

Abstract
Osteoarthritis (OA) is the most common human joint disease, characterized by loss and/or remodeling of joint synovium, cartilage, and bone. Here, we describe a genomewide linkage analysis of patients with idiopathic hand OA who were carefully phenotyped for involvement of either or both the distal interphalangeal (DIP) joints and the first carpometacarpal (CMC1) joints. The best linkage peaks were on chromosomes 4q and 3p and on the short arm of chromosome 2. Genomewide significance was reached for a locus on chromosome 2 for patients with affected CMC1 joints (LOD = 4.97); this locus was also significant for patients with OA in both CMC1 and DIP joints (LOD = 4.44). The peak LOD score at this locus coincides with a gene, MATN3, encoding the noncollagenous cartilage extracellular matrix protein, matrilin-3. Subsequent screening of the genomic sequence revealed a missense mutation, of a conserved amino acid codon, changing threonine to methionine in the epidermal growth factor–like domain in matrilin-3. The missense mutation cosegregates with hand OA in several families. The mutation frequency is slightly more than 2% in patients with hand OA in the Icelandic population and has a relative risk of 2.1.
Introduction
Osteoarthritis (OA [MIM 165720]) is the most common form of musculoskeletal disability in the developed countries (Lawrence et al. 1998). Clinical problems include pain and joint stiffness often leading to significant disability and joint replacement (Creamer and Hochberg 1997). OA exhibits a clear predilection for specific joints; it appears most commonly in the hip and knee joints and lumbar and cervical spine, as well as in the distal interphalangeal (DIP) and the first carpometacarpal (CMC1) (base of thumb) and proximal interphalangeal joints of the hand; however, patients with OA may have one, a few, or all of these sites affected. According to a conservative estimate, >40 million people in the United States alone are affected with OA, with >70% of the population at age ⩾65 years being affected by the disease, reflecting its age dependence (Roberts and Burch 1966).
Pathologically, OA is characterized by a dynamic process of destruction and repair of joint tissues. Its hallmark is decreased volume and disorganization of the articular cartilage. Synovial-layer hyperplasia and capsular thickening also occur. Dramatic bone expansion, especially near the edges of the joint (osteophyte formation), is also characteristic and may represent a reaction to the loss of articular cartilage.
Articular cartilage is composed of chondrocytes and the extracellular matrix (ECM). The chondrocytes synthesize and maintain the ECM. The ECM is primarily composed of collagen fibrils (mainly collagen II, with less of types IX and XI) and proteoglycans (especially aggrecan). The intersecting networks of collagens and proteoglycans are linked together with a set of noncollagenous matrix proteins. These networks are also tied to the chondrocytes either directly or, more commonly, through hyaluronic acid and noncollagenous matrix-linking proteins. The collagen network provides the tensile and shear strength to the articular cartilage, whereas the proteoglycan network, with its water-absorbing properties, provides the compressile strength to absorb force perpendicular to the joint surface. Normal articular cartilage may display balance, perhaps regulated by the noncollagenous matrix proteins, between these two opposing networks.
In OA, the earliest histological changes involve edema in the articular cartilage, suggesting an alteration in the balance between the proteoglycan and collagen networks, tilted toward the water-absorbing properties of the former. Indeed, in OA, there is gradual disorganization of the collagen fibril network, leading to loss of integrity of the matrix networks, evident pathologically in moderate and severe OA as cartilage delaminations and clefts. This process advances to include the deep layer of cartilage near the endochondral bone. The joint appears to respond with reparative and compensatory processes—such as chondrocyte proliferation and hypertrophy, as well as fibrocartilage production at the articular bone surface, sometimes leading to bony projections called “osteophytes.”
OA has been considered as a degenerative disease that results from everyday wear and tear or trauma of the joint. Indeed, damaged joint integrity owing to trauma or inflammation will often lead to secondary OA. In addition, large body weight increases the risk of knee OA (Cicuttini et al. 1997). However, numerous epidemiological studies have showed increased risk to relatives of patients with idiopathic OA. These studies include hip OA (Ingvarsson et al. 2000) and hand OA (Stecher 1941). Although there are rare families of patients with OA that have a Mendelian inheritance pattern, these patients have an unusual form of OA usually associated with dwarfism, congenital or early-onset anatomic abnormalities of joint, and osteochondrodysplasia. In general, the common forms of OA appear to skip generations, with unclear patterns of inheritance. Thus, OA most likely represents the confluence of environmental factors and multiple disease genes.
Several studies have suggested that OA of the hand has a significant genetic component. Previous studies found Heberden nodes (osteophytes of the DIP joint) to be three times more common in sisters of affected individuals than in the general population (Stecher 1941), and a genetic contribution has been estimated to be as much as 65% in patients with OA of the hand or knee (Spector et al. 1996; Bijkerk et al. 1999). Furthermore, sisters of patients with OA in the CMC1 joint have an almost sevenfold risk of the development of OA in the same joint (Jonsson et al. 2003).
Several genomewide linkage scans attempting to map genes that contribute to the common forms of OA have been reported. These have focused on either OA of the hand or OA of the large joints. None of the reported scans meet the criteria for genomewide significance. However, these studies reveal several suggestive loci for OA. Hip OA shows suggestive linkage to 2q, 4q (female hip OA), 4q35 (early-onset OA), 6p, 6q, 11q (female OA), 16p, and 16q (both the common form and a familial early-onset form) (Chapman et al. 1999; Leppävuori et al. 1999; Loughlin et al. 1999, 2002; Roby et al. 1999; Ingvarsson et al. 2001). Hand OA shows suggestive linkage to 1p, 2p, 2q12-13, 2q23-35, 4q26-27, 7p15-21, 9q, 11q, 12q, 13q, and X-cen (Wright et al. 1996; Leppävuori et al. 1999; Demissie et al. 2002). Of interest, 2q and 16p have appeared in more than one population. Together, these studies demonstrate that stratification of OA by phenotype appears to bring out loci that may have correlation to particular joints. However, there are probably also genes that contribute to OA in general.
