

Abstract
The presence of four lysosomal storage diseases (LSDs) at increased frequency in the Ashkenazi Jewish population has suggested to many the operation of natural selection (carrier advantage) as the driving force. We compare LSDs and nonlysosomal storage diseases (NLSDs) in terms of the number of mutations, allele-frequency distributions, and estimated coalescence dates of mutations. We also provide new data on the European geographic distribution, in the Ashkenazi population, of seven LSD and seven NLSD mutations. No differences in any of the distributions were observed between LSDs and NLSDs. Furthermore, no regular pattern of geographic distribution was observed for LSD versus NLSD mutations—with some being more common in central Europe and others being more common in eastern Europe, within each group. The most striking disparate pattern was the geographic distribution of the two primary Tay-Sachs disease mutations, with the first being more common in central Europe (and likely older) and the second being exclusive to eastern Europe (primarily Lithuania and Russia) (and likely much younger). The latter demonstrates a pattern similar to two other recently arisen Lithuanian mutations, those for torsion dystonia and familial hypercholesterolemia. These observations provide compelling support for random genetic drift (chance founder effects, one ∼11 centuries ago that affected all Ashkenazim and another ∼5 centuries ago that affected Lithuanians), rather than selection, as the primary determinant of disease mutations in the Ashkenazi population.
Introduction
The Ashkenazi Jewish (AJ) population is subject to a variety of genetic diseases (Goodman 1979), primarily owing to historical founder effects that have occurred in the Jewish population over the past two millennia. Much attention originally focused on lysosomal storage diseases (LSDs), four of which—Tay-Sachs disease (TSD [MIM 272800]), Gaucher disease (GD [MIM 230800]), Niemann-Pick disease (NPD [MIM 257200]), and mucolipidosis type IV (MLIV [MIM 252650])—occur at increased frequency in the AJ population (Goodman and Motulsky 1979). However, it has also become apparent that mutations for numerous other types of genetic disorders (nonlysosomal storage diseases [NLSDs]) also occur in this population at increased frequency, including Bloom syndrome (BLM [MIM 210900]), Fanconi anemia type C (FACC [MIM 227645]), Canavan disease (CAN [MIM 271900]), familial dysautonomia (DYS [MIM 223900]), factor XI deficiency (F11 [MIM 264900]), familial hyperinsulinism (HI [MIM 256450]), familial hypercholesterolemia (FH [MIM 143890]), cystinuria (CSNU [MIM 220100]), familial neurosensory deafness (NSRD1 [MIM 220290]) due to connexin 26 (CX26 [MIM 121011]), glycogen storage disease type VII (GLY7 [MIM 232800]), and torsion dystonia (DYT1 [MIM 128100]), among others (Goodman and Motulsky 1979; Motulsky 1995).
Over the past decade, developments in molecular biology have allowed for the identification of the specific mutations underlying most of these disorders. Typically, multiple different mutations have been identified for each, although, in most cases, it is a single mutation that appears to predominate.
The anomalous presence of four different LSDs in the AJ population has been the source of long-standing controversy. Many have argued that the low likelihood of four such diseases—particularly when all four are involved in the storage of glycosphingolipids—must reflect past selective advantage for heterozygous carriers of these conditions (Myrianthopoulos and Aronson 1966; Chakravarti and Chakraborty 1978; Motulsky 1979; Yokoyama 1979; Rotter and Diamond 1987; Zlotogora et al. 1988; Jorde 1992; Beutler et al. 1993; Diamond 1994; Boas 2000), whereas others have argued for the predominant role of genetic drift and founder effects (Chase and McKusick 1972; Rao and Morton 1973; Fraikor 1977; Wagener et al. 1978; Cavalli-Sforza 1979; Risch et al. 1995).
The large number of mutations that have been found, in the AJ population, for LSDs has provided some with an argument in favor of the selection hypothesis (Diamond 1994). In particular, statistical analyses have been presented, to confirm the low probability of seeing multiple different mutations for the same disease at increased frequency (Jorde 1992). If selection was the driving force, however, questions remain as to the timing, location, and agent of such selection.
