

Abstract
To control for hidden population stratification in genetic-association studies, statistical methods that use marker genotype data to infer population structure have been proposed as a possible alternative to family-based designs. In principle, it is possible to infer population structure from associations between marker loci and from associations of markers with the trait, even when no information about the demographic background of the population is available. In a model in which the total population is formed by admixture between two or more subpopulations, confounding can be estimated and controlled. Current implementations of this approach have limitations, the most serious of which is that they do not allow for uncertainty in estimations of individual admixture proportions or for lack of identifiability of subpopulations in the model. We describe methods that overcome these limitations by a combination of Bayesian and classical approaches, and we demonstrate the methods by using data from three admixed populations—African American, African Caribbean, and Hispanic American—in which there is extreme confounding of trait-genotype associations because the trait under study (skin pigmentation) varies with admixture proportions. In these data sets, as many as one-third of marker loci show crude associations with the trait. Control for confounding by population stratification eliminates these associations, except at loci that are linked to candidate genes for the trait. With only 32 markers informative for ancestry, the efficiency of the analysis is ∼70%. These methods can deal with both confounding and selection bias in genetic-association studies, making family-based designs unnecessary.
Introduction
Associations between genotype and outcome may be confounded by unrecognized population stratification. Family-based designs are accepted as the definitive method of controlling for this confounding in studies of qualitative (Thomson 1995) and quantitative (Allison 1997) traits. On this basis, many reviewers and journal editors require that genetic associations observed in population-based studies be confirmed in family-based designs (Anonymous 1999). In practice, family-based designs have serious limitations: large collections are difficult to assemble, especially for late-onset diseases, and they yield less information about association than do case-control studies of equivalent size (Morton and Collins 1998). Although sibling controls can be used when parents are not available (Spielman and Ewens 1998), this study design is even more inefficient (because of overmatching), and siblings of cases may not be available for study.
In general, population stratification exists when the total population has been formed by admixture between subpopulations and when admixture proportions (defined as the proportions of the genome that have ancestry from each subpopulation) vary between individuals. Stratification into discrete subpopulations is a special case of this general model, in which the admixture proportions of each individual are specified by a vector of 0s and 1s. If the risk of disease varies with admixture proportions, this will confound associations of disease with genotype at any locus where allele frequencies vary between subpopulations. A recent example is the association of prostate cancer with a CYP3A4 polymorphism in African Americans (Kittles et al. 2002)
If the confounder—admixture proportions—can be measured accurately, control for it can be achieved in a straightforward manner by modeling its effects in the analysis. When the ancestral subpopulations and ancestry-specific allele frequencies (the frequencies of each allele, given the subpopulation from which the gene copy has ancestry) are known for a set of marker loci, the admixture proportions of an individual can be estimated from marker genotypes (Elston 1971; Chakraborty 1975). In principle, this approach can be extended to situations in which the ancestral subpopulations and marker allele frequencies are unknown. In a population in which admixture proportions vary between individuals and in which allele frequencies at marker loci vary with locus ancestry, there will be allelic association between unlinked markers. With a sample of individuals typed at these marker loci, it is possible to exploit these allelic associations to learn about the ancestry-specific allele frequencies at each locus and the admixture proportions of each individual. This is the basis of the structured-association approach pioneered by Pritchard and colleagues, which uses Bayesian methods to learn about admixture from marker data (Pritchard et al. 2000; Pritchard and Donnelly 2001).
Although the “structured association” approach has been used to detect population stratification (Wilson et al. 2001), applications to the control of confounding by population stratification in real data sets have not been reported. Several difficulties arise in current implementations of this approach. One problem is to determine how many subpopulations should be specified in the model. Another is to allow for uncertainty in the estimates of individual admixture proportions: if such allowance is not made, the effect of confounding will be underestimated. More fundamentally, unless individuals of known ancestry are included in the sample, the subpopulations are not identifiable in the model, and point estimates of admixture proportions (obtained by averaging over the posterior distribution) will be meaningless. Finally, we require a method of assessing the adequacy with which the marker set has extracted information about the confounder.
In the present article, we describe methods that we have developed to overcome these problems, and we demonstrate their application to three populations in which there is extreme confounding of trait-genotype associations by population stratification because the trait under study—skin pigmentation—varies with individual admixture proportions. The population samples consisted of 232 African Americans in Washington, DC, 173 African Caribbean residents in England, and 446 Hispanic Americans in Colorado. For comparison, we examined a sample of 185 European Americans residing in Pennsylvania. These individuals were typed for polymorphisms in three candidate genes for skin pigmentation (TYR, OCA2, and MC1R) and at 19–30 other marker loci chosen to have large allele frequency differentials between European, West African, and Native American subpopulations. Although estimates of the allele frequencies in these subpopulations were available, this information was not used in the analyses reported here, because the objective was to evaluate the performance of our methods in a situation in which the demographic background of the population under study is unknown.
Methods
Populations and Data Sets
Details of the populations sampled, the trait measurements, and the markers typed are given elsewhere for the African American, African Caribbean, and European American samples (Shriver et al., 2003), as well as the Hispanic American sample (C.B., F.J.P., C. L. Pfaff, S. Dios, K. Hiester, J. A. Marshall, R. F. Hamman, R. E. Ferrell, C.J.B., P.M.K., and M.D.S., unpublished data). The studies were approved by the institutional review boards of Pennsylvania State University and Howard University. Skin reflectance was measured on the inner surface of the arm. With the African Caribbean, African American, and European American samples, a CyberDerm DermaSpectrometer was used, and a measure of skin melanin content was scored as the trait value. With the Hispanic American sample, a Photovolt 575 spectrophotometer was used, and a measure of skin lightness, which does not directly measure melanin content, was scored as the trait value.
Table 1 lists the marker loci and their estimated map distances between linked loci (in centimorgans). All markers were SNPs or insertion/deletion polymorphisms, selected on the basis of large allele frequency differentials between samples of modern European, West African, and Native American subpopulations by searching published reports and on-line databases of allele frequencies in diverse populations. In comparison with the marker panels used for the other three population samples, the marker panel for the Hispanic American sample included more loci with large allele frequency differentials between Europeans and Native Americans, and fewer loci with large allele-frequency differentials between Europeans and West Africans. Detailed information about the markers is available in dbSNP, under the submitter name PSU-ANTH; the locus names used in the present article are the submitter identifications used in dbSNP. The loci named “TYR-192,” “OCA2,” and “MC1R-314” are in the TYR, OCA2, and MC1R genes, respectively. All markers except GC (three alleles) were diallelic. Markers were typed by PCR-RFLP, with melting-curve analysis (Akey et al. 2001) or agarose gel electrophoresis of the PCR product.
