |
| “−21M13 forward”-tagged Ex5/6frw | 5′-tgtaaaacgacggccagtATGCCTCCTAAGTGACTTG-3′ |
| “M13 reverse”-tagged Ex5/6rev | 5′-caggaaacagctatgaccAAAGGAAGGGCAGAGGAGC-3′ |
| +2284HADH2 frw | 5′-TGGGCACCTTCAATGAGATC-3′ |
| BamHI-tagged forward primer | 5′-aaaggatccaaaATGGCAGCAGCGTGTCGGAG-3′ |
| HindIII-tagged reverse primer | 5′-aaaaagcttTCAAGGCTGCATACGAATGGC-3′ |
Note.— On the basis of the nucleotide sequence of the human gene, primers were selected and used for the amplification of HADH2 from both cDNA and genomic DNA. The +2284HADH2frw primer contains a mismatch (single underlined) which creates a BglII-restriction site in combination with the HADH2 388C→T mutation. The Ex3/4rev primer has an internal BglII restriction site (bold italics) to function as a control. Restriction sites for BamHI and HindIII are shown in italics.
Figure 2.

Detection of HADH2 388C→T mutation by RFLP analysis. The zygosity of the 388C→T mutation found in patients 2 and 3 (table 1) was studied using RFLP analysis. Genomic DNA was amplified using the primers 2284HADH2frw plus “M13 reverse”-tagged Ex3/4rev. PCR conditions were as described for the other genomic DNA amplifications. After amplification, the PCR products were incubated for 2 h at 37°C in the presence (+) or absence (−) of 10 units of BglII, and, afterward, the restriction products were separated on a 2% agarose gel.
Figure 3.

Detection of the HADH2 364C→G mutation by RFLP analysis in a family with MHBD deficiency. The presence of the 364C→G mutation found in patient 4 (table 1) was studied in other family members (father, mother, brother, and sister) and two unrelated control subjects. Therefore, fragment 2 was amplified from genomic DNA, followed by an incubation in the presence (+) or absence (−) of the restriction enzyme HinfI. Finally, the restriction fragments were separated on a 2% agarose gel.
To determine the effect of these mutations on enzyme stability or activity, we first performed immunoblot analysis of MHBD in cultured skin fibroblasts from our group of patients. For antigen preparation, wild-type human MHBD was expressed in E. coli as a maltose-binding protein (MBP) fusion protein. To this end, the complete ORF of human wild-type HADH2 was amplified by use of Taq DNA polymerase, with the BamHI-tagged forward primer and HindIII-tagged reverse primer (table 3). The PCR program used for amplification started with 2 min of denaturation at 94°C; followed by 25 cycles of 30 s at 94°C, 30 s at 55°C, and 1.5 min at 72°C; and a final extension step at 72°C for 2 min. The PCR product was cloned downstream of the isopropyl-1-thio-D-galactopyranoside (IPTG)-inducible PTAC promoter into the BamHI and HindIII sites of the bacterial expression vector pMAL-C2X (New England Biolabs) to express MHBD as a fusion protein with MBP. The complete ORF was sequenced to exclude sequence errors introduced during PCR. Transformed bacteria (INVα) were grown in 100-ml LB medium, supplemented with 100 μg/ml ampicillin to an OD600 of ∼0.5, and IPTG was added to a final concentration of 1 mM to induce protein expression. After 4 h at 37°C, cells were pelleted and resuspended in 20 ml 10-mM sodium phosphate buffer pH 7.4, containing 140 mM NaCl, 0.1% (wt/vol) Triton X-100, and protease inhibitors (2 tablets Completemini [Boehringer Mannheim]). The fusion protein was subsequently purified from the supernatant, according to the protocol of the manufacturer (New England BioLabs), and stored at −20 °C.
