

Abstract
One of the most striking findings to emerge from the study of genomic patterns of variation is that regions with lower recombination rates tend to have lower levels of intraspecific diversity but not of interspecies divergence. This uncoupling of variation within and between species has been widely interpreted as evidence that natural selection shapes patterns of genetic variability genomewide. We revisited the relationship between diversity, divergence, and recombination in humans, using data from closely related species and better estimates of recombination rates than previously available. We show that regions that experience less recombination have reduced divergence to chimpanzee and to baboon, as well as lower levels of diversity. This observation suggests that mutation and recombination are associated processes in humans, so that the positive correlation between diversity and recombination may have a purely neutral explanation. Consistent with this hypothesis, diversity levels no longer increase significantly with recombination rates after correction for divergence to chimpanzee.
Introduction
A recent meta-analysis of 22 regions of the human genome found a positive correlation between recombination rates and nucleotide diversity among humans and no relationship between recombination rates and human-chimpanzee divergence (Nachman 2001). This finding mirrors evidence from Drosophila melanogaster that diversity, but not synonymous site divergence to D. simulans, increases with the recombination rate (Begun and Aquadro 1992; Betancourt and Presgraves 2002), as well as more tentative reports for a wide variety of species, from tomato to mouse (reviewed in Andolfatto and Przeworski 2001).
If mutation and recombination are associated processes and most mutations have no fitness effect, both intraspecies and interspecies differences should increase with recombination rates. Since no correlation was found between divergence levels and recombination rates, researchers discounted neutral explanations linking genetic exchange to mutation and, instead, focused on models of variation-reducing selection (Begun and Aquadro 1992; Charlesworth et al. 1993). In humans, it was suggested that repeated episodes of selection for strongly favorable alleles could account for the variation in diversity levels (Nachman 2001; Payseur and Nachman 2002b). However, humans differ from Drosophila in a number of the parameters that determine the effects of natural selection; in particular, their effective population size is roughly 2 orders of magnitude smaller. As a consequence, a large fraction of mutations would have to be advantageous for positive selection alone to account for variation in diversity levels (Andolfatto 2001).
To date, there have been little appropriate data with which to evaluate the relative strength of these arguments for humans, so that analyses had to pool polymorphism data collected according to a wide variety of sampling schemes or to use distant relatives of humans to estimate divergence rates. Three studies recently reported a positive correlation between human recombination rates and fourfold degenerate site divergence (or ancestral repeat divergence) between human and mouse (Lercher and Hurst 2002; Waterston et al. 2002; Hardison et al. 2003). However, estimates of substitution rates for distantly related species are highly sensitive to the assumptions of the method used (Nei and Kumar 2000; Castresana 2002), specifically, to mutation rate variation among sites and differences in mutation rates among lineages (Hardison et al. 2003). In addition, the genomic landscape has changed dramatically between mouse and human (Waterston et al. 2002), so it is unclear why substitutions on the mouse lineage would be associated with recombination rates estimated in humans. These relationships are better examined with data from more closely related species. We therefore revisited these issues using sequence data from close relatives of humans and the most recent estimates of the human recombination rates.
Materials and Methods
Recombination Rate Estimates
Recombination rate estimates stem from a comparison of the physical map of the human genome with a recent, high-resolution genetic map that is based on 1,257 meiosis and 5,136 microsatellite markers (Kong et al. 2002). Sex-averaged estimates of recombination rates are provided for 4,690 markers that could be placed in sequence contigs of the August 2001 freeze of the Human Genome Project Working Draft at the University of California, Santa Cruz. Although the markers are slightly less densely spaced than in a previous map (Yu et al. 2001), the sampling error associated with estimates of genetic distance is much smaller (Weber 2002). The estimates of recombination rates represent average rates for a window of 3 Mb centered on the marker. To find the closest markers to the regions for which we had divergence and diversity estimates, we repositioned the sequences on the August 2001 freeze. We assigned each region to the closest marker and included only regions for which this marker is within 1.5 Mb.