Table 1.
Marker Loci
|
Scored in |
|||||
| PSU-ANTHSubmitter ID in dbSNP | Position | Distance fromPrevious Marker(cM) | AfricanAmerican | AfricanCaribbeans | HispanicAmericans |
| MID-575 | 1p34.3 | Yes | Yes | Yes | |
| MID-187 | 1p32 | 10 | Yes | Yes | |
| FY-null | 1q23.2 | Yes | Yes | Yes | |
| AT3-indel | 1q25.1 | 22 | Yes | Yes | |
| F13B | 1q31.3 | 18 | Yes | Yes | Yes |
| TSC1102055 | 1q32.1 | 10 | Yes | Yes | Yes |
| WI-11392 | 1q42.2 | 30 | Yes | ||
| WI-16857 | 2p16.1 | Yes | Yes | ||
| WI-11153 | 3p12.1 | Yes | Yes | ||
| GC (3 alleles) | 4q13.3 | Yes | Yes | ||
| MID-52 | 4q24 | Yes | |||
| SGC-30610 | 5q11.2 | Yes | Yes | Yes | |
| SGC-30055 | 5q22.1 | Yes | Yes | ||
| WI-17163 | 5q33.1 | 40 | Yes | ||
| WI-9231 | 7p22.3 | Yes | Yes | ||
| WI-4019 | 7q21.3 | Yes | |||
| LPL | 8p21.3 | Yes | Yes | ||
| WI-11909 | 9q21.31 | Yes | Yes | Yes | |
| D11S429 | 11q13.3 | Yes | Yes | Yes | |
| TYR-192 | 11q14.3 | 8 | Yes | Yes | Yes |
| DRD2a | 11q23.2 | 18 | Yes | Yes | Yes |
| APOA1-Alu | 11q23.3 | 2 | Yes | Yes | |
| GNB3 C825T | 12p13.31 | Yes | Yes | Yes | |
| RB1 | 13q14.2 | Yes | Yes | ||
| OCA2 | 15q12 | Yes | Yes | ||
| WI-14319 | 15q14 | 15 | Yes | Yes | Yes |
| CYP19-E2 | 15q21.2 | 20 | Yes | Yes | Yes |
| PV92-Alu | 16q23.3 | Yes | Yes | ||
| MC1R-314 | 16q24.3 | 15 | Yes | Yes | |
| WI-14867 | 17p13.2 | Yes | Yes | ||
| WI-7423 | 17p13.1 | 10 | Yes | Yes | Yes |
| Sb19.3-Alu | 19p13.11 | Yes | Yes | ||
| CKM | 19q13.2 | Yes | Yes | Yes | |
| MID-154 | 20q11.21 | Yes | Yes | ||
| MID-161 | 20q11.21 | 1 | Yes | ||
| MID-93 | 22q13.2 | Yes | Yes | Yes | |
Consists of two SNPs as four haplotypes.
Modeling Admixture
In a total population formed by admixture between k subpopulations, the admixture proportions of each gamete are defined by a vector M with k coordinates. The distribution of this proportion vector in the population is modeled as a Dirichlet distribution specified by a parameter vector with k coordinates. The ancestry of the gamete at each locus is modeled as a random variable with k states. The number of subpopulations is fixed when the model is specified. Inference about k is based on comparing the fit of models with different values of k, as described below. The stochastic variation of states of ancestry over all chromosomes in each gamete is modeled as a Markov process, with stationary distribution equal to the gamete admixture M, in which transitions to new states of ancestry are generated by k independent Poisson arrival processes, with intensity parameters that sum to a value s. For two loci separated by map distance x morgans, the transition matrix of this Markov process is a function of x, s, and M. The probabilities of the observed pair of alleles at each locus are specified by the ancestry of the two gene copies at this locus and the ancestry-specific allele frequencies.
Under the null hypothesis of no effect of alleles or haplotypes at the locus under study, the dependence of the trait value Y upon parental admixture is specified as a generalized linear model of the form 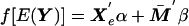 , where f is a link function, Xe is a vector of environmental covariates,
, where f is a link function, Xe is a vector of environmental covariates, 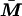 is the mean of the admixture proportions of the two parental gametes, and α and β are vectors of regression parameters. For a quantitative trait, the link function is the identity function. Vague prior distributions are specified for the k parameters of the population distribution of admixture, the sum-of-intensities parameter s, the ancestry-specific allele frequencies at each of n loci in each of k subpopulations (proportion vectors q11, …qkn) and the regression parameters α and β. Figure 1 shows the model in graphical form. The dependence of locus ancestry between adjacent loci is not shown in this figure but is included in the model. Where two or more polymorphisms in the same gene have been typed, additional nodes are introduced for the unobserved haplotype pairs, and haplotype frequencies are specified instead of allele frequencies.
is the mean of the admixture proportions of the two parental gametes, and α and β are vectors of regression parameters. For a quantitative trait, the link function is the identity function. Vague prior distributions are specified for the k parameters of the population distribution of admixture, the sum-of-intensities parameter s, the ancestry-specific allele frequencies at each of n loci in each of k subpopulations (proportion vectors q11, …qkn) and the regression parameters α and β. Figure 1 shows the model in graphical form. The dependence of locus ancestry between adjacent loci is not shown in this figure but is included in the model. Where two or more polymorphisms in the same gene have been typed, additional nodes are introduced for the unobserved haplotype pairs, and haplotype frequencies are specified instead of allele frequencies.
Figure 1.

Directed graphical model for dependence-of-trait measurement and genotypes on admixture proportions and locus ancestry. Observed data (double-edged rectangles), stochastic nodes (ellipses), stochastic dependence (continuous arrows), and strata (single-edged rectangles) of individuals and loci, are shown.