For the present study, we chose to focus on a subtype of OA, namely hand OA, that fulfills rigorous inclusion criteria. We used the genealogical approach to cluster large numbers of patients with well-defined hand OA, and we mapped hand OA to three prominent locations on chromosomes 2, 3, and 4. The present article also describes a mutation in MATN3, the gene that encodes matrilin-3 (MIM 602109), with an association in a minority of patients with hand OA.
Patients and Methods
Patients
A list of patients with OA of the hand was obtained on the basis of patients’ records at hospitals and health care centers in Iceland. The encrypted patient list was cross-referenced with our comprehensive Icelandic genealogy database (Gulcher and Stefansson 1998; Gulcher et al. 2000), and pedigrees with two or more affected individuals related at or within five meioses were identified. Patients within these families and as many as three first-degree relatives were recruited and examined by a rheumatologist (H.J.) or an orthopedic surgeon (T.I.). Additionally, a group of patients and their relatives from another ongoing study of hip OA and knee OA also had their hands examined for the present study. Individuals were classified as having idiopathic hand OA if they met either or both of the following two criteria: (1) OA with a minimum of two nodes at the DIP joints on each hand (hereafter, “DIP phenotype”), or (2) OA of the thumb with squaring or dislocation of a CMC1 joint (hereafter, “CMC1 phenotype”). Only individuals with idiopathic OA were included in the patient cohorts. Included in the linkage analysis were 1,143 affected individuals, along with 939 of their relatives. For the mutational analysis, 1,077 of the affected individuals from the linkage group, along with an additional 1,085 affected individuals and a group of 873 unrelated Icelandic control individuals, participated. The present study was approved by the Data Protection Commission of Iceland and the National Bioethics Committee of Iceland. Informed consent was obtained from all participants.
Microsatellite Markers and Maps
Our framework genomewide scan uses a 1,000-microsatellite-marker set that contains markers from the ABI Linkage Marker (version 2) screening and intercalating sets, in combination with 500 custom-made markers. All markers were extensively tested for robustness, ease of scoring, and efficiency in multiplex PCR. Marker positions were obtained from our genetic map (Kong et al. 2002). All reported marker positions are in Kosambi centimorgans. In our framework set, the average spacing between markers is ∼4 cM. PCR amplifications were set up, run, and pooled on Gilson Cyberlab robots. The reaction volume was 5 μl, and, for each PCR, 20 ng genomic DNA was amplified in the presence of 2 pmol each primer, 0.25 U AmpliTaq Gold, 0.2 mM dNTPs, and 2.5 mM MgCl2 (buffer was supplied by the manufacturer [Applera]). Cycling conditions were as follows: 95°C for 10 min, followed by 37 cycles of 94°C for 15 s, annealing at 55°C for 30 s, and extension at 72°C for 1 min. The PCR products were supplemented with the internal size standard, and the pools were separated and detected on ABI Prism 3700 sequencers by use of Genescan (version 3.0) peak-calling software (Applera). Alleles were automatically called using DAC, an allele-calling program developed at deCode Genetics (Fjalldal et al. 2001), and the program DecodeGT was used to fractionate the called genotypes according to quality and to edit when necessary (Pálsson et al. 1999).
Statistical Methods for Linkage Analysis
We used multipoint, affected-only allele-sharing methods to assess the evidence for linkage. All results, including LOD and nonparametric linkage scores, were obtained using the program Allegro (Gudbjartsson et al. 2000). We used the Spairs scoring function (Whittemore and Halpern 1994; Kruglyak et al. 1996) and an exponential allele-sharing model (Kong and Cox 1997) to generate the relevant 1-df statistics. When combining the family scores to obtain an overall score, instead of weighting the families equally (the default of Genehunter [Kruglyak et al. 1996]) or weighting the affected pairs equally, we used a weighting scheme that is halfway between the two in the log scale; our family weights are the geometric means of the weights of the two schemes. Although not identical, this weighting scheme tends to give similar results to that proposed by Weeks and Lange (1988) as an extension of a weighting scheme of Hodge (1984) designed for sibships. We computed the P value in two different ways and report the less significant one. The first P value was computed on the basis of large sample theory; 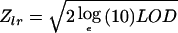 is distributed approximately as a standard normal random variable under the null hypothesis of no linkage (Kong and Cox 1997). Furthermore, because of the concern with small-sample behavior, we computed a second P value by comparing the observed LOD score to its complete data sampling distribution under the null hypothesis (Gudbjartsson et al. 2000). When a data set consists of more than a handful of families, which is the case here, these two P values tend to be very similar. To ensure that the results were a true reflection of the information contained in the material (i.e., for us to consider a linkage result significant), we required not only that the P value be <2×10-5 (Lander and Kruglyak 1995) but also that the information content in the region be ⩾85%. For the families in the present study, an information content of 85% corresponded to a marker density of approximately one marker every centimorgan. The information measure that we used has been defined elsewhere (Nicolae 1999) and is implemented in Allegro. This measure is closely related to a classical measure of information (Dempster et al. 1977), having the property that it is between 0, if the marker genotypes are completely uninformative, and 1, if the genotypes determine the exact amount of allele sharing by descent among the affected relatives.
is distributed approximately as a standard normal random variable under the null hypothesis of no linkage (Kong and Cox 1997). Furthermore, because of the concern with small-sample behavior, we computed a second P value by comparing the observed LOD score to its complete data sampling distribution under the null hypothesis (Gudbjartsson et al. 2000). When a data set consists of more than a handful of families, which is the case here, these two P values tend to be very similar. To ensure that the results were a true reflection of the information contained in the material (i.e., for us to consider a linkage result significant), we required not only that the P value be <2×10-5 (Lander and Kruglyak 1995) but also that the information content in the region be ⩾85%. For the families in the present study, an information content of 85% corresponded to a marker density of approximately one marker every centimorgan. The information measure that we used has been defined elsewhere (Nicolae 1999) and is implemented in Allegro. This measure is closely related to a classical measure of information (Dempster et al. 1977), having the property that it is between 0, if the marker genotypes are completely uninformative, and 1, if the genotypes determine the exact amount of allele sharing by descent among the affected relatives.