Antibodies were raised in rabbits using 100 μg fusion protein per injection, as described elsewhere (Jansen et al. 2000). The result of the immunoblot (fig. 4) revealed that, in all patients with the 388C→T mutation, the amount of MHBD protein is decreased. Apparently, the R130C substitution has a strong effect on MHBD stability and probably results in a more rapid degradation of the enzyme. In the patient with the 364C→G mutation, no significant decrease in the amount of MHBD protein could be observed. Next, heterologous expression studies in E. coli were performed to determine the effect of the substitutions on enzyme activity. The result, depicted in figure 5, shows that the R130C mutation leads to a fully inactive enzyme, whereas some residual activity amounting to 2%–3% of the wild-type enzyme was found in the case of the L122V mutant. These results indicate that the R130C substitution affects both enzyme stability and activity, whereas the L122V substitution mainly effects enzyme activity. This explains the high residual MHBD activity (∼25% of the controls) found in the patient with the L122V substitution and could account for the milder clinical presentation of MHBD deficiency in this patient, compared with the male patients with the R130C mutation. The clinical phenotype of the female patient with the R130C mutant is also milder, compared with the male patients with this mutation, although residual MHBD activity in their cultured skin fibroblasts is comparable. However, the MHBD activity measured in the female patient does not properly reflect the amount of MHBD activity present in other tissues. We believe that, because her clinical phenotype is less severe than that of the male patients, her wild-type allele is silenced, owing to X-chromosome inactivation, in most but not all body tissues.
Figure 4.

Immunological detection of MHBD in cultured skin fibroblasts from patients with MHBD deficiency. For immunoblot analysis, 50 μg protein of cultured skin fibroblasts from control subjects and patients with MHBD deficiency was applied on a 10% polyacrylamide gel and separated, as described elsewhere (Laemmli 1970). After electrophoresis, proteins were transferred to a nitrocellulose membrane by semidry blotting, probed with the polyclonal antibody raised against recombinant human MHBD, and developed (Wanders et al. 1995). After staining of the blot with NBT/BCIP, the result was digitalized by use of a desktop scanner and the amount of MHBD protein quantified, using the National Insitutes of Health Image 1.62 software program. CRIM = cross reactive immunological material. Control-mean value of three control subjects represented as 100% (±SD); 1 = patient 1; 2 = patient 2; 3 = patient 3, and 5 = patient 5 (see table 1).
Figure 5.

Heterologous expression of human MHBD in E. coli. The complete ORF of HADH2 from a control subject and from patients with MHBD deficiency were amplified and ligated into the bacterial expression vector pMAL-C2X, as described. Each ORF was sequenced to exclude sequence errors introduced during PCR. Transformed bacteria (INVα) were grown in 20-ml LB medium supplemented with 100 μg/ml ampicillin to an OD600 of ∼0.5, and IPTG was added to a final concentration of 1 mM to induce protein expression. After 4 h at 37°C, cells were pelleted and resuspended in 1 ml 10 mM sodium phosphate buffer pH 7.4, containing 140 mM NaCl, 0.1% (wt/vol) Triton X-100 and protease inhibitors (1 tablet Completemini [Boehringer Mannheim] in 10-ml solution). Lysis was achieved by sonication at 9 W for 10 s. The bacterial lysate was centrifuged for 10 min at 14,000×gav, and the pellet was discarded. The supernatant was used for protein measurement and MHBD activity, as described. Expression levels of the fusion protein in the bacterial extracts were equal, on the basis of immunoblot analysis (data not shown). pMAL = expression with empty pMAL-c2x vector; pHADH2 = expression with vector containing the wild-type human MHBD; pHADH2 388C→T = expression of human MHBD mutant with R130C; pHADH2 364C→G = expression of human MHBD mutant with L122V.