With advances in molecular genetics and large-scale screening, the opportunity to provide further evidence regarding these alternative hypotheses now presents itself. The AJ population is subject to a variety of different inherited medical conditions, including cancers, blood diseases, biochemical disorders, and neuropathies, in addition to the LSDs. It is unlikely that all these conditions have been the subject of carrier selective advantage (especially the dominant conditions—e.g., DYT1, breast cancer, and FH) or at least the subject of the same selective forces, should they exist. Thus, it is informative to compare mutations for the LSDs and NLSDs in several respects, to see if differences exist. In particular, we contrast (1) the number and frequency of mutations for the LSDs and NLSDs; (2) the estimated coalescence times for various mutations obtained, through haplotype analysis (Risch et al. 1995), from LSDs and NLSDs; and (3) the geographic distribution in Europe—in particular, central Europe (CE) versus eastern Europe (EE)—for LSD versus NLSD mutation frequencies. Clear differences in the above distributions between LSDs and NLSDs would support the hypothesis that special selective forces have operated on the former. By contrast, lack of differences between them supports genetic drift as the primary force.
Material and Methods
Analysis of Mutation Frequencies
The published literature was scanned for disease mutations found in the AJ population, and allele frequencies were extracted. For multiple reports of the same mutation, an overall allele frequency was obtained by combining the estimates weighted by sample size. A list of the diseases and mutations included in this analysis are given in appendix A (see table A1).
Analysis of Mutation Ages
Estimates for coalescence times for various mutations were obtained from published reports when available. Coalescence dates not previously published but for which haplotype data were available for such estimation were derived from published haplotype data by use of previously derived formulas (Risch et al. 1995; Goldstein et al. 1999).
Analysis of Geographic Distributions
Analysis of geographic distributions of mutation frequencies was based on new data derived from the Dor Yeshorim genetic-testing program (see Broide et al. 1993).
Study subjects.—Study subjects were generally high-school-age students from the Orthodox Jewish community who agreed to anonymous testing for recessive disease mutations found at increased frequency in the AJ population. Most subjects were self-identified as AJ. All subjects were questioned about country or region of origin of all four grandparents. Approximately 2% of grandparents were identified as Jews of either Sephardic or Asian origin. Informed consent forms were received either from study subjects or from the parents of minors. The number of subjects tested varies depending on the date when analysis of the disease in question was initiated—1992 (for TSD and GD), 1993 (for CF), 1995 (for FACC), or 2000 (for BLM and NPD). Because of uncertain counseling for homozygotes for the most common GD mutation (1226G), GD testing was limited to an initial pilot screening project plus subjects requesting this test.
Dor Yeshorim maintains two intake centers, one in Brooklyn, NY, and the other in Jerusalem. Approximately 45% of subjects were from the New York metropolitan area, and 5% were from the rest of North America; for these subjects, blood samples were drawn and were delivered to the Brooklyn site. Another 45% of subjects were from Israel, and another 5% were from Europe; blood samples from these subjects were sent to the center in Israel.
DNA analysis.—Coded and anonymized blood samples were sent from the two Dor Yeshorim intake centers, in New York and Israel, to various laboratories for DNA analysis. Mutation testing was performed for seven recessive diseases and 14 mutations, by 12 independent quality-controlled laboratories: Mt. Sinai School of Medicine (New York); Albert Einstein College of Medicine (Bronx, NY); Baylor College of Medicine Kleeberg DNA Laboratory (Houston); University of Pittsburgh (Pittsburgh); Kingsbrook Jewish Medical Center (Brooklyn, NY); New York University (New York); Hadassah Hebrew University Hospital (Jerusalem); National Hospital, Institute of Neurology (London); Thomas Jefferson Hospital (Philadelphia); Scripps Research Laboratory (San Diego, CA); Integrated Genetics (Framingham, MA); and Rockefeller University (New York). Different laboratories tested different mutations, and several laboratories received aliquots from the same individual for testing. Results from participating laboratories were entered into a secured computer file by identification number. Double data entry was employed, to eliminate data-entry errors. Random control samples were included in most shipments of samples to each laboratory, to evaluate and rectify any laboratory genotyping problems.