Mutational and Association Analyses
To identify polymorphisms within the MATN3 gene, primers were designed for PCR amplification of all known exons and the promoter sequence of the MATN3 gene (table 1). DNA from 76 patients from families that scored positive in a nonparametric linkage analysis for the markers under the peak of the LOD score and DNA from 18 control individuals were initially sequenced for polymorphism within the gene. Both the forward and reverse strands were sequenced on an ABI Prism 3700 DNA analyzer. Some of the variants identified were further examined for association with hand OA, usually by using a SNP assay applied to a follow-up cohort of 745 patients and 368 control individuals and applying the fluorescent polarization (FP) method (Chen et al. 1999). For the genotyping of SNPs, markers were designed adjacent to each polymorphic site (table 2) and were genotyped using the FP method. One variant was further assessed by FP in all patients with hand OA and in a total of 873 control individuals. A microsatellite marker, Indel1, was designed around a polymorphic repeat in the purported Sp1 promoter element (Wagener et al. 2000), resulting in one or two putative Sp1 elements.
Table 1.
Primers Used for PCR Amplification of MATN3 Exons and Neighboring Regions for Detection of Nucleotide Variation
| Position | Forward Primer(5′→ 3′) | Reverse Primer(5′→ 3′) |
| Exon 1 | AGTTTCCCCCACAGGATTCA | CCCAGACATTCCGTTTCTGC |
| Exon 2 | TGTTTAGGTTGGTCGCACACAC | GTCGAATCACACCCTTGTCAAC |
| Exon 2 | GTCGAATCACACCCTTGT | GCCTCCCATCTGTAACAA |
| Exon 2 | GCCTCCCATCTGTAACAATGAT | ATGTTGGGGTCAGGAGATGC |
| Exon 3 | TTCAGGGAAAACTGCTGA | AGCATTGCTAATAAGCCAG |
| Exon 3 | TTCAGGGAAAACTGCTGATGAA | AGCATTGCTAATAAGCCAGAGG |
| Exon 4 | TTTGGGCCGAGATACATACA | GCCCCTCCCTTGTTTCATTAG |
| Exon 5 | TGAGCAATGTGTGCCTTCATCT | CAGTGCCAATGGCTTTGAATAG |
| Exon 6 | TCTGACAGGGAGAAAGAATCACA | GGCCTGTTGCTGACTACTGACT |
| Exon 7 | TAGGGGACAAAATAGGCAGCTT | CCTCTTGGAAAATGCACTTAGG |
| Exon 8 | TTGAAGCCTTTCTTTTGAACCT | TTTGCTTCAGGATCCAAGTGAC |
| Exon 8 | AAAAAGTTAGACTTTCAAGCCTGT | TGACTGTGAAATGATTGGTAGGAA |
| Exon 8 | AGACCTAAAGTTTCCATTGTGAAT | TGTGCTCTCTGAAGAACAGGATTT |
| Indel1 | GTTTCTGCCGCTGGAATG | CCAGAGCAGCAGGAGGAG |
Table 2.
Sequence Polymorphisms in MATN3, along with Estimated Relative Risks and Patient and Control Frequencies—in an Initial Screening Set of 78 Patients and 18 Control Individuals and in a Follow-Up Cohort of 745 Patients and 368 Control Individuals[Note]
|
Initial Screening Set |
Follow-Up Cohort |
||||||||
| Frequency among |
Frequency among |
||||||||
| Marker (Position in AC079145a) | Polymorphism | PredictedAmino Acid Change | Allele | RelativeRisk | AffectedIndividuals(N=78) | ControlIndividuals(N=18) | RelativeRisk | AffectedIndividuals(N=745) | ControlIndividuals(N=368) |
| Indel1 (38383) | gcgggg(cggggcgggg)cccgag | Insertion | 1.15 | .535 | .500 | 1.16 | .697 | .665 | |
| SNP1 (38496) | gcctcYcggga | Ser/Pro | T | ∞ | .007 | .000 | |||
| SNP2 (45010) | caggcYgtggg | T | 1.10 | .412 | .389 | 1.17 | .424 | .387 | |
| SNP3 (45178) | gaatgaRgtggc | A | 1.10 | .412 | .389 | 1.16 | .425 | .390 | |
| SNP4 (45317) | gtcattRagaaa | Glu/Lys | A | 2.11 | .987 | .972 | 1.24 | .977 | .980 |
| SNP5 (47928) | agaaaaYgtgtt | Met/Thr | T | ∞ | .067 | .000 | 4.34 | .017 | .004 |
| SNP6b (47929) | gaaaacRtgttc | G | 1.62 | .787 | .694 | ||||
| SNP6b (47929) | gaaaacRtgttc | A | 1.32 | .279 | .226 | ||||
Note.— For details on the sequence polymorphisms shown here, see the IUPAC Ambiguity Codes Web site.
For details, see GenBank (sequences AC079145, 7844257, 7844258, 7844259, 7844260, 7844261, 7844262, and 7844263).
For SNP6, the at-risk allele differed in the screening set and in the follow-up cohort.
Haplotype Analysis
To handle missing genotypes and uncertainty as to phase, we applied our own implementation of the likelihood approach (Stefansson et al. 2002), using the expectation-maximization algorithm as a computational tool, to estimate the haplotype frequencies (Dempster et al. 1977). Under the null hypothesis, the affected individuals and control individuals are assumed to have identical frequencies of all haplotypes. Under the alternative hypothesis, the candidate at-risk haplotype is allowed to have a higher frequency in affected individuals than in control individuals, whereas the ratios of the frequencies of all other haplotypes are assumed to be the same in both groups. Likelihoods are maximized under both hypotheses, and a corresponding 1-df likelihood-ratio statistic is used to evaluate statistical significance.
Results
Genome Scan
A genomewide linkage scan was performed for 329 families—altogether containing 1,143 individuals with idiopathic hand OA, along with 939 genotyped relatives. Each family contained at least two affected individuals related to each other at or within five meioses. The results of the linkage scan are displayed in figure 1. The highest LOD score was observed on chromosome 4 (LOD = 2.61 [D4S3046, at 155.9 cM]). The next highest LOD scores were obtained on chromosomes 3 (LOD = 1.79 [D3S3551, at 96.8 cM]) and 2 (LOD = 1.48 [D2S146, at 51.5 cM]). Other peaks with LOD scores >1 were obtained on chromosomes 6 and 7.