Human Polymorphism Data
We considered three sets of polymorphism data (Stephens et al. [2001], National Institute of Environmental Health Sciences [NIEHS SNPs], and SeattleSNPs). Diversity levels were summarized by the average pairwise difference in the sample (Tajima 1983), π, as reported by the authors. The data from Stephens et al. (2001) contain a high proportion of exons, whereas the data from SeattleSNPs and NIEHS SNPs are mainly noncoding. For the analyses of the surveys of diversity, we excluded X-linked loci; there are not enough X-linked polymorphism data sets to consider them separately, and there is no clear way to combine X-linked and autosomal loci, given uncertainty about their relative effective population sizes and mutation rates (Payseur and Nachman 2002b). We did not combine data from different studies, because the sampling designs differ (cf. Ptak and Przeworski 2002).
Chimpanzee Shotgun Library Data
Divergence estimates came from two sources: a shotgun library of chimpanzee (Ebersberger et al. 2002) and available BAC sequences. The 8,652 reads from the chimpanzee shotgun library were mapped to the August 2001 freeze of the human genome to estimate divergence and to identify the closest marker on the genetic map. We excluded exons, as well as sequences that mapped to markers for which the map order was uncertain (as noted in Kong et al. [2002]). Chimpanzee sequences within 1.5 Mb of the closest marker were combined to estimate human-chimpanzee divergence for a 3-Mb window. We restricted our analyses to those windows where the sum of the lengths of the reads was at least 1 kb (median: 1,130 bp). Divergence was estimated as the number of differences divided by the number of base pairs compared. There is no correction for multiple hits, since the species are closely related (Nei and Kumar 2000; Smith et al. 2002). Our approach provides us with divergence estimates for 480 3-Mb windows.
Chimpanzee and Baboon BAC Data
We used all chimpanzee and baboon sequences from genomic DNA cloned into bacterial artificial chromosomes (BACs) available from GenBank. The BAC sequences were cut into fragments of 1 kb, then aligned and analyzed in the same way as the chimpanzee shotgun sequences. Since BAC data provide us with more sequences within a given 3-Mb window than does a shotgun library, thereby improving the precision of our divergence estimates, we include windows only with a minimum of 10 kb of sequence. Using this approach, we have divergence estimates for 103 3-Mb windows for chimpanzee and for 115 3-Mb windows for baboon.
SeattleSNPs Chimpanzee Divergence Data
A chimpanzee sequence was available for the loci in the SeattleSNPs but not for the regions sequenced in the two other diversity surveys. To estimate human-chimpanzee divergence for the SeattleSNPs data, a human sequence was retrieved from GenBank, and the chimpanzee sequence was retrieved from the project Web site. The chimp sequence was aligned to the homologous human sequence by use of BLAST (Altschul et al. 1990). Divergence was estimated as the number of sites that differed between the two species over the total number of base pairs in the alignment. Each locus was considered separately, even when two or more mapped to the same recombination marker. The mean length of sequence for a locus is ∼13 kb.
Univariate Analyses
We used both a parametric (Pearson’s correlation coefficient) and a nonparametric (Kendall’s coefficient of rank correlation) approach to examine the relationship between π (or the divergence d or π/d) and recombination rate estimates. The test of significance for Pearson’s correlation coefficient assumes that both variables are normally distributed. We therefore transformed π (or d or π/d) and recombination rates by x → ln(x + 0.001) when these transformations improved the fit of the variable to a normal distribution (as assessed by a Kolmogorov-Smirnoff test and by visual inspection). Qualitative conclusions are similar if the variables are not transformed or are always transformed (results not shown). We reported two-tailed P values and assessed significance at the 5% level.
Multivariate Analysis
We performed a multiple linear regression of transformed divergence values on three aspects of sequence content (see below) and on recombination rates. For the regression analyses, the divergence values, d, were transformed by d→ln(d+0.001) to be approximately normally distributed. Examination of the residuals suggests that the assumption of a linear relationship between dependent and independent variables is appropriate (results not shown).