Definition of geographic ancestry.—Grandparents were classified into 12 country/region-of-origin categories, on the basis of sufficient numbers of subjects. These included the countries of Austria, Czechoslovakia, Germany, Hungary, Lithuania, Poland, Romania, and Russia. Grandparents from Galicia were included as Polish, and those from the Ukraine were included as Russian. Grandparents from South Africa were included as Lithuanian, since South African Jews are primarily of Lithuanian origin (Meiner et al. 1991). It must be recognized that these categories are only approximate, since national/regional borders have shifted during the past century. Because of small numbers, Sephardic and North African Jews were combined into a single category, designated as “Mediterranean,” which included all North African countries plus Spain, Portugal, Gibraltar, Greece, Cyprus, Rhodes, Turkey, and Italy. Another combined group, designated as “Mideast,” included those from all countries in the Middle East, India, and southern republics of the former Soviet Union (Georgia, Azerbaijan, Kazakhstan, Armenia, Dagestan, and Uzbekistan).
A mixed, primarily Ashkenazi group, designated as “mixed AJ,” was defined on the basis of other countries of origin with mostly Ashkenazi representation (the United States, Canada, Australia, the United Kingdom, other countries of western Europe, Switzerland, and Latin America). Finally, those with no known country or region of origin were included in the category designated as “unknown.”
Several prior studies have shown a northeastern–south-central cline, in Europe, of allele frequencies for some Ashkenazi diseases and mutations (Petersen et al. 1983; Meiner et al. 1991; Peleg et al. 1994; Risch et al. 1995; Shahrabani-Gargir et al. 1998). Accordingly, we also created a category designated as “CE,” composed of Austria, Czechoslovakia, Hungary, and Romania, and a category designated as “EE,” composed of Poland, Russia, and Lithuania. The former group is primarily Hungarian, and the latter group is primarily Polish. Germany was not included in the CE category, because a prior study of TSD (Petersen et al. 1983) suggested that Germany is more typical of EE countries, possibly owing to migrations from EE in modern times.
Statistical analysis.—To extract the maximum amount of information from our data, we estimated mutation frequencies by country/region of origin through use of maximum likelihood and tested for differences between countries/regions through use of likelihood-ratio tests. The probability model is as follows: For I countries/regions, we define I parameters pi, i=1,…,I, with pi representing the allele frequency for a given mutation in the ith country/region. The probability π that an individual is a carrier is a linear combination of the pi values based on the individual’s four grandparents. For example, if the four grandparents are from categories 2, 4, 5, and 2, then π=p2+(p4+p5)/2. For I countries of origin, there are A=I+3I(I-1)/2+I(I-1)(I-2)/2 + I(I − 1)(I − 2)(I − 3)/24 possible country/region-of-ancestry combinations for four grandparents. The four terms in A, above, correspond to the number of combinations involving 1, 2, 3, or 4 countries/regions, respectively. In our case, for I=12, A=1,365; for I=7, A=210. We enumerate each possible ancestry combination by use of the subscript letter “a,” a=1,…,A. The carrier probability for ancestry combination a is denoted πa.
The data consist of a set of variables Na and Ya, a=1,…,A, where Na is the total number of individuals with ancestry combination a and Ya is the number of such individuals who are mutation carriers. As a close approximation to the binomial, we model the carrier counts Ya by a Poisson distribution with mean Naπa. The log likelihood for the data Ya, aside from a constant, is given by
where each πa is a different linear combination of the pi values. The maximum of lnL as a function of the pi values and the corresponding maximum-likelihood estimates of the pi values were obtained using Matlab (Mathworks).
Differences between CE and EE were tested as follows: First, the seven-parameter model, allowing separate estimates for CE and EE, was fit, and log likelihood (eq. [1]) was estimated. Two of the parameters were for the CE frequency and the EE frequency; the remaining five parameters represented the allele frequency for the unknown, mixed AJ, German, Mediterranean, and Mideast groups. Next, a six-parameter model—allowing a single estimate for CE and EE combined, along with the other five frequencies described above—was fit, and equation (1) was reestimated. Twice the difference in log likelihood was assumed to have a χ2 distribution with 1 df.