This model is fitted using Markov chain–Monte Carlo (MCMC) simulation to generate the posterior distribution of all unobserved variables, conditional on observed genotypes and trait values (McKeigue et al. 2000). To test whether the model has been specified with a sufficient number of subpopulations, we constructed a diagnostic as follows. Admixture from a subpopulation not represented in the model will give rise to residual allelic associations between unlinked loci. The latent variable underlying these associations can be detected by a principal components analysis of the covariance matrix, equivalent to computing the eigenvalues. For all pairs [j, k] of unlinked loci, we calculate a matrix of covariances of allele values (scored as 0 or 1), conditional on the gamete admixture proportion vector M and ancestry-specific allele frequencies qj, qk. The test statistic Tobs is calculated as the ratio of the largest eigenvalue to the sum of the eigenvalues, representing the proportion of variance accounted for by the latent variable. For each realization of the complete data, Tobs is compared with a value Trep, calculated from a replicate data set that is generated by drawing for each gamete at each locus a simulated allele conditional on M and qj, qk. The posterior predictive check probability P is the frequency with which Trep exceeds Tobs in the posterior distribution (Rubin 1984; Gelman et al. 1995). If the model is true, Tobs and Trep are generated from the same probability distribution, and P has expectation 0.5 in hypothetical repetitions of the experiment. A value of P<0.5 (Tobs more extreme than Trep over the posterior distribution) suggests that the model is inadequate. However, P cannot be interpreted in the same way as a conventional P value, since Tobs varies over the posterior distribution of the missing data; thus, the distribution of P in hypothetical repetitions of the experiment when the model is true is not uniform on the interval [0–1]. Because this test is a model diagnostic (for an aspect of the model that is not of direct interest) rather than a formal hypothesis test, standards for rejecting the model need not be rigorous.
Testing for Association
To test loci for association with the trait, we construct score tests based on the missing-data likelihood. We specify the alternative to the null hypothesis as a model of the form 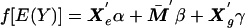 , where Xg is a vector of observed alleles or haplotypes at the locus. In this example, Xg is coded as 0, 1, or 2 copies of the allele (or haplotype). For each realization of the complete data, we calculate the score (gradient of the log-likelihood) and the information (curvature of the log-likelihood) at γ=0. The score U is evaluated as the posterior mean of the realized score, and the observed information V is evaluated by subtracting the missing information (posterior variance of the realized score) from the complete information (posterior mean of the realized information) (Little and Rubin 1987). From standard theory, U′V−1U had a χ2 distribution.
, where Xg is a vector of observed alleles or haplotypes at the locus. In this example, Xg is coded as 0, 1, or 2 copies of the allele (or haplotype). For each realization of the complete data, we calculate the score (gradient of the log-likelihood) and the information (curvature of the log-likelihood) at γ=0. The score U is evaluated as the posterior mean of the realized score, and the observed information V is evaluated by subtracting the missing information (posterior variance of the realized score) from the complete information (posterior mean of the realized information) (Little and Rubin 1987). From standard theory, U′V−1U had a χ2 distribution.
If the model is parameterized so that X and 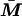 are centered about their sample means, the covariance between α, β, and γ is zero, and the ratio of observed to complete information can be interpreted as the proportion of Fisher information at γ = 0 extracted by the analysis, relative to the information that would be available from a complete data set in which
are centered about their sample means, the covariance between α, β, and γ is zero, and the ratio of observed to complete information can be interpreted as the proportion of Fisher information at γ = 0 extracted by the analysis, relative to the information that would be available from a complete data set in which 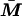 was known for each individual.
was known for each individual.
This procedure for constructing score tests by averaging over the posterior distribution of the missing data has been applied in other recently developed programs for testing for genetic associations (Clayton 1999; Schaid et al. 2002). The procedure can be viewed as a hybrid of Bayesian and classical approaches in which Bayesian methods are used to compute the gradient and curvature of the log likelihood surface. It is straightforward to extend the regression model to estimate the effect of alleles or haplotypes at a particular locus by adding the variables Xg to the regression model and generating the posterior distribution of the regression coefficient γ. However, the score test procedure has several advantages for genetic-association studies. It is computationally efficient, allowing all loci to be tested for association in a single run of the MCMC sampler. It can be extended to problems to which a fully Bayesian approach is not applicable because of ascertainment problems that arise when considering hypotheses that are distant from the null hypothesis. It also yields a useful estimate of the adequacy with which the marker set has extracted information about the confounder, based on the ratio of observed to complete information, as defined above. Furthermore, to combine studies in a meta-analysis, we simply add the score and the observed information from each study.
Results
Fitting Models for Admixture
Table 2 compares, for each data set, the fit of models specifying one or more subpopulations, evaluated by the posterior predictive check probability P. A value of P<0.5 is evidence of residual stratification (model specified with too few subpopulations). When a model with a single population was specified, there was evidence of stratification in the three admixed populations but not in the European American sample. Subsequent analyses were based on the “best-fitting” model for each data set, defined as the most parsimonious model that achieves P⩾0.5; these were specified as two, two, and three subpopulations for the African American, African Caribbean, and Hispanic American samples, respectively.
Table 2.
Tests of Fit of Models with Different Numbers of Subpopulations
|
Posterior Predictive Check Probability (P) among |
||||
| No. ofSubpopulations | African Americans(Washington, DC) | African Caribbeans(England) | Hispanic Americans(Colorado) | European Americans(Pennsylvania) |
| 1 | .30 | .37 | .16 | .50 |
| 2 | .55 | .61 | .46 | … |
| 3 | .55 | .60 | .54 | … |
| 4 | … | … | .53 | … |
The estimated proportion of the gene pool accounted for by the largest subpopulation was 84% in the African American sample, 90% in the African Caribbean sample, and 61% in the Hispanic American sample. For comparison, when the analyses were repeated specifying subpopulation allele frequencies estimated from modern Europeans, West Africans, and Native Americans (Shriver et al. 2003; C. Bonilla, unpublished data), the estimated proportion of the gene pool accounted for by the largest subpopulation was 78% African in the African American sample, 87% African in the African Caribbean sample, and 62% European in the Hispanic American sample.
Testing for Association
Figures 2–4 and tables 3–5 show the results of score tests for association of skin pigmentation with allele or haplotype (coded as 0, 1, or 2 copies). Without adjustment for admixture proportions, 11 of the 32 loci in the African American sample showed associations with skin pigmentation significant at P<.05. With adjustment for confounding by admixture proportions, associations with skin pigmentation in this population were significant for only two candidate loci: TYR-192 and OCA2. In the African Caribbean sample, crude associations with skin pigmentation were significant at P<.05 for 7 of the 30 loci. With adjustment for confounding, associations significant at P<.05 were observed only for TYR-192, for markers WI-14319 (which is linked to OCA2) and WI-11909. In the Hispanic American sample, 5 of 21 loci showed associations with skin reflectance significant at P<.05 in the unadjusted analysis. With adjustment for confounding, only one of these five loci (CYP19-E2) showed a significant association (P=.004) with skin reflectance. This locus lies 1 cM from MYO5A, a gene that we had specified in a list of candidate genes for skin pigmentation before these studies were undertaken. Mutations in MYO5A cause an autosomal recessive condition (Griscelli disease) characterized by reduced pigmentation among other features (Pastural et al. 1997). An association with MID-93 significant at P=.01 was observed in the adjusted analysis but not in the unadjusted analysis. To estimate the effect of CYP19-E2, genotype at this locus (coded as number of copies of allele 1) was added to the regression model. The posterior mean of the regression coefficient was 1.10 (95% credible interval 0.55–1.68) reflectance units, compared with an SD = 3.8 in this population sample.