We measured three aspects of the sequence content at three scales. First, we tabulated the GC content, CpG content, and polyAT content of the sequences used to estimate divergence (referred to as “local”). We also characterized the sequence content of 100-kb neighborhoods of each read (Reich et al. 2002) as follows: If there were k sequences in a given 3-Mb window, we weighted the count of sequence motifs for the 100-kb window centered on sequence i of length Li by 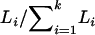 (“100 Kb”). To take into account possible long-range influences of sequence motifs on divergence rates, we recorded the three aspects of sequence content for the 3-Mb window centered on the recombination marker (“3 Mb”).
(“100 Kb”). To take into account possible long-range influences of sequence motifs on divergence rates, we recorded the three aspects of sequence content for the 3-Mb window centered on the recombination marker (“3 Mb”).
Finally, we performed a linear regression of diversity on divergence and recombination rates for the SeattleSNPs data. The diversity values were transformed by π→ln(π+0.001) to be approximately normally distributed.
Results and Discussion
Diversity, Divergence, and Recombination
We first related diversity (as summarized by π) to sex-averaged recombination rates for three newly available sets of loci (Stephens et al. [2001], NIEHS SNPs, and SeattleSNPs) where the same individuals were sequenced across loci (fig. 1A). As can be seen in table 1, diversity levels increase weakly but significantly with the recombination rate in all three sets of data. This is also true if diversity levels are estimated from the number of segregating sites in the sample (results not shown). Thus, diversity levels are indeed positively correlated with recombination rates in humans.
Figure 1.

A, Human diversity levels (π) increase with human recombination rates. B, Human-chimpanzee divergence levels increase with human recombination rates. Note that the lines are for illustrative purposes only; the regression analyses used transformed values (see “Materials and Methods” section).
Table 1.
Significance of the Association between Diversity (or Divergence) Levels and Estimates of Human Recombination Rates
| Data Set | Kendall’s τa (P)b | Pearson’s rc (P)b | n |
| Diversity: | |||
| Stephens et al. (2001) | .111 (.010) | .127 (.039) | 263 |
| SeattleSNPs | .171 (.037) | .257 (.025) | 76 |
| NIEHS SNPs | .178 (.032) | .249 (.032) | 74 |
| Divergence: | |||
| SeattleSNPs | .164 (.037) | .259 (.024) | 76 |
| Chimp shotgun | .149 (10−6) | .214 (2×10-6) | 480 |
| Chimp BAC | .235 (4×10-4) | .289 (.003) | 103 |
| Baboon BAC | .204 (.001) | .264 (.004) | 115 |
τ is Kendall’s coefficient of rank correlation.
P values are two-tailed.
r is Pearson’s correlation coefficient.
Next, we considered the relationship of human-chimpanzee divergence levels to human recombination rates in three sets of data. We mapped the nucleotide sequence reads obtained from a recently constructed chimpanzee shotgun library (Ebersberger et al. 2002) to the human genome to estimate divergence. We also estimated divergence levels for available chimpanzee BAC data and for data from the SeattleSNPs project (see “Materials and Methods” section). The divergence estimates obtained from these three sources are plotted against recombination rate estimates in figure 1B. In all three sets of data, divergence levels and recombination rates are significantly positively correlated (see table 1). Thus, in humans, not only diversity but also divergence increases with recombination rates. This finding contrasts with a number of earlier reports based on many fewer data and less accurate estimates of the recombination rate (Nachman et al. 1998; Przeworski et al. 2000; Nachman 2001; Payseur and Nachman 2002a). However, it is consistent with the observation that a region of the genome with elevated rates of genetic exchange, the pseudoautosomal region, exhibits high levels of divergence and diversity in humans (Perry and Ashworth 1999; Schiebel et al. 2000).