Because genetic testing at Dor Yeshorim is anonymized, it is impossible to determine relatedness of study subjects. It is likely that some related individuals are included in the database, and, thus, their carrier status should not technically be considered to be statistically independent. The most serious potential violation of independence is likely to occur through sibships in which more than one sib from the same family has been tested. To assess the potential impact that such nonindependence has on our statistical analyses, we consider the following model. Suppose that there are a total of N families, each with s sibs tested. Let Xij be 1 if the jth sib in the ith family is a carrier and 0 otherwise. Then, the allele-frequency estimate is given by
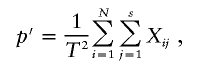 |
where T=2Ns. The variance of p′ is given by
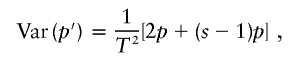 |
where the first term corresponds to the individual variance for each sib and the second term corresponds to the covariance (nonindependence effect) for multiple sibs. Here, we ignore terms with higher orders of p, assuming that p is small. Then, the information ratio (inverse of variance), defined as the actual information divided by the information obtained assuming that sibs are independent, is given by 2/(s+1). If we assume (very conservatively) that the sample contains families with three tested sibs, then the information ratio is 2. Thus, a conservative adjustment to our statistical (χ2) tests would be to divide the obtained χ2 values by 2.
Results
Mutation Frequencies
Table 1 presents mutations for LSDs and NLSDs in descending order of frequency, summarized from the literature. There are no striking differences in terms of the number of mutations or their frequencies between the two groups. The highest frequency among LSDs, 0.032 (for the GD 1226G mutation), is similar to the adenomatous polyposis coli (APC [175100]) I1307K mutation. Both of these mutations have low penetrance and are generally nonlethal. Although TSD and GD both have second mutations with frequency >0.002, the same is true for breast cancer, CAN, F11, NSRD1 due to CX26, and CF.
Table 1.
Ordered Mutation Frequencies for LSDs and NLSDs in the Ashkenazi Population—Data from the Literature
| Disease/Locus (Mutation) | Frequency | Reference(s) |
| LSDs: | ||
| GD (1226) | .032 | Beutler et al. 1993 |
| TSD (1277) | .013 | Paw et al. 1990; Triggs-Raine et al. 1990; Grebner and Tomczak 1991 |
| NPD (1302) | .005 | Caganna et al. 1994 |
| MLIV (IVS3) | .004 | Bargal et al. 2001; Edelmann 2002 |
| TSD (1421) | .003 | Paw et al. 1990; Triggs-Raine et al. 1990; Grebner and Tomczak 1991 |
| GD (84GG) | .002 | Beutler et al. 1993 |
| Other diseases: | ||
| APC (I1307K) | .035 | Laken et al. 1997; Woodage et al. 1998; Rozen et al. 1999; Drucker et al. 2000 |
| F11 (type III) | .025 | Shpilberg et al. 1995 |
| F11 (type II) | .022 | Shpilberg et al. 1995 |
| CX26 (167delT) | .020 | Morrell et al. 1998; Dong et al. 2001 |
| DYS (IVS20) | .016 | Dong et al. 2002 |
| CAN (854) | .012 | Kronn et al. 1995; Matalon et al. 1995 |
| CSNU (808C→T) | .008 | Pras et al. 1995 |
| CF (1282) | .008 | Kerem et al. 1995 |
| FACC (IVS4) | .006 | Verlander et al. 1995; Peleg et al. 2002 |
| BRCA1a (185delAG) | .006 | Roa et al. 1997; Streuwing et al. 1997 |
| BRCA2a (6174delT) | .006 | Oddoux et al. 1997; Roa et al. 1997; Streuwing et al. 1997 |
| HI (3992) | .006 | Nestorowicz et al. 1996 |
| BLM (2281) | .005 | Shahrabani-Gargir et al. 1998; Peleg et al. 2002 |
| CF (F508) | .005 | Kerem et al. 1995 |
| CX26 (35delG) | .004 | Morrell et al. 1998; Dong et al. 2001 |
| FHa (G197del) | .003 | Meiner et al. 1991 |
| CAN (693) | .002 | Kronn et al. 1995; Matalon et al. 1995 |
| GLY7 (del5) | .002 | Sherman et al. 1994 |
Autosomal dominant mutations; all others are autosomal recessive.