Figure 2.

Effect of adjustment for population stratification on P values for association with skin pigmentation in African American. Loci for which unadjusted associations are significant at P<.05 are shown as shaded squares.
Figure 3.

Effect of adjustment for population stratification on P values for association with skin pigmentation in African Caribbeans. Loci for which unadjusted associations are significant at P<.05 are shown as shaded squares.
Figure 4.

Effect of adjustment for population stratification on P values for association with skin reflectance in Hispanic Americans. Loci for which unadjusted associations are significant at P<.05 are shown as shaded squares.
Table 3.
Tests for Association with Skin Pigmentation in African American Sample
|
Unadjusted |
Adjusted for Admixture Proportions |
||||||
| Locus (SubmitterLocal SNP ID) | Distance fromPrevious Marker(cM) | ta | Pb | ObservedInformation | % of InformationExtracted | StandardizedNormal Deviate | Pb |
| MID-575 | −1.22 | .22 | .71 | 81 | −.49 | .62 | |
| MID-187 | 10 | −1.54 | .13 | .98 | 69 | 1.07 | .28 |
| FY-null | 5.02 | .000001 |
.71 | 50 | 1.28 | .20 | |
| AT3-indel | 22 | −3.70 | .0003 |
.90 | 66 | −1.38 | .17 |
| F13B | 18 | −2.10 | .04 |
1.19 | 76 | −.79 | .43 |
| TSC1102055 | 10 | .17 | .87 | 1.33 | 80 | .21 | .84 |
| WI-11392 | 30 | −2.15 | .03 | .80 | 64 | −.62 | .53 |
| WI-16857 | .25 | .80 | 1.33 | 72 | .41 | .68 | |
| WI-11153 | −2.53 | .01 |
1.17 | 71 | −.59 | .56 | |
| GCc | .38 | .47 | |||||
| SGC-30610 | −2.19 | .03 |
1.46 | 76 | −1.73 | .08 | |
| SGC-30055 | 3.43 | .0007 |
.51 | 61 | .73 | .47 | |
| WI-9231 | −.33 | .74 | .59 | 73 | −1.48 | .14 | |
| LPL | −3.58 | .0004 |
.47 | 59 | −.29 | .77 | |
| WI-11909 | −1.65 | .10 | .97 | 69 | −.82 | .41 | |
| D11S429 | .91 | .36 | .60 | 71 | −.88 | .38 | |
| TYR-192 | 8 | 3.00 | .003 |
.31 | 62 | 2.66 | .008 |
| DRD2d | 18 | .004 |
.92 | 71 | −.60 | .55 | |
| APOA1-Alu | 2 | 1.09 | .28 | .51 | 67 | −2.05 | .04 |
| GNB3 C825T | −.07 | .95 | .10 | ||||
| RB1 | −.90 | .37 | .52 | 67 | .87 | .39 | |
| OCA2 | 4.28 | .00003 |
.77 | 60 | 2.48 | .01 |
|
| WI-14319 | 15 | .74 | .46 | 1.13 | 75 | −.02 | .98 |
| CYP19-E2 | 20 | −.61 | .54 | 1.15 | 78 | .05 | .96 |
| PV92-Alu | 2.24 | .03 |
.82 | 68 | .72 | .47 | |
| MC1R-314 | 15 | −.46 | .65 | 1.48 | 77 | .03 | .98 |
| WI-14867 | 1.69 | .09 | .59 | 67 | −.78 | .43 | |
| WI-7423 | 10 | 1.35 | .18 | .44 | 64 | −1.73 | .08 |
| Sb19.3-Alu | 1.03 | .30 | 1.43 | 77 | .41 | .68 | |
| CKM | .00 | 1.00 | .77 | 76 | .17 | .87 | |
| MID-154 | −1.40 | .16 | 1.11 | 71 | −1.58 | .11 | |
| MID-93 | 1.71 | .09 | 1.13 | 72 | .83 | .40 | |
t = Student's t deviate.
Significant values are underlined.
Score and information are not scalars for GC (three alleles)
Score and information are not scalars for DRD2 (four haplotypes defined by TaqD and BclI SNPs).
Table 4.
Tests for Association with Skin Pigmentation in African Caribbean Sample
|
Unadjusted |
Adjusted for Admixture Proportions |
||||||
| Locus (SubmitterLocal SNP ID) | Distance fromPrevious Marker(cM) | ta | Pb | ObservedInformation | % InformationExtracted | StandardizedNormal Deviate | Pb |
| MID-575 | 1.32 | .19 | .27 | 65 | .62 | .54 | |
| MID-187 | 10 | −1.07 | .28 | .79 | 75 | −.13 | .90 |
| FY-null | 2.35 | .02 |
.30 | 55 | −.27 | .78 | |
| AT3-Indel | 22 | .00 | 1.00 | .66 | 78 | .60 | .55 |
| F13B | 18 | −1.92 | .06 | .88 | 74 | −.13 | .89 |
| TSC1102055 | 10 | 1.13 | .26 | 1.00 | 81 | .49 | .62 |
| WI-16857 | .15 | .88 | .69 | 70 | 1.86 | .06 | |
| WI-11153 | −2.05 | .04 |
.72 | 75 | −.84 | .40 | |
| GC | … | .09 | … | … | … | .15 | |
| SGC-30610 | −1.27 | .21 | .98 | 81 | −.41 | .68 | |
| SGC-30055 | 2.20 | .03 | .28 | 61 | .52 | .60 | |
| WI-9231 | .46 | .64 | .45 | 72 | −.40 | .69 | |
| LPL | −3.99 | .0001 |
.16 | 49 | −1.39 | .16 | |
| WI-11909 | −1.17 | .24 | .83 | 71 | −2.03 | .04 |
|
| D11S429 | 1.73 | .08 | .38 | 75 | .86 | .39 | |
| TYR-192 | 8 | 4.98 | .000002 |
.14 | 49 | 2.97 | .003 |
| DRD2 | 18 | … | .11 | … | … | … | .99 |
| APOA1-Alu | 2 | 2.20 | .03 |
1.05 | 78 | .96 | .34 |
| GNB3 C825T | −2.65 | .009 |
.70 | 68 | −1.05 | .29 | |
| RB1 | .52 | .60 | .41 | 70 | .81 | .42 | |
| OCA2 | .04 | .97 | .55 | 79 | .25 | .80 | |
| WI-14319 | 15 | −2.78 | .006 |
.89 | 73 | −2.94 | .003 |
| CYP19-E2 | 20 | .93 | .36 | .80 | 74 | 1.32 | .19 |
| MC1R-314 | −.57 | .57 | .94 | 80 | .56 | .57 | |
| WI-14867 | .88 | .38 | .15 | 68 | −.78 | .44 | |
| WI-7423 | 10 | 1.64 | .10 | .12 | 69 | .35 | .73 |
| Sb19.3-Alu | .07 | .94 | 1.08 | 75 | −.66 | .51 | |
| CKM | −.98 | .33 | .49 | 76 | −1.68 | .09 | |
| MID-154 | −1.01 | .32 | .82 | 69 | .22 | .82 | |
| MID-93 | 1.57 | .12 | .90 | 73 | .49 | .62 | |
t = Student's t deviate.