Humans and chimpanzees are sufficiently closely related that divergence between species reflects not only substitutions accumulated since the two species split, but also the diversity present in the ancestral species. This raises the possibility that human-chimpanzee divergence increases with the recombination rate because diversity levels increased with the recombination rate in the ancestor of humans and chimpanzees; that is, because of variation-reducing selection in the ancestral species. To evaluate this possibility, we compared human with baboon sequences. The common ancestor of humans and baboons is estimated to have lived 25–30 million years ago (Goodman et al. 1998; Yoder and Yang 2000), so that the contribution of ancestral polymorphism to human-baboon divergence should be minor, assuming plausible values for the generation time and the ancestral effective population size of the species (Wall 2003). However, since recombination rates could have changed substantially between the two species, the baboon may be a less suitable choice to detect an association between divergence and recombination. Nonetheless, when we plotted human-baboon divergence against our estimates of human recombination rates (fig. 2), we found a highly significant correlation (see table 1). Since the association between recombination and divergence is evident in a comparison of human and chimpanzee as well as of human and baboon, and since similar observations have been reported for human and mouse (e.g., Waterston et al. 2002), we conclude that it is not due to variation in ancestral polymorphism levels.
Figure 2.

Human-baboon divergence levels increase with human recombination rates. Note that the line is for illustrative purposes only; the regression analyses used transformed values of divergence (see “Materials and Methods” section).
Rather, the variation in divergence levels likely reflects changes in the underlying mutation rate. Indeed, the divergence estimates are mostly for intergenic and intronic regions, where it is plausible to assume that most mutations have no fitness effect. If most mutations were, instead, slightly deleterious, divergence levels would be expected to decrease in regions of high recombination, owing to the increased efficiency of purifying selection (Li 1987). The positive correlation between divergence levels and recombination rates suggests that mutation rates increase with the frequency of genetic exchange—in other words, that these two processes are associated.
This points to a nonselective explanation for the positive correlation between diversity and recombination observed in humans. Consistent with a purely neutral explanation, intraspecific (π) and interspecific (d) variation levels are positively correlated for the 76 loci in the SeattleSNPs survey (fig. 3A) (τ=0.179, P=.028; r=0.252, P=.028). Further, π/d no longer increases with recombination (τ=0.068, P=.387; r=0.073, P=.529), as can be visualized in figure 3B. Indeed, recombination is no longer a significant predictor of diversity in a regression of diversity on recombination rates and divergence (F=2.88; df 1, 73; P=.094; using the method described in the legend of table 2). In general, there is no evidence that the magnitude of the effect of recombination rates on diversity is stronger than on divergence (contrast fig. 1A and 1B). Thus, it appears that the correlation of diversity and recombination rates can be accounted for entirely by recombination-associated variation in mutation rates, at least for these data.
Figure 3.

A, Human diversity levels (π) versus human-chimpanzee divergence (d) for the 76 loci in the SeattleSNPs survey. B, π/d versus human recombination rate estimates for the 76 loci in the SeattleSNPs survey. Note that the lines are for illustrative purposes only (see “Materials and Methods” section).
Table 2.
Multiple Linear Regression of Transformed Divergence Values on Three Aspects of Sequence Content and Recombination Rates
| Dependent Variable | Independent Variablesa | Adjusted R2 b | Test Statistic(F)c | df | P |
| Shotgun human-chimp divergence values | |||||
| 3 Mb | .083 | 8.07 | 1, 475 | .005 | |
| 100 kb | .072 | 24.71 | 1, 475 | 9×10-7 | |
| Local | .060 | 26.76 | 1, 475 | 3×10-7 | |
| BAC human-chimp divergence values | |||||
| 3 Mb | .159 | 4.59 | 1, 98 | .035 | |
| 100 kb | .181 | 8.57 | 1, 98 | .004 | |
| Local | .130 | 7.58 | 1, 98 | .007 | |
| SeattleSNP human-chimp divergence values | |||||
| 3 Mb | .071 | 1.63 | 1, 71 | .205 | |
| 100 kb | .072 | 4.86 | 1, 71 | .031 | |
| Local | .078 | 6.37 | 1, 71 | .014 | |
| Human-baboon divergence values | |||||
| 3 Mb | .203 | 3.38 | 1, 110 | .069 | |
| 100 kb | .136 | 7.72 | 1, 110 | .006 | |
| Local | .395 | 18.35 | 1, 110 | 4×10-5 |
“3 Mb,” “100 kb,” and “local” refer to the CpG, GC, and polyAT content at each scale (see “Materials and Methods” section).