In conclusion, we have resolved the molecular basis of 2-methyl-3-hydroxybutyric aciduria through the identification of mutations in the HADH2 gene coding for MHBD. This is the first known organic aciduria that is inherited as an X-chromosomal trait. The exact functions of the MHBD protein, apart from its role in isoleucine metabolism, are unclear. The protein was first identified during a two-hybrid screen by its ability to bind to amyloid-β peptide (Aβ), a neurotoxic peptide implicated in the pathogenesis of Alzheimer disease (MIM 104300) (Yan et al. 1997). The protein, was named “endoplasmic reticulum–associated Aβ-binding protein” (ERAB), since it was found predominantly at the endoplasmic reticulum. Its expression was found to be increased in the brain of patients with Alzheimer disease, and ERAB facilitated Aβ cytotoxicity in neuroblastoma and transfected COS cells in vitro. Independent studies by He et al. (1998) had led to the identification of the same protein as the human homolog of bovine L-3-hydroxyacyl-CoA dehydrogenase type II, which was purified and cloned by Hashimoto and coworkers (Kobayashi et al. 1996; Furuta et al. 1997).
The exact role of this 3-hydroxyacyl-CoA dehydrogenase in mitochondrial fatty acid β-oxidation remained unclear at that time, especially since mitochondria are known to already contain two different 3-hydroxyacyl-CoA dehydrogenases for short- and long-chain substrates (see Wanders et al. [1999] and Rinaldo et al. [2002] for review), which are expressed in all tissues. It has also been shown that the enzyme also harbors 17β/3α-hydroxysteroid dehydrogenase activity toward steroid hormones, and, accordingly, the enzyme has been referred to as “17β-hydroxysteroid dehydrogenase type 10” (He et al. 1999). The reactivity of the enzyme with 2-methyl-3-hydroxyacyl-CoA esters, as reported in the present study, adds another function to this protein. The subcellular localization of the protein in mitochondria, rather than in the endoplasmic reticulum, as demonstrated convincingly by several groups (Frackowiak et al. 2001), is in line with the role of MHBD/ERAB in isoleucine metabolism.
The link among MHBD, Alzheimer disease, and the neurodegeneration observed in patients with MHBD deficiency remains unclear. It has been reported that the enzymatic activity of ERAB is central to its capacity to potentiate Aβ toxicity; indeed, a catalytically crippled form of ERAB was ineffective (Yan et al. 1999). In contrast, it has also been reported that deposition of Alzheimer vascular amyloid-β is associated with decreased rather than increased expression of brain ERAB (Ghaedi et al. 1999). On the other hand, high levels of the enzyme were found in hippocampal synaptic mitochondria in transgenic mice that overexpress β-amyloid protein precursor, a model organism of Alzheimer disease, suggesting a possible pathogenetic link between the enzyme and disturbed metabolism of intraneuronal steroid hormones (He et al. 2000). Further work is required to clarify the functional role of MHBD. It appears likely that the multifunctional capacities of the enzyme are at the basis of the unusual clinical abnormalities observed in MHBD-deficient patients as compared with β-ketothiolase deficient patients.
Note added in proof.— More detailed information on patient 5 can be found in the paper entitled “2-Methyl-3-Hydroxybutyryl-CoA Dehydrogenase Deficiency: Impaired Catabolism of Isoleucine Presenting as Neurodegenerative Disease,” by Jörn Oliver Sass, Rosemarie Forstner, and Wolfgang Sperl, which was accepted for publication in Brain and Development after acceptance of the present paper.
Acknowledgments
The authors gratefully acknowledge Mrs. M. Festen and Mrs. S. M. Gersen-van Zadel for expert preparation of the manuscript. We thank H. R. Waterham for critically reading this manuscript.