Significant values are underlined.
Score and information are not scalars for GC (three alleles).
Score and information are not scalars for DRD2 (two-locus haplotype).
Table 5.
Tests for Association with Skin Reflectance in Hispanic American Sample[Note]
|
Unadjusted |
Adjusted for Admixture Proportions |
||||||
| Locus (Submitter Local SNP ID) | Distance fromPrevious Marker(cM) | ta | Pb | ObservedInformation | % InformationExtracted | StandardizedNormal Deviate | Pb |
| MID-575 | .91 | .36 | 3.24 | 25.2 | .51 | .61 | |
| FY-null | −.13 | .90 | 2.35 | 49.3 | .55 | .59 | |
| F13B | 40 | −.36 | .72 | 3.02 | 56 | .55 | .58 |
| TSC-1102055 | 10 | −2.53 | .01 |
8.99 | 36.4 | .92 | .36 |
| WI-11153 | 2.66 | .008 |
7.29 | 37.1 | .71 | .47 | |
| MID-52 | 2.30 | .02 |
6.44 | 31.5 | .33 | .74 | |
| SGC-30610 | 1.92 | .06 | 7.46 | 36.6 | 1.21 | .23 | |
| WI-17163 | 1.82 | .07 | 5.77 | 27.4 | 1.86 | .06 | |
| WI-4019 | −.08 | .94 | 7.61 | 35.6 | .46 | .64 | |
| WI-11909 | −1.84 | .07 | 7.76 | 43.6 | 1.08 | .28 | |
| D11S429 | −1.11 | .27 | 10.1 | 46.8 | .36 | .72 | |
| TYR-192 | 8 | −2.73 | .007 |
9.3 | 53.1 | 1.78 | .08 |
| DRD2 | 18 | … | .29 | … | … | … | .10 |
| GNB3 C825T | .03 | .98 | 8.45 | 44.1 | .39 | .69 | |
| WI-14319 | 1.32 | .19 | 8.78 | 46.1 | .29 | .78 | |
| CYP19-E2 | 20 | 5.05 | .0000006 |
9.35 | 45.7 | 2.88 | .004 |
| PV92-Alu | 1.74 | .08 | 7.28 | 33.2 | .10 | .92 | |
| WI-7423 | −.88 | .38 | 9.81 | 51.9 | .08 | .93 | |
| CKM | 1.65 | .10 | 12.3 | 19.6 | .33 | .74 | |
| MID-161 | 1.18 | .24 | 6.9 | 32.4 | .98 | .32 | |
| MID-93 | −.42 | .68 | 8.32 | 32.7 | 2.48 | .01 |
|
Note.— Associations with reflectance are in opposite direction to those of associations with pigmentation.
t = Student's t deviate.
Significant values are underlined.
In the European American sample, the standard deviation of skin pigmentation was smaller than in the African Caribbean and African American samples (4.2 compared with 9.7 and 9.6 units, respectively), and none of the unadjusted score tests for association of pigmentation with allele or haplotype were significant at P<.05. With the “best-fitting” model, the average proportion of information extracted by the score test (defined above as the ratio of observed to complete information, ignoring loci where there was evidence against the null hypothesis) was 70% in the African American sample, 71% in the African Caribbean sample, and 39% in the Hispanic American sample, in which only 21 loci were typed. When the analyses were repeated specifying a model with one subpopulation more than the number that gave the “best-fitting” model, the average proportion of information extracted by the score test was slightly lower (60%, 63%, and 31% for the African American, African Caribbean, and Hispanic American samples, respectively). When the model was specified with more than the “best-fitting” number of subpopulations, the P values for the associations with TYR-192, OCA2, and CYP19-E2 were changed only slightly. For example, in the African American sample, the P values for association with TYR-192 were .008, .011, and .015 when the model was specified with two, three, and four subpopulations, respectively. The corresponding P values for association with OCA2 were .013, .015, and .016.
For comparison, the score tests for association of skin pigmentation with allele or haplotype were repeated with allele frequencies specified in the models as known constants, estimated from sampling modern European, African American, and Native American populations, as described elsewhere (Shriver et al. 2003). Results of these analyses were generally similar to the results obtained with models in which the allele frequencies were specified as unknown. Thus, for instance, in the African American sample, the P values for association with TYR-192 were .10 and .008 in models specified with known and unknown allele frequencies, respectively. The corresponding P values for association with OCA2 were .012 and .013, respectively.
With fixed allele frequencies, analysis of the Hispanic American samples showed significant association with skin pigmentation at CYP19-E2, as in the model with unknown allele frequencies, but not at MID-93. For the African Caribbean sample, the loci that showed significant associations did not differ between the analyses with fixed and those with unknown allele frequencies.