The adjusted R2 is the proportionate reduction of the variance in transformed divergence values achieved by the introduction of recombination rates and sequence motifs in a given regression model. Note that R2 values are not comparable across data sets. In particular, they depend on the variance of the error terms, which will differ across data sets, because of varying precision of divergence estimates (see “Materials and Methods” section). They also depend on the range of the independent variables.
By use of a partial F test, we examine whether adding the recombination rate to the regression model explains a larger proportion of the variance in divergence values than do the three aspects of sequence content alone.
Why Are Mutation and Recombination Associated?
These observations raise the possibility of a causal relationship between mutation and recombination processes. The association between mutation and recombination could reflect a mutagenic effect of recombination or a recombination-stimulating effect of mutation. Most evidence, however, suggests that high levels of diversity decrease the rate of repair of double-strand breaks by recombination (e.g., Resnick et al. 1989), so that regions with higher mutation rates would tend to have lower recombination rates. On the other hand, in Saccaromyces cerevisiae, there is evidence that double-strand-break repair by recombination is mutagenic: in one experiment, DNA synthesis associated with mitotic double-strand-break repair led to a high error rate at a marker 0.3 kb from the double-strand break (Strathern et al. 1995; Rattray et al. 2001). Most of these repair-associated errors appear to be point mutations (Rattray et al. 2001) made by an error-prone translesion polymerase (Rattray et al. 2002). Thus, evidence from S. cerevisiae would suggest that divergence rates increase because of increasing recombination rates.
However, the association between mutation and recombination in humans could also be due to one or more factors that shape both mutation and recombination rates. Three sequence motifs, CpG, GC, and PolyAT content, were recently identified as strong predictors of recombination rates (Kong et al. 2002). There also appears to be a correlation between peaks of GC content and recombination hot spots in S. cerevisiae (Gerton et al. 2000; Petes and Merker 2002). Further, CpG dinucleotides are known to have a higher rate of transitions than other bases (Cooper and Krawczak 1989), and synonymous substitution rates in mammals appear to increase with GC content (Castresana 2002). Thus, some of these sequence motifs could influence both recombination and mutation rates, leading to an association between the two rates that is not causal. To tease apart the direct effects of recombination on mutation from those mediated by sequence content, we performed a multiple regression with the three sequence motifs and recombination rates as explanatory variables and divergence as the dependent variable (see table 2 for details).
The scale at which sequence motifs predict divergence is unknown, so we tried to correct for the content of the sequences themselves, as well as for possible longer range effects (see “Materials and Methods” section). At small scales (“local” and “100 kb”), recombination rates remained a significant predictor of human-chimpanzee and human-baboon divergence levels after adjustment for sequence motifs (see table 2). In other words, after correction for a number of potentially confounding factors, mutation rates remain higher in regions of higher recombination. However, when we controlled for sequence motifs at a 3-Mb scale, recombination no longer explained a significant proportion of the variance (at the 5% level) of the SeattleSNPs human-chimpanzee divergence data or the human-baboon data. Similarly, recombination is no longer a significant predictor of diversity levels after correction for 3-Mb sequence motifs (results not shown). It may be that a large number of regions from very closely related species are required to detect the effect of recombination. Alternatively, the association of mutation and recombination may not be causal but, instead, it may be mediated by large-scale sequence motifs or by additional, unknown variables, such as global features of chromosome structure (Petes 2001; Petes and Merker 2002).
In this light, it is interesting to speculate as to what distinguishes humans from D. melanogaster, where there is no evidence of a correlation between synonymous site divergence with D. simulans and recombination rates in a survey of 254 genes (Betancourt and Presgraves 2002). One possibility is that a feature of the recombination process or a confounding factor differs from humans, such that recombination and mutation are not associated processes in fruit flies. If so, the variation in diversity levels in Drosophila reflects the effects of variation-reducing selection.