Figure 1.

Framework genomewide scan for 329 families with 1,143 individuals with idiopathic hand OA, using a 1,000-marker screening set. The allele-sharing LOD score is given on the Y-axis, and the genetic location (in Kosambi cM) is given on the X-axis.
To study the effects that the subphenotypes had on the linkage results, we performed three additional genomewide scans, in which individuals were considered affected if they had the DIP phenotype (hereafter, “DIP cohort”), the CMC1 phenotype (hereafter, “CMC1 cohort”), or both the DIP and CMC1 phenotypes (hereafter, “DIP/CMC1 cohort”). The DIP cohort scan included 944 affected individuals in 274 families, the CMC1 cohort scan included 558 affected individuals in 204 families, and the DIP/CMC1 cohort scan included 382 affected individuals in 142 families.Table 3 indicates the location and size of all peaks with a LOD score >1 for these four genomewide scans. Only three locations—one on each of chromosomes 2, 3, and 4—achieved a LOD score ⩾2 in at least one of the four scans. The LOD scores in these locations are 2.23 at D2S2168, on chromosome 2, for the CMC1 cohort; 2.20 at D3S1566, on chromosome 3, for the DIP cohort; and 2.61 at D4S3046 and 2.58 at D4S2980, on chromosome 4, for the hand-OA and DIP cohorts, respectively. Some of our peaks may overlap with the previously reported location for linkage (see table 4). In particular, our linkage to the p arm of chromosome 2 is close to a peak that has recently been reported by Demissie et al. (2002). We added markers in these three regions to increase the information content on allele-sharing among affected relatives. The results of the linkage scans with the additional markers on chromosomes 2, 3, and 4 are displayed in figure 2. These fine-mapping results show that the evidence for linkage on chromosome 3 changed very little—decreasing slightly for the hand-OA and DIP cohorts and increasing slightly for the other two cohorts. Chromosome 4 exhibited slightly decreased evidence for linkage in the CMC1 cohort but increasing evidence in the other cohorts, with a LOD score >3 in the DIP cohort (LOD = 3.29 [D4S2982]). The most striking result, however, was the increased evidence for linkage on chromosome 2 for all cohorts, with a LOD score of 4.97 between D2S175 and D2S2201 for the CMC1 cohort (P=8.5×10-7). This latter LOD score remains significant even after correction for the four genomewide scans. For the CMC1 cohort, the size of the region, on chromosome 2, that has a LOD score within one of the peak LOD scores is a little more than 5 cM, from D2S175 (at 41.9 cM) to D2S1324 (at 47.1 cM). Table 5 summarizes the fine-mapping linkage results for these three loci, indicating the peak marker(s) and its genetic locations.
Table 3.
Summary of LOD Scores >1 for the Framework Genomewide Scans of Four Hand-OA Cohorts
| Cohort and LOD Score | Chromosome (Marker) | Location(cM) |
| Hand OA: | ||
| 1.48 | 2 (D2S146) | 51.5 |
| 1.14 | 2 (D2S2277) | 160.4 |
| 1.79 | 3 (D3S3551) | 96.8 |
| 1.00 | 3 (D3S1279) | 160.2 |
| 2.61 | 4 (D4S3046) | 155.9 |
| 1.05 | 6 (D6S270) | 134.9 |
| 1.08 | 7 (D7S664) | 26.0 |
| 1.14 | 7 (D7S559) | 183.0 |
| DIP: | ||
| 1.13 | 2 (D2S2324) | 160.8 |
| 2.20 | 3 (D3S1566) | 94.8 |
| 2.58 | 4 (D4S2980) | 153.9 |
| 1.33 | 6 (D6S460) | 90.6 |
| 1.20 | 6 (D6S270) | 134.9 |
| 1.02 | 7 (D7S798) | 169.9 |
| 1.18 | 11 (D11S987) | 72.2 |
| CMC1: | ||
| 2.23 | 2 (D2S2168) | 48.0 |
| 1.15 | 3 (D3S3614) | 99.1 |
| 1.70 | 4 (D4S3046) | 155.9 |
| DIP/CMC1: | ||
| 1.17 | 3 (D3S3614) | 99.1 |
| 1.09 | 3 (D3S1565) | 177.9 |
| 1.24 | 4 (D4S3046) | 155.9 |
Table 4.
Summary of Three Genomewide Linkage Scans for Various OA Phenotypes
| Chromosome (Marker[s]a) | Locationb(cM) | Evidencec | Referenced |
| 1 (D1S1665) | 99.6 | LOD = 2.96 | Demissie et al. 2002 |
| 2 (D2S405) | 51.5 | LOD = 2.23 | Demissie et al. 2002 |
| 2 (D2S1399) | 155 | P = .007 | Leppävuori et al. 1999 |
| 2 (IL1R1) | 116.1 | P = .0001 | Leppävuori et al. 1999 |
| 4 (D4S2394) | 128.1 | P = .0001 | Leppävuori et al. 1999 |
| 7 (D7S664–D7S673) | 13–39.5 | P = .001 | Leppävuori et al. 1999 |
| 7 (D7S817) | 52.2 | LOD = 2.32 | Demissie et al. 2002 |
| 9 (D9S1122) | 74.3 | LOD = 2.29 | Demissie et al. 2002 |
| 11 (D11S901) | 88.5 | MLS = 3.51 | Chapman et al. 1999 |
Marker names as reported in the respective studies.
Our estimates (in Kosambi cM).
All linkage peaks with multipoint LOD score (MLS) >2 or P value <.01, as reported in the three genomewide linkage scans.
Chapman et al. studied families with at least two siblings with total knee or total hip replacement, Leppävuori et al. studied families with members affected with radiographic OA in DIP joints, and Demissie et al. used Kellgren/Lawrence grading, osteophytes, and/or joint-space narrowing in hand joints.