Discussion
Modeling Admixture and Controlling for Confounding
These data sets have been chosen as extreme examples of confounding by population stratification, in which as many as one-third of marker loci show evidence of association with the trait. We have demonstrated that, in these data sets, it is possible to learn from marker data about the number of subpopulations that have undergone admixture and the admixture proportions of each individual, without supplying any prior information about subpopulations or allele frequencies. Estimates of the proportion of the gene pool contributed by the largest subpopulation agree well with direct estimates based on specifying allele frequencies estimated from modern descendants of these subpopulations. Our criterion for model choice is based on the least number of subpopulations for which the test statistic yields no evidence of residual population stratification. According to this criterion, the African Caribbean and African American samples are adequately modeled by two-way admixture, and the Hispanic American sample is adequately modeled by three-way admixture. Although the test statistic does not provide a direct estimate of the strength of the evidence for three-way rather than two-way admixture in the Hispanic American sample, the choice between these two models has little effect on the results. Specifying a model with more subpopulations than required to account for the associations between loci will introduce random “noise” but not systematic errors. Only a few markers with large allele frequency differentials between West African and non-African subpopulations were typed in the Hispanic American sample. For adequate modeling of three-way admixture in this population, it would be preferable to type more markers informative for West African ancestry.
Adjusting for mean parental admixture proportions eliminates almost all associations of the trait with alleles or haplotypes, except at two loci that are in candidate genes and two other loci that are linked to candidate genes. Thus, as theory predicts (McKeigue 1998), conditioning on admixture appears to eliminate the associations with unlinked loci that result from population stratification. In a recently admixed population, covariance of ancestry between linked loci may give rise to associations of the trait with alleles or haplotypes at loci ⩾20 cM from a trait locus (Parra et al. 1998). Conditioning on admixture does not eliminate these associations with linked loci. This is not a limitation of our approach, because the same associations would be observed in a family-based design (McKeigue 1997). For fine mapping, it would be necessary to eliminate these long-range associations generated by recent admixture. This could be achieved by scoring additional markers within the region of interest, to extract more information about locus ancestry, and then testing for association conditional on locus ancestry. When the objective is to exploit admixture to localize genes to a broad region (the purpose for which our program was originally developed), we can test instead for association of the trait with locus ancestry conditional on admixture proportions (McKeigue 1998; Shriver et al. 2003).
Classical methods of adjustment for confounding in epidemiological studies assume that the confounder is measured without error. Because this assumption is not realistic when the confounder is individual admixture proportions that are estimated from a relatively small set of markers, it is necessary to allow for error in measurement of individual admixture when testing for association. By averaging over the posterior distribution of individual admixture proportions, the score test correctly allows for uncertainty in the measurement of this confounder and for lack of identifiability of the subpopulations in the model. The use of a small panel of markers reduces the efficiency of the analysis but will not affect the type I error rate if the model is adequate. Even with only 32 marker loci, we have ∼70% of the information (about the adjusted effect of the allele) that we would have if individual admixture proportions were measured without error. By specifying a model with more subpopulations than the minimum required to account for residual associations between unlinked loci, we reduce the efficiency of the analysis slightly but do not otherwise affect the results. Although this example is based on a cross-sectional study of a quantitative trait, the method is easily applied to case-control studies by specifying a logistic link function in the linear model. As we would expect with a sample of this size, a fully Bayesian analysis for the effect at the CYP-19E2 locus corresponds well with the classical score test for an effect at this locus. Although the present article presents only hypothesis tests, parameter estimates can be obtained by specifying the Bayesian model, to include the effect of alleles at the locus under study.
Selection of Markers
The ability to control for confounding by population stratification depends critically upon the use of markers that are informative for ancestry (in that allele frequencies vary between the subpopulations that make up the gene pool of the total population). The number of markers required to extract a given proportion of information about the (adjusted) effect of genotype or haplotype will depend upon the strength of the confounding effect and upon the ancestry information content of the marker panel. For this application, in which the ancestral subpopulations are known to the level of continental groups (Europeans, West Africans, and Native Americans), we used markers that were preselected to have large allele frequency differentials between modern descendants of these subpopulations. The procedures by which these markers were identified have been described elsewhere (Parra et al. 1998; Shriver et al., 2003). Large numbers of such markers can now be identified using allele frequency data in the public domain. However, even when the ancestral subpopulations are unknown or unavailable for study, it is possible to preselect markers that are informative for ancestry by typing a large panel of markers in individuals from the stratified population and selecting those loci that show the strongest allelic associations with other unlinked loci. This approach could be used to define panels of markers suitable for use in various populations—or even to define a generic panel for worldwide use.
Calculations based on the large-sample variance of the maximum likelihood estimator of individual admixture (when allele frequencies are known) show that, in a population formed by two-way admixture, ∼40 biallelic markers with average ancestry-specific allele frequency differentials of 0.6 are required to measure the admixture proportions of each individual with SE⩽0.1. When it is not possible to identify markers with such extreme allele frequency differentials, when allele frequencies are not known in advance, or when there are more than two subpopulations, the number of markers required to measure each individual’s admixture proportions will be greater. We note, however, that when confounding is weak, tests for association and estimates of the adjusted association may be efficient, even if (as in these studies) the marker set is not adequate for accurate estimation of individual admixture proportions.
Comparison with Other Approaches
We cannot evaluate the ability of other statistical programs that have been developed to control for population stratification against these data sets, because these other programs do not provide tests for association with a quantitative trait. Our approach uses both the associations between markers and the associations of the trait with markers to infer stratification. Satten and colleagues (2001) have proposed a similar approach, using classical likelihood-based methods to model a total population made up of discrete subpopulations without admixture. When there is admixture, as in the populations studied here, a model based on discrete subpopulations cannot allow for the dependence of ancestry between linked loci and is likely to be less efficient than a more general model that allows for admixture. The Structure program (Pritchard et al. 2000) is based on a Bayesian model of population admixture similar to the one we have specified, but it uses only the associations between markers to infer stratification. This necessitates a two-stage analysis, in which estimates of admixture are “plugged in” to a second step that tests for association with the trait. As noted earlier, this does not allow for uncertainty in the estimates of individual admixture or for the lack of identifiability of subpopulations in the model.
We now discuss the question of identifiability in more detail. Unless individuals of known ancestry are included in the sample and their admixture proportions are specified in the model, the labeling of the k subpopulations in the model is arbitrary and the posterior distribution will have k! symmetrical modes. Thus, for instance, in a population formed by admixture between two subpopulations in proportions [0.6, 0.4] the posterior distribution of the admixture proportion vector is expected to have two modes: one at [0.4, 0.6] and one at [0.6, 0.4]. If the sampler explores both modes, the posterior mean of the admixture proportion vector will be [0.5, 0.5] in all individuals, which would provide no information about the confounder. In practice, an MCMC sampler based on updating locus ancestry states and allele frequencies one at a time is likely to remain stuck in one mode after the first few iterations, unless the sampling algorithm includes special measures to improve mixing. The two-stage analysis relies on this defect in the sampler. With the approach described here, it does not matter whether the sampler explores more than one mode: swapping between symmetrical modes will permute the labels of the subpopulations but will also reverse the signs of the regression parameters for admixture proportions, so that the adjusted effects associated with an allele or haplotype are not affected.