Ages of Mutations
Table 2 contains the coalescence times (in generations) for LSD and NLSD mutations reported in the literature, for GD (Diaz et al. 2000), F11 (Goldstein et al. 1999), DYT1 (Risch et al. 1995), BRCA1 (Neuhausen et al. 1996), BRCA2 (Neuhausen et al. 1998), and FH (Durst et al. 2001). Times for other mutations were obtained from published haplotype data and were estimated by use of published formulas (Risch et al. 1995; Goldstein et al. 1999): BLM (Ellis et al. 1998), MLIV (Slaugenhaupt et al. 1999), and DYS (Blumenfeld et al. 1993). The numbers of generations fall approximately into three founder-event categories: >100 generations (founding and expansion of the Jewish population in the Middle East), 50 generations (founding and expansion of Ashkenazi Jews in CE), and 12 generations (founding and expansion of Lithuanian Jews). Although some mutations of the first two groups are found in non-Ashkenazi Jews—in particular, F11 type II (Peretz et al. 1997) and BRCA1 185delAG (Bar-Sade et al. 1997, 1998)—others are restricted primarily to Ashkenazim. The two recent mutations (in DYT1 and FH) are specific to Lithuanian Jews (Meiner et al. 1991; Risch et al. 1995). The three LSD mutations date approximately to the founding of the Ashkenazim, as do most of the NLSD mutations.
Table 2.
Estimated Coalescence Times of Various Mutations
| Disease/Locus (Mutation) | Generations | Reference |
| LSDs: | ||
| GD (1226) | 48 | Diaz et al. 2000 |
| MLIV (IVS3) | 57 | Slaugenhaupt et al. 1999 |
| GD (84GG) | 55 | Diaz et al. 2000 |
| Other diseases: | ||
| F11 (type III) | 31 | Goldstein et al. 1999 |
| F11 (type II) | 120 | Goldstein et al. 2000 |
| DYS (IVS20) | 52 | Blumenfeld et al. 1993 |
| BLM (2281) | 50 | Ellis et al. 1998 |
| BRCA1a (185delAG) | 46 | Neuhausen et al. 1996 |
| BRCA2a (6174delT) | 29 | Neuhausen et al. 1996 |
| FHa (G197del) | 12 | Durst et al. 2001 |
| DYT1 (delGAG) | 12 | Risch et al. 1995 |
Autosomal dominant mutations; all others are autosomal recessive.
Geographic Distribution of Mutations
Here, we present new data on the geographic distribution of 14 mutations for seven diseases, three of which (GD, NPD, and TSD) are LSDs and four of which (BLM, CAN, CF, and FACC) are not. These data derive from the Dor Yeshorim carrier-testing program, initiated in Brooklyn, NY, in 1983, with the purpose of testing marriage-age individuals from the Orthodox Ashkenazi community (Broide et al. 1993). The original program was based on enzyme testing for TSD. In 1992, mutation testing was initiated. Since that time, mutation testing for the six other diseases listed above has been introduced (see the “Material and Methods” section). Study subjects were queried regarding country/region of origin of each of their four grandparents. These responses were condensed into 12 different geographic categories and were used for mutation-frequency estimation by maximum likelihood (see the “Material and Methods” section).
The number of grandparents that study subjects identified by geographic region for each of these seven diseases is given in table 3. The number of subjects tested is one-fourth the total number of grandparents listed in the bottom of the table and varies by year of introduction of the test. Thus, the numbers range from 5,221 subjects tested for NPD to 84,421 subjects tested for CF. The numbers of grandparents listed may include the same individual counted more than once, because there is the possibility that related individuals (e.g., sibs) were included in the database (see the “Material and Methods” section). As can be seen in table 3, ∼41% of grandparents are of unknown origin, and 13% are classified as mixed AJ. Among the remainder, the highest percentages are for Hungary (14%) and Poland (12%), followed by Russia (5%), Romania (4%), Germany (3%), Czechoslovakia (3%), Lithuania (1%), and Austria (1%). Mediterranean and Mideast grandparents each constitute ∼1% of the sample. Because of the particular Orthodox groups included in this testing program, a higher proportion derives from CE (Hungary) than is true for other Ashkenazi samples (Petersen et al. 1983).