Figure 2.

Fine-mapping results for chromosomes 2, 3, and 4 for the cohorts with idiopathic hand OA and the DIP, CMC1, and DIP/CMC1 phenotypes. The allele-sharing LOD score is given on the Y-axis, and the genetic location (in Kosambi cM) is given on the X-axis.
Table 5.
Summary of Peak LOD Scores after Fine Mapping in Four Hand-OA Cohorts
| Locus and Cohort | LOD Score | Peak Location (Marker[s]) |
| Chromosome 2: | ||
| Hand OA | 2.42 | 44.0 (D2S175, D2S2201) |
| DIP | 2.44 | 44.0 (D2S175, D2S2201) |
| CMC1 | 4.97 | 44.0 (D2S175, D2S2201) |
| DIP/CMC1 | 4.44 | 44.0 (D2S175, D2S2201) |
| Chromosome 3: | ||
| Hand OA | 1.62 | 96.1 (D3S1562) |
| DIP | 1.84 | 96.1 (D3S1562) |
| CMC1 | 1.42 | 107.2 (D3S1274) |
| DIP/CMC1 | 1.27 | 107.2 (D3S3049) |
| Chromosome 4: | ||
| Hand OA | 2.91 | 154.1 (D4S2982) |
| DIP | 3.29 | 154.1 (D4S2982) |
| CMC1 | 1.60 | 155.9 (D4S3046) |
| DIP/CMC1 | 1.42 | 155.5 (D4S2993) |
Mutational Analysis
On the basis of these results—and, primarily, the result in the CMC1 cohort—we have an ongoing association analysis at the chromosome 2 locus. In addition, we have also searched for possible nearby candidate genes. Six publicized genes were found to be within a 4-Mb region centered on the chromosome 2 peak (see the Human Genome Browser Gateway Web site). One of the genes, MATN3, is located within 100 kb of the LOD-score peak, and a recent publication implicated mutations in this gene in a class of dysplasias of large joints with associated early-onset OA (Chapman et al. 2001). MATN3 encodes matrilin-3, a noncollagenous ECM protein with restricted expression in articular cartilage and bone tissues (Klatt et al. 2000). We screened 76 patients and 18 control individuals for mutations in the exons of MATN3 and identified six exonic SNPs and a putative promoter polymorphism that is biallelic—consisting of an insertion or deletion of two pentanucleotide repeats (table 2, “Initial Screening Set”). The initial screening indicated one very rare SNP (namely SNP1, with allele frequency <1% in patients) and one promising SNP (namely SNP5, with allele frequency >6% in patients and nearly nonexistent in control individuals). We decided to test five of the SNPs and the promoter in a larger cohort, of 745 patients and 368 control individuals. Only one of the variants, SNP5, showed more than marginal excess in patients (table 2, “Follow-Up Cohort”) and involves a nucleotide change from cytidine to thymidine in the third exon of the gene, predicting a substitution of a threonine by a methionine residue at position 303 (hereafter, “T303M”) in the first epidermal growth factor (EGF) domain of the protein (fig. 3). This threonine residue is conserved in all four of the EGF domains in the coding sequences for MATN3 in human, chicken, and mouse (fig. 4). The observed frequency of just under 2% for this SNP seemed more likely than the previously observed frequency for the screening set, since the screening set was weighted toward the patients in families with linkage. To more fully investigate the contribution that this mutation makes to hand-OA risk in Iceland, we decided to type the entire patient set. A total of 2,162 patients and 873 control individuals were typed for this coding SNP. Among the patients, 1,312 had the CMC1 phenotype. The results of the mutation screening are displayed in table 6. Of the 873 control individuals, 9 were heterozygous and 0 were homozygous for the mutation, whereas, of the 2,162 patients, 43 were heterozygous and 2 were homozygous for the mutation. The estimated relative risk of hand OA for carriers of a single copy of the mutation as compared to the noncarrier under the multiplicative model is 2.12. Both of the homozygous carriers and 31 of the 43 patients heterozygous for the mutation had the CMC1 phenotype. This led to an estimated relative risk for the CMC1 phenotype of 2.61, which is slightly higher than that for idiopathic hand OA.
Figure 3.

Allelic variation in MATN3. The top chromatogram shows the nucleotide sequence for an individual homozygous for the allele for the predicted missense mutation resulting in methionine residue (ATG codon). The center chromatograph depicts the sequence for an individual heterozygous for the allele, and the bottom chromatograph shows the sequence for an individual homozygous for the threonine residue (ACG codon). The nucleotide highlighted in red depicts the polymorphic allele.
Figure 4.

Alignment of amino acid residues for all four EGF domains of matrilin-3 from human (HuEGF1–4), mouse (MouEGF1–4), and chicken (ChEGF1–4). Residues conserved in all EGF domains are highlighted in green. The predicted missense mutation at position 303 in MATN3 from human protein sequence changes the threonine residue (boldface) to methionine.
Table 6.
Association Analysis of a MATN3 Mutation, SNP5, in Four Hand-OA Cohorts
| Cohort | Relative Risk (95% CI)a | No. of Affected Individuals | No. of Mutations among Affected Individuals | No. of Control Individuals | No. of Mutations among Control Individuals |
| Hand OA | 2.12 (.92–4.86) | 2,162 | 47 | 873 | 9 |
| DIP | 2.06 (.85–4.97) | 1,801 | 38 | 873 | 9 |
| CMC1 | 2.61 (1.05–6.48) | 1,312 | 35 | 873 | 9 |
| DIP/CMC1 | 2.67 (1.07–6.68) | 951 | 26 | 873 | 9 |
Adjusted for family relationships.
We observed that 30 of the 45 carriers of this mutation were in the families with linkage, including both homozygous carriers. We then performed a linkage analysis for the CMC1 cohort less the mutation carriers, to assess the effect that these carriers had on the locus. The LOD score at the chromosome 2 peak dropped to 3.80, which demonstrates that, although these carriers have a significant impact on the linkage, there are likely to be as-yet-undetected associations between hand OA and either MATN3 or other genes in the region.