On the basis of our findings, we can gain a very rough sense of the proportion of mutations that are associated with recombination in humans. To do so, we used the regression of transformed divergence values on recombination rates, controlling for the 3-Mb sequence motifs (since it is at this scale that the sequence motifs explain the largest proportion of the variance in divergence levels). Using the regression estimates for the chimp BAC data and the median value for all independent variables (recombination rates and three sequence motifs), the predicted divergence per bp is 0.0124. In the absence of recombination, the predicted divergence is 0.0116. Thus, slightly >6% of mutations are associated with the median recombination rate of 1.2 cM/Mb; this proportion changes with the recombination rate and the sequence content. Similar estimates are obtained using other chimpanzee data (results not shown). However, the estimate from baboon data is lower (∼3%). This is consistent with the notion that recombination rates differ across species, so that rates estimated in humans are poorer predictors of human-baboon divergence than of human-chimpanzee divergence. Whether 6% is a plausible proportion depends on the fidelity of recombination-associated repair and the distribution of branch migration lengths, parameters about which little is currently known. Note further that error in the estimates of recombination rates will tend to bias our estimate of the regression coefficients, so even these crude values should be interpreted with caution.
These estimates do raise an interesting question, however. Given that humans and mouse last had a common ancestor 70–110 million years ago (Kumar and Hedges 1998; Eizirik et al. 2001) and that approximately two-thirds of all substitutions are thought to have occurred on the mouse lineage (Waterston et al. 2002), most substitutions occurred in a genomic landscape very different from that of extant humans. Why, then, is a significant positive correlation detected between human recombination rates and human-mouse fourfold degenerate site divergence (Lercher and Hurst 2002, Waterston et al. 2002; Hardison et al. 2003)? Lercher and Hurst (2002) suggest that “any significant correlations found above this random noise could be signs of underlying strong relationships” between recombination and mutation processes. However, using chimpanzee, we estimate that only 6% of mutations are associated with recombination, and, using baboon, this proportion drops to 3%. The detection of a correlation between human-mouse divergence and human recombination rates would, therefore, imply a remarkable conservation of recombination rates across 140–220 million years of evolution. This seems unlikely, given that >200 genome rearrangements are estimated to have occurred between humans and mice (Waterston et al. 2002). Such conservation of recombination rates also seems implausible, given what is known about species more closely related than human and mouse—for example, the fact that the genetic map is shorter in baboons than in humans (Rogers et al. 2000) and that sibling species of Drosophila have different recombination landscapes (True et al. 1996). As mentioned above, a second possibility is that a confounding factor accounts for the apparent relationship between recombination and human-mouse divergence. Thus, although these and previous results from mouse are consistent with a mutagenic effect of recombination, such an interpretation is premature.
What is clear from our findings is that, whatever the determinants of mutation rates in humans, they seem to have comparable effects on diversity and divergence. Thus, there is currently no need to assume that natural selection has shaped genomewide patterns of variability in humans, although it has undoubtedly shaped patterns of variation in some genomic regions.
Acknowledgments
Thanks to P. Andolfatto, C. Aquadro, B. Charlesworth, A. Di Rienzo, Y. Gilad, H. Kaessmann, M. Lachmann, M. Nachman, A. Przeworski, G. Smith, J. Strathern, J. Wall, and an anonymous reviewer for helpful discussions and/or comments on the manuscript.