Electronic-Database Information
Accession numbers and URLs for data presented herein are as follows:
- GenBank and National Center for Biotechnology Information (NCBI), http://www.ncbi.nlm.nih.gov/ (for ERAB [accession numbers U96132 and AF037438] and HADH2 [NM_004493])
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for 2-methylacetoacetyl-CoA thiolase deficiency, 2-methylbutyryl-CoA dehydrogenase deficiency, 2-methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency, and Alzheimer disease)
- Swiss-Prot Protein Knowledgebase, http://www.ebi.ac.uk/swissprot/ (for a protein sequence database)
References
- Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254 [DOI] [PubMed] [Google Scholar]
- Ensenauer R, Niederhoff H, Ruiter JP, Wanders RJ, Schwab KO, Brandis M, Lehnert W (2002) Clinical variability in 3-hydroxy-2-methylbutyryl-CoA dehydrogenase deficiency. Ann Neurol 51:656–659 [DOI] [PubMed] [Google Scholar]
- Frackowiak J, Mazur-Kolecka B, Kaczmarski W, Dickson D (2001) Deposition of Alzheimer’s vascular amyloid-β is associated with decreased expression of brain L-3-hydroxyacyl-coenzyme A dehydrogenase (ERAB). Brain Res 907:44–53 [DOI] [PubMed] [Google Scholar]
- Furuta S, Kobayashi A, Miyazawa S, Hashimoto T (1997) Cloning and expression of cDNA for a newly identified isozyme of bovine liver 3-hydroxyacyl-CoA dehydrogenase and its import into mitochondria. Biochim Biophys Acta 1350:317–324 [DOI] [PubMed] [Google Scholar]
- Ghaedi K, Itagaki A, Toyama R, Tamura S, Matsumura T, Kawai A, Shimozawa N, Suzuki Y, Kondo N, Fujiki Y (1999) Newly identified Chinese hamster ovary cell mutants defective in peroxisome assembly represent complementation group A of human peroxisome biogenesis disorders and one novel group in mammals. Exp Cell Res 248:482–488 [DOI] [PubMed] [Google Scholar]
- Gibson KM, Burlingame TG, Hogema B, Jakobs C, Schutgens RB, Millington D, Roe CR, Roe DS, Sweetman L, Steiner RD, Linck L, Pohowalla P, Sacks M, Kiss D, Rinaldo P, Vockley J (2000) 2-Methylbutyryl-coenzyme A dehydrogenase deficiency: a new inborn error of L-isoleucine metabolism. Pediatr Res 47:830–833 [DOI] [PubMed] [Google Scholar]
- He XY, Merz G, Mehta P, Schulz H, Yang SY (1999) Human brain short chain L-3-hydroxyacyl coenzyme A dehydrogenase is a single-domain multifunctional enzyme. Characterization of a novel 17β-hydroxysteroid dehydrogenase. J Biol Chem 274:15014–15019 [DOI] [PubMed] [Google Scholar]
- He XY, Merz G, Yang YZ, Pullakart R, Mehta P, Schulz H, Yang SY (2000) Function of human brain short chain L-3-hydroxyacyl coenzyme A dehydrogenase in androgen metabolism. Biochim Biophys Acta 1484:267–277 [DOI] [PubMed] [Google Scholar]
- He XY, Schulz H, Yang SY (1998) A human brain L-3-hydroxyacyl-coenzyme A dehydrogenase is identical to an amyloid β-peptide-binding protein involved in Alzheimer’s disease. J Biol Chem 273:10741–10746 [DOI] [PubMed] [Google Scholar]
- IJlst L, Wanders RJ, Ushikubo S, Kamijo T, Hashimoto T (1994) Molecular basis of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: identification of the major disease-causing mutation in the alpha-subunit of the mitochondrial trifunctional protein. Biochim Biophys Acta 1215:347–350 [DOI] [PubMed] [Google Scholar]