We also assessed the common haplotype background of the gene for association to idiopathic hand OA. Using the three exonic SNPs with minor-allele frequency >5% and the promoter polymorphism, five haplotypes with estimated allelic frequency of >1% in the population were identified, accounting for >99% of all haplotypes in the region. The haplotypes and their estimated risks are displayed in table 7. Only one of these haplotypes shows a slight and insignificant excess risk. Interestingly, it is the second least frequent haplotype that is the background for the mutation that we report. Considering only the subset of patients with the CMC1 phenotype has no appreciable effect on these results. We continue to investigate this region for other at-risk variants.
Table 7.
Association Study of MATN3 Haplotypes Common to Idiopathic Hand OA in a Follow-Up Cohort of 745 Patients and 368 Control Individuals
|
Frequency among |
Allele at |
||||||
| Label | RelativeRisk | AffectedIndividuals(N=745) | ControlIndividuals(N=368) | Indel1 | SNP1 | SNP2 | SNP6 |
| Hap1 | .92 | .288 | .306 | Insertion | C | G | G |
| Hap2 | .88 | .284 | .310 | Deletion | C | G | G |
| Hap3 | 1.31 | .273 | .223 | Insertion | T | A | A |
| Hap4a | .93 | .132 | .140 | Insertion | T | A | G |
| Hap5 | .49 | .012 | .016 | Deletion | T | A | G |
Haplotype background of reported mutation.
Discussion
MATN3 is an interesting candidate gene for OA. It is a noncollagenous ECM protein that is one of a class of four related proteins, termed “matrilin-1” through “matrilin-4.” All four matrilins are expressed in the developing skeletal system, but matrilin-3 exhibits the expression pattern most restricted to developing cartilage, especially the epiphyseal cartilage. The matrilins are composed of von Willebrand factor A (vWFA) domains, EGF-like repeats, and a C-terminal α-helical coiled-coil domain. Matrilin-3 is composed of a single N-terminal vWFA domain followed by four EGF repeats and the coiled-coil domains, whereas the other matrilins are each composed of two vWFA domains, separated by 1–10 EGF repeats, followed by the C-terminal coiled-coil domain. The coiled-coil domains mediate covalent multimer formation among the matrilins, through their heptad repeats and two cysteines. The matrilins form homomultimers and heteromultimers in almost every combination with each other in proportion to the concentration of each subunit. Matrilin-3 forms heteromultimers only with matrilin-1, and these are heterotetramers with two subunits each. The vWFA domain is a collagen binding domain in other proteins, and matrilin-1 has been shown to bind to type II collagen fibrils in cartilage in a periodic pattern. Matrilin-1 also interacts with aggrecan and may also bind to integrin α1β1 (Makihira et al. 1999). Therefore, matrilin-1 may represent the link between the collagen fibril network and the proteoglycan network, as well as a connection to the chondrocytes. Indeed, mice without matrilin-1 develop normally but show ultrastructural changes in fibril networks in cartilage (Huang et al. 1999). The gene that encodes matrilin-1 would be an excellent candidate gene for OA on the basis of its interaction with the collagen and proteoglycan networks. Several groups have analyzed the association between hip OA and a microsatellite polymorphism in the 3′ UTR of the gene that encodes matrilin-1. A Dutch cohort, stratified on males, found a significant association between an allelic polymorphism and hip OA. In contrast, British and Argentinean cohort studies failed to replicate these results in their population (Meulenbelt et al. 1997; Loughlin et al. 2000; Strusberg et al. 2002).
Matrilin-3 forms heterotetramers with matrilin-1, with higher affinity than either of the respective homomultimers, and therefore may play a modulating role in the cross-linking function of matrilin-1. Whereas matrilin-1 and matrilin-3 expression patterns overlap in cartilage, matrilin-3 expression is higher than matrilin-1 expression in the proliferation zone, where there are mainly noncollagenous filaments, whereas matrilin-1 has the higher expression in the mature zone, where the collagen-proteoglycan network is more extensive. If matrilin-1 facilitates collagen fibril formation and binding to proteoglycan, then matrilin-3 may play an inhibitory role in normal development and maintenance of cartilage and bone. If there is too much matrilin-3 because of increased synthesis or decreased breakdown, then these matrilin-1 roles may be impaired. It has recently been shown that there is increased expression of matrilin-3 in joint cartilage of patients with OA, both at the RNA level and at the protein level (Pullig et al. 2002). This could certainly be a reaction of the chondrocytes to an underlying process in OA. However, our data suggest that matrilin-3 may be primarily involved in OA, at least in some patients with OA. Although we have found the T303M mutation in only a small percentage of patients, there may be other mutations affecting regulatory sequences of matrilin-3. Other genes controlling expression, processing, and turnover of matrilin-3 may lead to the increased matrilin-3 observed in OA joints. The T303M mutation in the first EGF repeat may favor heterotetramer formation of both matrilin-1 and matrilin-3 over homomultimer formation of each. Recent work has showed that the number of EGF repeats in matrilin-3 does not affect the formation of homotrimers; however, that study did not address a possible regulatory role for EGF repeats on heterotetramer formation (Zhang and Chen 2000). Previously characterized mutations in the coding region for the vWFA domain of matrilin-3 cause a rare autosomal dominant form of multiple epiphyseal dysplasia and short stature with early onset of OA in adulthood (Chapman et al. 2001). These mutations have a high penetrance and mainly affect the hips and knees (Mortier et al. 2001), suggesting that a mutation in the presumed collagen II binding domain of matrilin-3 causes developmental abnormalities in the skeletal system. In contrast, the T303M mutation reported here may represent a more subtle variant of matrilin-3, since it does not appear to cause skeletal deformities, epiphyseal dysplasia, or short stature; instead, it appears to contribute to an adult-onset OA that affects the hand joints preferentially, and it is not associated with an early onset or unusual form of OA. The full population-attributable risk of matrilin-3 mutations will require more-extensive screening of the gene in the Icelandic population and in other populations, but, if confirmed, this mutation would represent the first mutation that contributes to a common form of OA.