Electronic-Database Information
URLs for data presented herein are as follows:
- GenBank, http://www.ncbi.nih.gov/Genbank/ (accessed, September 2002)
- NIEHS SNPs, http://egp.gs.washington.edu/ (accessed, October 2002)
- SeattleSNPs, http://pga.mbt.washington.edu/ (accessed, October 2002)
- UCSC Genome Bioinformatics, http://genome.ucsc.edu/
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410 [DOI] [PubMed] [Google Scholar]
- Andolfatto P (2001) Adaptive hitchhiking effects on genome variability. Curr Opin Genet Dev 11:635–641 [DOI] [PubMed] [Google Scholar]
- Andolfatto P, Przeworski M (2001) Regions of lower crossing over harbor more rare variants in African populations of Drosophila melanogaster. Genetics 158:657–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begun DJ, Aquadro CF (1992) Levels of naturally occurring DNA polymorphism correlate with recombination rates in D. melanogaster. Nature 356:519–520 [DOI] [PubMed] [Google Scholar]
- Betancourt AJ, Presgraves DC (2002) Linkage limits the power of natural selection in Drosophila. Proc Natl Acad Sci USA 99:13616–13620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castresana J (2002) Estimation of genetic distances from human and mouse introns. Nucleic Acids Res 30:1751–175611937628 [Google Scholar]
- Charlesworth B, Morgan MT, Charlesworth D (1993) The effect of deleterious mutations on neutral molecular variation. Genetics 134:1289–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DN, Krawczak M (1989) Cytosine methylation and the fate of CpG dinucleotides in vertebrate genomes. Hum Genet 83:181–188 [DOI] [PubMed] [Google Scholar]
- Ebersberger I, Metzler D, Schwarz C, Pääbo S (2002) Genomewide comparison of DNA sequences between humans and chimpanzees. Am J Hum Genet 70:1490–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eizirik E, Murphy WJ, O’Brien SJ (2001) Molecular dating and biogeography of the early placental mammal radiation. J Hered 92:212–219 [DOI] [PubMed] [Google Scholar]
- Gerton JL, DeRisi J, Shroff R, Lichten M, Brown PO, Petes TD (2000) Inaugural article: global mapping of meiotic recombination hotspots and coldspots in the yeast Saccharomyces cerevisiae. Proc Natl Acad Sci USA 97:11383–11390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman M, Porter CA, Czelusniak J, Page SL, Schneider H, Shoshani J, Gunnell G, Groves CP (1998) Toward a phylogenetic classification of primates based on DNA evidence complemented by fossil evidence. Mol Phylogenet Evol 9:585–598 [DOI] [PubMed] [Google Scholar]
- Hardison RC, Roskin KM, Yang S, Diekhans M, Kent WJ, Weber R, Elnitski L, Li J, O’Connor M, Kolbe D, Schwartz S, Furey TS, Whelan S, Goldman N, Smit A, Miller W, Chiaromonte F, Haussler D (2003) Covariation in frequencies of substitution, deletion, transposition, and recombination during eutherian evolution. Genome Res 13:13–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong A, Gudbjartsson DF, Sainz J, Jonsdottir GM, Gudjonsson SA, Richardsson B, Sigurdardottir S, Barnard J, Hallbeck B, Masson G, Shlien A, Palsson ST, Frigge ML, Thorgeirsson TE, Gulcher JR, Stefansson K (2002) A high-resolution recombination map of the human genome. Nat Genet 31:241–247 [DOI] [PubMed] [Google Scholar]
- Kumar S, Hedges SB (1998) A molecular timescale for vertebrate evolution. Nature 392:917–920 [DOI] [PubMed] [Google Scholar]
- Lercher MJ, Hurst LD (2002) Human SNP variability and mutation rate are higher in regions of high recombination. Trends Genet 18:337–340 [DOI] [PubMed] [Google Scholar]
- Li WH (1987) Models of nearly neutral mutations with particular implications for nonrandom usage of synonymous codons. J Mol Evol 24:337–345 [DOI] [PubMed] [Google Scholar]
- Nachman MW (2001) Single nucleotide polymorphisms and recombination rate in humans. Trends Genet 17:481–485 [DOI] [PubMed] [Google Scholar]
- Nachman MW, Bauer VL, Crowell SL, Aquadro CF (1998) DNA variability and recombination rates at X-linked loci in humans. Genetics 150:1133–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Kumar S (2000) Molecular evolution and genetics. Oxford University Press, Oxford, UK [Google Scholar]
- Payseur BA, Nachman MW (2002a) Gene density and human nucleotide polymorphism. Mol Biol Evol 19:336–340 [DOI] [PubMed] [Google Scholar]