The genomic control approach uses only the associations of the trait with markers to infer stratification. Instead of modeling subpopulations, this approach models confounding as a random effect distributed over marker and candidate gene polymorphisms that inflates the variance of the test statistic (Devlin and Roeder 1999; Reich and Goldstein 2001). This random-effects model implicitly assumes that marker and candidate polymorphisms have been chosen at random from some larger set of polymorphisms. This assumption is not valid when, as in this and other studies (Kittles et al. 2002), markers and candidate gene polymorphisms have been selected on the basis of allele frequency differentials between subpopulations (Satten et al. 2001). Measurement of the confounding variable, as well as adjustment for it, is of course more efficient than simply modeling confounding effects as random “noise”: for instance, the genomic control approach would be unable to detect a true association between trait and genotype that was obscured by a confounding effect in the opposite direction.
Implications for Study Design
Although the importance of hidden stratification as a source of false-positive results in genetic-association studies has been questioned (Morton and Collins 1998; Wacholder et al. 2002; Cardon and Palmer 2003), it remains a source of difficulty when studying recently admixed populations, such as African Americans and Hispanic Americans (Thomas and Witte 2002), in which variation of admixture proportions between individuals is maintained by continuing gene flow or socioeconomic stratification (Parra et al. 2001). On the basis of our results, it is reasonable for geneticists to design their studies on the assumption that the technical problems of controlling for stratification in population-based association studies have been solved. The additional effort required to collect family-based controls is unlikely to be justified unless parents are readily available for study (as in diseases with childhood onset) or when studying parent-of-origin effects is a key objective. Although family-based designs enable haplotypes of individuals to be inferred, it is unnecessary in epidemiological studies to assign haplotypes to each individual. To estimate haplotype effects on disease risk, we require only haplotype frequencies in cases and controls, which can be estimated efficiently from samples of unrelated individuals (Fallin and Schork 2000; McKeigue 2000; Kirk and Cardon 2002; Schaid 2002; Xu et al. 2002).
More generally, it is possible to rethink some of the basic principles of designing genetic-association studies. Although the importance of minimizing selection bias has been emphasized (Cardon and Bell 2001; Wacholder et al. 2002), this is difficult to achieve in case-control studies. Selection bias will not affect associations between genotype and disease unless there is population stratification. When the variables affecting selection do not lie in the causal pathway between exposure and disease and when they have been measured on all individuals in the study, selection bias can be controlled by adjusting for these variables as if they were confounders (Greenland 1998). Thus, if selection bias gives rise to mismatching of subpopulation admixture between cases and controls, this can be dealt with in the analysis by the approach we have described. This approach allows geneticists to focus on collecting large numbers of cases and controls at low cost, without the strict population-based sampling protocols that are required to minimize selection bias in case-control studies of environmental exposures. For instance, one could establish a single large collection of population controls—typed with markers informative for ancestry—for use in multiple case-control studies. We are not suggesting that every genetic case-control study should be analyzed by the approach described in the present article. Instead, researchers could test initially for genetic distance between cases and controls, to determine whether control for population stratification is necessary (Schork et al. 2001). The biggest problem in genetic-association studies is to exclude the role of chance, and this requires larger sample sizes, which are achievable only in population-based association studies (Dahlman et al. 2002; Cardon and Palmer 2003; Colhoun et al. 2003).
Acknowledgments
We thank Sonia Dios, Richard Hamman, Robert E. Ferrell, and Julie A. Marshall for allowing us to use their data. This work was supported by National Institutes of Health grants DK53958 and HG-02154 (both to M.D.S.) and MH60343 (to P.M.M.). D.G.C. is a Juvenile Diabetes Research Foundation/Wellcome Trust Principal Research Fellow.
Electronic-Database Information
The URLs for data presented herein are as follows:
- dbSNP Home Page, http://www.ncbi.nlm.nih.gov/SNP/ [Google Scholar]
- Genetic Epidemiology Group, http://www.lshtm.ac.uk/eu/genetics (for AdmixMap program)
References
- Akey JM, Sosnoski D, Parra E, Dios S, Hiester K, Su B, Bonilla C, Jin L, Shriver MD (2001) Melting curve analysis of SNPs (McSNP): a simple gel-free low-cost approach to SNP genotyping and DNA fragment analysis. Biotechniques 30:358–362 [DOI] [PubMed] [Google Scholar]
- Allison DB (1997) Transmission/disequilibrium tests for quantitative traits. Am J Hum Genet 60:676–690 [PMC free article] [PubMed] [Google Scholar]
- Anonymous (1999) Freely associating. Nat Genet 22:1–2 [DOI] [PubMed] [Google Scholar]
- Cardon LR, Bell JI (2001) Association study designs for complex diseases. Nat Rev Genet 2:91–99 [DOI] [PubMed] [Google Scholar]
- Cardon LR, Palmer LJ (2003) Population stratification and spurious allelic association. Lancet 361:598–604 [DOI] [PubMed] [Google Scholar]
- Chakraborty R (1975) Estimation of race admixture: a new method. Am J Phys Anthropol 42:507–511 [DOI] [PubMed] [Google Scholar]
- Clayton D (1999) A generalization of the transmission/disequilibrium test for uncertain-haplotype transmission. Am J Hum Genet 65:1170–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colhoun H, McKeigue P, Davey-Smith G (2003) Problems of reporting genetic associations with complex outcomes: can we avoid being swamped by spurious findings. Lancet 361:865–872 [DOI] [PubMed] [Google Scholar]
- Dahlman I, Eaves IA, Kosoy R, Morrison VA, Heward J, Gough SC, Allahabadia A, Franklyn JA, Tuomilehto J, Tuomilehto-Wolf E, Cucca F, Guja C, Ionescu-Tirgoviste C, Stevens H, Carr P, Nutland S, McKinney P, Shield JP, Wang W, Cordell HJ, Walker N, Todd JA, Concannon P (2002) Parameters for reliable results in genetic association studies in common disease. Nat Genet 30:149–150 [DOI] [PubMed] [Google Scholar]