Acknowledgments
We thank the patients and their families for their contribution to making this project possible. Furthermore, we thank the DNA Isolation and Genotyping Facility service groups at deCode Genetics for their work.
Electronic-Database Information
Accession numbers and URLs for data presented herein are as follows:
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for aligned matrilin-3 sequences [accession numbers AJ224741, Y10521, and AJ000055], matrilin-3 SNPs and exons [accession number AC079145], and STSs with polymorphic nucleotides used for haplotype analysis [accession numbers 7844257, 7844258, 7844259, 7844260, 7844261, 7844262, and 7844263])
- Human Genome Browser Gateway, http://genome.ucsc.edu/cgi-bin/hgGateway?db=hg12
- IUPAC Ambiguity Codes, http://www.ncbi.nlm.nih.gov/SNP/iupac.html
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for OA and matrilin-3)
References
- Bijkerk C, Houwing-Duistermaat JJ, Valkenburg HA, Meulenbelt I, Hofman A, Breedveld FC, Pols HA, van Duijn CM, Slagboom PE (1999) Heritabilities of radiologic osteoarthritis in peripheral joints and of disc degeneration of the spine. Arthritis Rheum 42:1729–1735 [DOI] [PubMed] [Google Scholar]
- Chapman KL, Mortier GR, Chapman K, Loughlin J, Grant ME, Briggs MD (2001) Mutations in the region encoding the von Willebrand factor A domain of matrilin-3 are associated with multiple epiphyseal dysplasia. Nat Genet 28:393–396 [DOI] [PubMed] [Google Scholar]
- Chapman K, Mustafa Z, Irven C, Carr AJ, Clipsham K, Smith A, Chitnavis J, Sinsheimer JS, Bloomfield VA, McCartney M, Cox O, Cardon LR, Sykes B, Loughlin J (1999) Osteoarthritis-susceptibility locus on chromosome 11q, detected by linkage. Am J Hum Genet 65:167–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Levine L, Kwok PY (1999) Fluorescence polarization in homogeneous nucleic acid analysis. Genome Res 9:492–498 [PMC free article] [PubMed] [Google Scholar]
- Cicuttini FM, Spector T, Baker J (1997) Risk factors for osteoarthritis in the tibiofemoral and patellofemoral joints of the knee. J Rheumatol 24:1164–1167 [PubMed] [Google Scholar]
- Creamer P, Hochberg MC (1997) Osteoarthritis. Lancet 350:503–508 [DOI] [PubMed] [Google Scholar]
- Demissie S, Cupples LA, Myers R, Aliabadi P, Levy D, Felson DT (2002) Genome scan for quantity of hand osteoarthritis: the Framingham Study. Arthritis Rheum 46:946–952 [DOI] [PubMed] [Google Scholar]
- Dempster AP, Laird NM, Rubin DB (1977) Maximum likelihood from incomplete data via the EM algorithm. J R Stat Soc B 39:1–38 [Google Scholar]
- Fjalldal JB, Sigurdsson J, Benediktsson K, Ellingssen LM (2001) Automated genotyping: combining neural networks and decision trees to perform robust allele calling. Proc Int Joint Conf Neural Networks A1–A6 [Google Scholar]
- Gudbjartsson DF, Jonasson K, Frigge ML, Kong A (2000) Allegro, a new computer program for multipoint linkage analysis. Nat Genet 25:12–13 [DOI] [PubMed] [Google Scholar]
- Gulcher JR, Kristjansson K, Gudbjartsson H, Stefansson K (2000) Protection of privacy by third-party encryption in genetic research in Iceland. Eur J Hum Genet 8:739–742 [DOI] [PubMed] [Google Scholar]
- Gulcher J, Stefansson K (1998) Population genomics: laying the groundwork for genetic disease modeling and targeting. Clin Chem Lab Med 36:523–527 [DOI] [PubMed] [Google Scholar]
- Hodge SE (1984) The information contained in multiple sibling pairs. Genet Epidemiol 1:109–122 [DOI] [PubMed] [Google Scholar]
- Huang X, Birk DE, Goetinck PF (1999) Mice lacking matrilin-1 (cartilage matrix protein) have alterations in type II collagen fibrillogenesis and fibril organization. Dev Dyn 216:434–441 [DOI] [PubMed] [Google Scholar]
- Ingvarsson T, Stefansson SE, Gulcher JR, Jonsson HH, Jonsson H, Frigge ML, Palsdottir E, Olafsdottir G, Jonsdottir T, Walters GB, Lohmander LS, Stefansson K (2001) A large Icelandic family with early osteoarthritis of the hip associated with a susceptibility locus on chromosome 16p. Arthritis Rheum 44:2548–2555 [DOI] [PubMed] [Google Scholar]
- Ingvarsson T, Stefansson SE, Hallgrimsdottir IB, Frigge ML, Jonsson H Jr, Gulcher J, Jonsson H, Ragnarsson JI, Lohmander LS, Stefansson K (2000) The inheritance of hip osteoarthritis in Iceland. Arthritis Rheum 43:2785–2792 [DOI] [PubMed] [Google Scholar]
- Jonsson H, Manolescu I, Stefansson SE, Ingvarsson I, Jonsson HH, Manolescu A, Gulcher J, Stefansson K (2003) The inheritance of hand osteoarthritis in Iceland. Arthritis Rheum 48:391–395 [DOI] [PubMed] [Google Scholar]
- Klatt AR, Nitsche DP, Kobbe B, Morgelin M, Paulsson M, Wagener R (2000) Molecular structure and tissue distribution of matrilin-3, a filament-forming extracellular matrix protein expressed during skeletal development. J Biol Chem 275:3999–4006 [DOI] [PubMed] [Google Scholar]
- Kong A, Cox NJ (1997) Allele-sharing models: LOD scores and accurate linkage tests. Am J Hum Genet 61:1179–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong A, Gudbjartsson DF, Sainz J, Jonsdottir GM, Gudjonsson SA, Richardsson B, Sigurdardottir S, Barnard J, Hallbeck B, Masson G, Shlien A, Palsson ST, Frigge ML, Thorgeirsson TE, Gulcher JR, Stefansson K (2002) A high-resolution recombination map of the human genome. Nat Genet 31:241–247 [DOI] [PubMed] [Google Scholar]
- Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES (1996) Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet 58:1347–1363 [PMC free article] [PubMed] [Google Scholar]
- Lander E, Kruglyak L (1995) Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11:241–247 [DOI] [PubMed] [Google Scholar]
- Lawrence RC, Helmick CG, Arnett FC, Deyo RA, Felson DT, Giannini EH, Heyse SP, Hirsch R, Hochberg MC, Hunder GG, Liang MH, Pillemer SR, Steen VD, Wolfe F (1998) Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum 41:778–799 [DOI] [PubMed] [Google Scholar]
- Leppävuori J, Kujala U, Kinnunen J, Kaprio J, Nissila M, Heliovaara M, Klinger N, Partanen J, Terwilliger JD, Peltonen L (1999) Genome scan for predisposing loci for distal interphalangeal joint osteoarthritis: evidence for a locus on 2q. Am J Hum Genet 65:1060–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughlin J, Dowling B, Mustafa Z, Smith A, Sykes B, Chapman K (2000) Analysis of the association of the matrillin-1 gene (CRTM) with osteoarthritis: comment on the article by Meulenbelt et al. Arthritis Rheum 43:1423–1425 [DOI] [PubMed] [Google Scholar]
- Loughlin J, Mustafa Z, Dowling B, Southam L, Marcelline L, Raina SS, Ala-Kokko L, Chapman K (2002) Finer linkage mapping of a primary hip osteoarthritis susceptibility locus on chromosome 6. Eur J Hum Genet 10:562–568 [DOI] [PubMed] [Google Scholar]
- Loughlin J, Mustafa Z, Irven C, Smith A, Carr AJ, Sykes B, Chapman K (1999) Stratification analysis of an osteoarthritis genome screen—suggestive linkage to chromosomes 4, 6, and 16. Am J Hum Genet 65:1795–1798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makihira S, Yan W, Ohno S, Kawamoto T, Fujimoto K, Okimura A, Yoshida E, Noshiro M, Hamada T, Kato Y (1999) Enhancement of cell adhesion and spreading by a cartilage-specific noncollagenous protein, cartilage matrix protein (CMP/matrilin-1), via integrin α1β1. J Biol Chem 274:11417–11423 [DOI] [PubMed] [Google Scholar]
- Meulenbelt I, Bijkerk C, de Wildt SC, Miedema HS, Valkenburg HA, Breedveld FC, Pols HA, Te Koppele JM, Sloos VF, Hofman A, Slagboom PE, van Duijn CM (1997) Investigation of the association of the CRTM and CRTL1 genes with radiographically evident osteoarthritis in subjects from the Rotterdam study. Arthritis Rheum 40:1760–1765 [DOI] [PubMed] [Google Scholar]
- Mortier GR, Chapman K, Leroy JL, Briggs MD (2001) Clinical and radiographic features of multiple epiphyseal dysplasia not linked to the COMP or type IX collagen genes. Eur J Hum Genet 9:606–612 [DOI] [PubMed] [Google Scholar]
- Nicolae DL (1999) Allele sharing models in gene mapping: a likelihood approach. PhD thesis, University of Chicago, Chicago [Google Scholar]
- Pálsson B, Pálsson F, Perlin M, Gudbjartsson H, Stefánsson K, Gulcher J (1999) Using quality measures to facilitate allele calling in high-throughput genotyping. Genome Res 9:1002–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullig O, Weseloh G, Klatt AR, Wagener R, Swoboda B (2002) Matrilin-3 in human articular cartilage: increased expression in osteoarthritis. Osteoarthritis Cartilage 10:253–263 [DOI] [PubMed] [Google Scholar]
- Roberts J, Burch TA (1966) Osteoarthritis prevalence in adults by age, sex, race, and geographic area. Vital Health Stat 1 11:1–27 [PubMed] [Google Scholar]
- Roby P, Eyre S, Worthington J, Ramesar R, Cilliers H, Beighton P, Grant M, Wallis G (1999) Autosomal dominant (Beukes) premature degenerative osteoarthropathy of the hip joint maps to an 11-cM region on chromosome 4q35. Am J Hum Genet 64:904–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector TD, Cicuttini F, Baker J, Loughlin J, Hart D (1996) Genetic influences on osteoarthritis in women: a twin study. BMJ 312:940–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stecher RM (1941) Heberdens nodes: heredity in hypertrophic arthritis of the finger joints. Am J Med Sci 210:801–809 [Google Scholar]
- Stefansson H, Sigurdsson E, Steinthorsdottir V, Bjornsdottir S, Sigmundsson T, Ghosh S, Brynjolfsson J, et al (2002) Neuregulin 1 and susceptibility to schizophrenia. Am J Hum Genet 71:877–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strusberg I, Sembaj A, Tabares S, Strusberg AM, del Castillo I, Villamar M, Barral JM (2002) Association analysis of genotypic frequencies of matrilin-1 gene in patients with osteoarthritis. Clin Exp Rheumatol 20:543–545 [PubMed] [Google Scholar]
- Wagener R, Kobbe B, Aszódi A, Liu Z, Beier DR, Paulsson M (2000) Structure and mapping of the mouse matrilin-3 gene (Matn3), a member of a gene family containing a U12-type AT-AC intron. Mamm Genome 11:85–90 [DOI] [PubMed] [Google Scholar]
- Weeks DE, Lange K (1988) The affected-pedigree-member method of linkage analysis. Am J Hum Genet 42:315–326 [PMC free article] [PubMed] [Google Scholar]
- Whittemore AS, Halpern J (1994) A class of tests for linkage using affected pedigree members. Biometrics 50:118–127 [PubMed] [Google Scholar]
- Wright GD, Hughes AE, Regan M, Doherty M (1996) Association of two loci on chromosome 2q with nodal osteoarthritis. Ann Rheum Dis 55:317–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen Q (2000) Changes of matrilin forms during endochondral ossification: molecular basis of oligomeric assembly. J Biol Chem 275:32628–32634 [DOI] [PubMed] [Google Scholar]