- ——— (2002b) Natural selection at linked sites in humans. Gene 300:31–42 [DOI] [PubMed] [Google Scholar]
- Perry J, Ashworth A (1999) Evolutionary rate of a gene affected by chromosomal position. Curr Biol 9:987–989 [DOI] [PubMed] [Google Scholar]
- Petes TD (2001) Meiotic recombination hot spots and cold spots. Nat Rev Genet 2:360–369 [DOI] [PubMed] [Google Scholar]
- Petes TD, Merker JD (2002) Context dependence of meiotic recombination hotspots in yeast: the relationship between recombination activity of a reporter construct and base composition. Genetics 162:2049–2052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przeworski M, Hudson RR, Di Rienzo A (2000) Adjusting the focus on human variation. Trends Genet 16:296–302 [DOI] [PubMed] [Google Scholar]
- Ptak SE, Przeworski M (2002) Evidence for population growth in humans is confounded by fine-scale population structure. Trends Genet 18:559–563 [DOI] [PubMed] [Google Scholar]
- Rattray AJ, McGill CB, Shafer BK, Strathern JN (2001) Fidelity of mitotic double-strand-break repair in Saccharomyces cerevisiae: a role for SAE2/COM1. Genetics 158:109–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattray AJ, Shafer BK, McGill CB, Strathern JN (2002) The roles of Rev3 and Rad57 in double-strand-break-repair-induced mutagenesis of Saccharomyces cerevisiae. Genetics 162:1063–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich DE, Schaffner SF, Daly MJ, McVean G, Mullikin JC, Higgins JM, Richter DJ, Lander ES, Altshuler D (2002) Human genome sequence variation and the influence of gene history, mutation and recombination. Nat Genet 32:135–142 [DOI] [PubMed] [Google Scholar]
- Resnick MA, Skaanild M, Nilsson-Tillgren T (1989) Lack of DNA homology in a pair of divergent chromosomes greatly sensitizes them to loss by DNA damage. Proc Natl Acad Sci USA 86:2276–2280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers J, Mahaney MC, Witte SM, Nair S, Newman D, Wedel S, Rodriguez LA, Rice KS, Slifer SH, Perelygin A, Slifer M, Palladino-Negro P, Newman T, Chambers K, Joslyn G, Parry P, Morin PA (2000) A genetic linkage map of the baboon (Papio hamadryas) genome based on human microsatellite polymorphisms. Genomics 67:237–247 [DOI] [PubMed] [Google Scholar]
- Schiebel K, Meder J, Rump A, Rosenthal A, Winkelmann M, Fischer C, Bonk T, Humeny A, Rappold G (2000) Elevated DNA sequence diversity in the genomic region of the phosphatase PPP2R3L gene in the human pseudoautosomal region. Cytogenet Cell Genet 91:224–230 [DOI] [PubMed] [Google Scholar]
- Smith NG, Webster MT, Ellegren H (2002) Deterministic mutation rate variation in the human genome. Genome Res 12:1350–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens JC, Schneider JA, Tanguay DA, Choi J, Acharya T, Stanley SE, Jiang R, et al (2001) Haplotype variation and linkage disequilibrium in 313 human genes. Science 293:489–493 [DOI] [PubMed] [Google Scholar]
- Strathern JN, Shafer BK, McGill CB (1995) DNA synthesis errors associated with double-strand-break repair. Genetics 140:965–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F (1983) Evolutionary relationship of DNA sequences in finite populations. Genetics 105:437–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- True JR, Mercer JM, Laurie CC (1996) Differences in crossover frequency and distribution among three sibling species of Drosophila. Genetics 142:507–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall JD (2003) Estimating ancestral population sizes and divergence times. Genetics 163:395–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, et al (2002) Initial sequencing and comparative analysis of the mouse genome. Nature 420:520–562 [DOI] [PubMed] [Google Scholar]
- Weber JL (2002) The Iceland map. Nat Genet 31:225–226 [DOI] [PubMed] [Google Scholar]
- Yoder AD, Yang Z (2000) Estimation of primate speciation dates using local molecular clocks. Mol Biol Evol 17:1081–1090 [DOI] [PubMed] [Google Scholar]
- Yu A, Zhao C, Fan Y, Jang W, Mungall AJ, Deloukas P, Olsen A, Doggett NA, Ghebranious N, Broman KW, Weber JL (2001) Comparison of human genetic and sequence-based physical maps. Nature 409:951–953 [DOI] [PubMed] [Google Scholar]