Table 3.
Number of Grandparents, as Reported by Tested Individuals, by Country/Region of Origin and Disease Tested
|
No. of Grandparents of Individuals with |
|||||||
| BLM | CAN | CF | FACC | GDa | NPD | TSD | |
| Unknown | 9,749 | 122,506 | 142,978 | 111,610 | 63,197 | 7,183 | 100,293 |
| Mixed AJb | 2,966 | 41,784 | 49,777 | 34,499 | 15,668 | 2,318 | 29,485 |
| Germany | 1,000 | 9,867 | 10,750 | 9,635 | 4,615 | 752 | 29,485 |
| CE: | 6,554 | 62,358 | 68,221 | 60,992 | 32,898 | 4,910 | 55,875 |
| Austria | 301 | 3,066 | 3,384 | 3,007 | 1,583 | 225 | 2,783 |
| Czech | 715 | 7,283 | 8,008 | 7,154 | 4,073 | 545 | 6,437 |
| Hungary | 4,255 | 39,666 | 43,076 | 39,052 | 21,269 | 3,171 | 35,945 |
| Romania | 1,283 | 12,343 | 13,753 | 11,779 | 5,973 | 969 | 10,710 |
| EE: | 6,086 | 55,651 | 59,429 | 54,778 | 26,008 | 4,514 | 50,027 |
| Lithuania | 488 | 4,027 | 4,225 | 3,988 | 1,552 | 409 | 3,718 |
| Polandc | 3,790 | 35,761 | 38,199 | 35,307 | 16,958 | 2,784 | 32,328 |
| Russiad | 1,808 | 15,862 | 17,004 | 15,482 | 7,498 | 1,321 | 13,980 |
| Mediterraneane | 839 | 3,611 | 4,088 | 3,320 | 1,953 | 703 | 2,941 |
| Mideastf | 558 |
2,219 |
2,441 |
2,098 |
1,197 |
504 |
1,896 |
| Total | 27,752 | 297,996 | 337,684 | 276,932 | 145,536 | 20,884 | 249,372 |
Only 48% were tested for the 1604 mutation.
Includes the United States, Canada, Australia, the United Kingdom, western Europe, Switzerland, and Latin America.
Includes Galicia.
Includes Ukraine.
Includes North Africa, Spain, Portugal, Gibraltar, Greece, Cyprus, Rhodes, Turkey, and Italy.
Includes Middle East countries, India, Georgia, Azerbaijan, Kazakhstan, Armenia, Dagestan, and Uzbekistan.
A total of 14 different mutations for seven diseases were studied: BLM (2281), CAN (693 and 854), CF (1282, F508, and 542), FACC (IVS4), GD (1226G, 1604, and 84GG), NPD (1302), and TSD (1277, 1421, and G269). Frequencies for the seven LSD mutations obtained from maximum-likelihood estimation (see the “Material and Methods” section) are given in table 4, and those for the seven NLSD mutations are given in table 5. The individual countries that constitute the CE and EE categories are listen in the tables, beneath those for the combined categories. The bottom row provides the total number of mutation carriers observed among all tested subjects. For the LSDs, four mutations are more common in CE, and three are more common in EE; four of these differences reached statistical significance. Because of the possibility that related individuals were included in the database, the two χ2 values, 6.1 and 6.6, should be considered as suggestive (see the “Material and Methods” section). The others are clearly significant. Similar to the results of Petersen et al. (1983) for enzyme detected carriers, the primary TSD mutation 1277 has a higher frequency in CE versus EE. The same is true for the first (1226G) and, especially, the second (84GG) most common mutations in GD. By contrast, the two less frequent TSD mutations were each more common in EE, especially the 1421 variant.
Table 4.