- Devlin B, Roeder K (1999) Genomic control for association studies. Biometrics 55:997–1004 [DOI] [PubMed] [Google Scholar]
- Elston RC (1971) The estimation of admixture in racial hybrids. Ann Hum Genet 35:9–17 [DOI] [PubMed] [Google Scholar]
- Fallin D, Schork NJ (2000) Accuracy of haplotype frequency estimation for biallelic loci, via the expectation-maximization algorithm for unphased diploid genotype data. Am J Hum Genet 67:947–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman A, Carlin DB, Stern HS, Rubin DB (1995) Bayesian data analysis. Chapman & Hall, London [Google Scholar]
- Greenland S (1998) Basic methods for sensitivity analysis and external adjustment. In: Rothman KJ, Greenland S (eds) Modern epidemiology, 2nd ed. Lippincott-Raven, Philadelphia [Google Scholar]
- Kirk KM, Cardon LR (2002) The impact of genotyping error on haplotype reconstruction and frequency estimation. Eur J Hum Genet 10:616–622 [DOI] [PubMed] [Google Scholar]
- Kittles RA, Chen W, Panguluri RK, Ahaghotu C, Jackson A, Adebamowo CA, Griffin R, Williams T, Ukoli F, Adams-Campbell L, Kwagyan J, Isaacs W, Freeman V, Dunston GM (2002) CYP3A4-V and prostate cancer in African Americans: causal or confounding association because of population stratification? Hum Genet 110:553–560 [DOI] [PubMed] [Google Scholar]
- Little RJA, Rubin DB (1987) Statistical analysis with missing data. Wiley, New York [Google Scholar]
- McKeigue PM (1997) Mapping genes underlying ethnic differences in disease risk by linkage disequilibrium in recently admixed populations. Am J Hum Genet 60:188–196 [PMC free article] [PubMed] [Google Scholar]
- ——— (1998) Mapping genes that underlie ethnic differences in disease risk: methods for detecting linkage in admixed populations by conditioning on parental admixture. Am J Hum Genet 63:241–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— (2000) Efficiency of estimating haplotype frequencies: use of marker phenotypes of unrelated individuals versus counting of phase-known gametes. Am J Hum Genet 67:1626–1627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeigue PM, Carpenter J, Parra EJ, Shriver MD (2000) Estimation of admixture and detection of linkage in admixed populations by a Bayesian approach: application to African-American populations. Ann Hum Genet 64:171–186 [DOI] [PubMed] [Google Scholar]
- Morton NE, Collins A (1998) Tests and estimates of allelic association in complex inheritance. Proc Natl Acad Sci USA 95:11389–11393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra EJ, Kittles RA, Argyropoulos G, Pfaff CL, Hiester K, Bonilla C, Sylvester N, Parrish-Gause D, Garvey WT, Jin L, McKeigue PM, Kamboh MI, Ferrell RE, Pollitzer WS, Shriver MD (2001) Ancestral proportions and admixture dynamics in geographically defined African-Americans living in South Carolina. Am J Phys Anthropol 114:18–29 [DOI] [PubMed] [Google Scholar]
- Parra EJ, Marcini A, Akey J, Martinson J, Batzer MA, Cooper R, Forrester T, Allison DB, Deka R, Ferrell RE, Shriver MD (1998) Estimating African-American admixture proportions by use of population-specific alleles. Am J Hum Genet 63:1839–1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastural E, Barrat FJ, Dufourcq-Lagelouse R, Certain S, Sanal O, Jabado N, Seger R, Griscelli C, Fischer A, de Saint BG (1997) Griscelli disease maps to chromosome 15q21 and is associated with mutations in the myosin-Va gene. Nat Genet 16:289–292 [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Donnelly P (2001) Case-control studies of association in structured or admixed populations. Theor Popul Biol 60:227–237 [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Rosenberg NA, Donnelly P (2000) Association mapping in structured populations. Am J Hum Genet 67:170–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich DE, Goldstein DB (2001) Detecting association in a case-control study while correcting for population stratification. Genet Epidemiol 20:4–16 [DOI] [PubMed] [Google Scholar]
- Rubin DB (1984) Bayesianly justifiable and relevant frequency calculations for the applied statistician. Ann Stat 12:1151–1172 [Google Scholar]
- Satten GA, Flanders WD, Yang Q (2001) Accounting for unmeasured population substructure in case-control studies of genetic association using a novel latent-class model. Am J Hum Genet 68:466–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaid DJ (2002) Relative efficiency of ambiguous vs. directly measured haplotype frequencies. Genet Epidemiol 23:426–443 [DOI] [PubMed] [Google Scholar]
- Schaid DJ, Rowland CM, Tines DE, Jacobson RM, Poland GA (2002) Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am J Hum Genet 70:425–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schork NJ, Fallin D, Thiel B, Xu X, Broeckel U, Jacob HJ, Cohen D (2001) The future of genetic case-control studies. Adv Genet 42:191–212 [DOI] [PubMed] [Google Scholar]
- Shriver MD, Parra EJ, Dios S, Bonilla C, Norton H, Jovel C, Pfaff C, Jones C, Massac I, Cameron N, Baron A, Jackson T, Argyropoulos G, Jin L, Hoggart CL, McKeigue PM, Kittles RA (2003) Skin pigmentation, biogeographical ancestry, and admixture mapping. Hum Genet 112:387–399 [DOI] [PubMed] [Google Scholar]
- Spielman RS, Ewens WJ (1998) A sibship test for linkage in the presence of association: the sib transmission/disequilibrium test. Am J Hum Genet 62:450–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DC, Witte JS (2002) Point: population stratification: a problem for case-control studies of candidate-gene associations? Cancer Epidemiol Biomarkers Prev 11:505–512 [PubMed] [Google Scholar]
- Thomson G (1995) Mapping disease genes: family-based association studies. Am J Hum Genet 57:487–498 [PMC free article] [PubMed] [Google Scholar]
- Wacholder S, Rothman N, Caporaso N (2002) Counterpoint: bias from population stratification is not a major threat to the validity of conclusions from epidemiological studies of common polymorphisms and cancer. Cancer Epidemiol Biomarkers Prev 11:513–520 [PubMed] [Google Scholar]
- Wilson JF, Weale ME, Smith AC, Gratrix F, Fletcher B, Thomas MG, Bradman N, Goldstein DB (2001) Population genetic structure of variable drug response. Nat Genet 29:265–269 [DOI] [PubMed] [Google Scholar]
- Xu CF, Lewis K, Cantone KL, Khan P, Donnelly C, White N, Crocker N, Boyd PR, Zaykin DV, Purvis IJ (2002) Effectiveness of computational methods in haplotype prediction. Hum Genet 110:148–156 [DOI] [PubMed] [Google Scholar]