- Jansen GA, Hogenhout EM, Ferdinandusse S, Waterham HR, Ofman R, Jakobs C, Skjeldal OH, Wanders RJ (2000) Human phytanoyl-CoA hydroxylase: resolution of the gene structure and the molecular basis of Refsum’s disease. Hum Mol Genet 9:1195–1200 [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Jiang LL, Hashimoto T (1996) Two mitochondrial 3-hydroxyacyl-CoA dehydrogenases in bovine liver. J Biochem 119:775–782 [DOI] [PubMed] [Google Scholar]
- Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- Luo MJ, Mao LF, Schulz H (1995) Short-chain 3-hydroxy-2-methylacyl-CoA dehydrogenase from rat liver: purification and characterization of a novel enzyme of isoleucine metabolism. Arch Biochem Biophys 321:214–220 [DOI] [PubMed] [Google Scholar]
- Mao LF, Chu C, Luo MJ, Simon A, Abbas AS, Schulz H (1995) Mitochondrial β-oxidation of 2-methyl fatty acids in rat liver. Arch Biochem Biophys 321:221–228 [DOI] [PubMed] [Google Scholar]
- Mitchell GA, Fukao T (2001) Inborn errors of ketone body metabolism. In: Scriver CR et al (eds) The metabolic and molecular basis of inherited disease, eighth ed. McGraw-Hill, New York: pp 2327–2356 [Google Scholar]
- Poll-The BT, Duran M, Ruiter JPN, Wanders RJA, Barth PG (2001) Mild cerebral white matter disease associated with 2-methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency. J Inherit Metab Dis 24:59 [Google Scholar]
- Rabilloud T, Carpentier G, Tarroux P (1988) Improvement and simplification of low-background silver staining of proteins by using sodium dithionite. Electrophoresis 9:288–291 [DOI] [PubMed] [Google Scholar]
- Rinaldo P, Matern D, Bennett MJ (2002) Fatty acid oxidation disorders. Annu Rev Physiol 64:477–502 [DOI] [PubMed] [Google Scholar]
- Sass JO, Sperl W (2001) 2-methyl-3-hydroxybutyryl-CoA dehydrogenase (MHBD) deficiency versus β-ketothiolase (MAT) deficiency. J Inherit Metab Dis 24:6011286384 [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, Mann M (1996) Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem 68:850–858 [DOI] [PubMed] [Google Scholar]
- Walsh PS, Metzger DA, Higuchi R (1991) Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques 10:506–513 [PubMed] [Google Scholar]
- Wanders RJ, Dekker C, Ofman R, Schutgens RB, Mooijer P (1995) Immunoblot analysis of peroxisomal proteins in liver and fibroblasts from patients. J Inherit Metab Dis 181:S101–S112 [DOI] [PubMed] [Google Scholar]
- Wanders RJA, Vreken P, Den Boer MEJ, Wijburg FA, Van Gennip AH, IJlst L (1999) Disorders of mitochondrial fatty acyl-CoA beta-oxidation. J Inherit Metab Dis 22:442–487 [DOI] [PubMed] [Google Scholar]
- Yan SD, Fu J, Soto C, Chen X, Zhu H, Al-Mohanna F, Collison K, Zhu A, Stern E, Saido T, Tohyama M, Ogawa S, Roher A, Stern D (1997) An intracellular protein that binds amyloid-β peptide and mediates neurotoxicity in Alzheimer’s disease. Nature 389:689–695 [DOI] [PubMed] [Google Scholar]
- Yan SD, Shi Y, Zhu A, Fu J, Zhu H, Zhu Y, Gibson L, Stern E, Collison K, Al-Mohanna F, Ogawa S, Roher A, Clarke SG, Stern DM (1999) Role of ERAB/L-3-hydroxyacyl-coenzyme A dehydrogenase type II activity in Aβ-induced cytotoxicity. J Biol Chem 274:2145–2156 [DOI] [PubMed] [Google Scholar]
- Zschocke J, Ruiter JP, Brand J, Lindner M, Hoffmann GF, Wanders RJ, Mayatepek E (2000) Progressive infantile neurodegeneration caused by 2-methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency: a novel inborn error of branched-chain fatty acid and isoleucine metabolism. Pediatr Res 48:852–855 [DOI] [PubMed] [Google Scholar]