Mutation Frequencies for LSDs, by Geographic Region
|
Frequency of Mutation in |
|||||||
| GD |
NPD |
TSD |
|||||
| 1226 | 1604 | 84GG | L302 | 1277 | 1421 | G269 | |
| Unknown | .0371 | .0026 | .0053 | .0031 | .0190 | .0009 | .0007 |
| Mixed AJ | .0284 | .0033 | .0016 | 0 | .0145 | .0041 | .0013 |
| Germany | .0367 | .0040 | .0080 | 0 | .0173 | .0019 | .0006 |
| CE: | .0382 | .0023 | .0050 | .0036 | .0218 | 0 | .0003 |
| Austria | .0163 | .0047 | .0027 | 0 | .0464 | 0 | .0058 |
| Czech | .0381 | 0 | .0034 | 0 | .0283 | 0 | 0 |
| Hungary | .0356 | .0023 | .0046 | .0058 | .0198 | 0 | 0 |
| Romania | .0528 | .0033 | .0101 | 0 | .0190 | 0 | 0 |
| EE: | .0303 | .0032 | .0006 | .0010 | .0175 | .0041 | .0010 |
| Lithuania | .0322 | 0 | 0 | 0 | .0211 | .0113 | .0022 |
| Poland | .0311 | .0043 | .0010 | .0016 | .0171 | .0022 | .0013 |
| Russia | .0287 | .0018 | .0001 | 0 | .0168 | .0067 | 0 |
| Mediterranean | .0028 | .0006 | 0 | 0 | 0 | 0 | 0 |
| Mideast | 0 | 0 | 0 | 0 | 0 | .0005 | 0 |
|
Result for Mutation in |
|||||||
| GD |
NPD |
TSD |
|||||
| 1226 | 1604 | 84GG | L302 | 1277 | 1421 | G269 | |
| χ2 (CE vs. EE) | 6.6** | .5 | 28.5*** | 2.8 | 6.1* | 89.2*** | 3.7 |
| No. of carriers | 2,504 | 93 | 289 | 22 | 2,289 | 215 | 88 |
P<.05.
P<.01.
P<10-7.
Table 5.
Mutation Frequencies for Other Diseases, by Geographic Region
|
Frequency of Mutation in |
|||||||
| BLM |
CAN |
CF |
FACC |
||||
| 2281 | 693 | 854 | 1282 | F508 | 542 | IVS4 | |
| Unknown | .0108 | .0020 | .0103 | .0104 | .0064 | .0013 | .0064 |
| Mixed AJ | .0119 | .0012 | .0082 | .0105 | .0068 | .0017 | .0068 |
| Germany | 0 | .0025 | .0049 | .0061 | 0 | .0017 | 0 |
| CE: | .0030 | .0023 | .0110 | .0117 | .0085 | .0007 | .0085 |
| Austria | 0 | .0010 | .0055 | .0210 | .0010 | .0009 | .0039 |
| Czech | 0 | .0029 | .0098 | .0126 | .0050 | 0 | .0065 |
| Hungary | .0044 | .0030 | .0090 | .0102 | .0086 | .0007 | .0095 |
| Romania | 0 | 0 | .0205 | .0136 | .0118 | .0018 | .0015 |
| EE: | .0181 | .0008 | .0107 | .0096 | .0056 | .0010 | .0056 |
| Lithuania | 0 | .0018 | .0200 | .0081 | .0055 | .0006 | .0114 |
| Poland | .0222 | .0009 | .0109 | .0099 | .0058 | .0012 | .0059 |
| Russia | .0152 | .0002 | .0080 | .0059 | .0054 | .0002 | .0038 |
| Mediterranean | 0 | 0 | 0 | 0 | .0085 | .0011 | 0 |
| Mideast | 0 | 0 | 0 | 0 | 0 | .0005 | 0 |
|
Result for Mutation in |
|||||||
| BLM |
CAN |
CF |
FACC |
||||
| 2281 | 693 | 854 | 1282 | F508 | 542 | IVS4 | |
| χ2 (CE vs. EE) | 18.3** | 10.3* | .1 | 3.2 | 9.3* | .5 | 3.3 |
| No. of carriers | 135 | 252 | 1,467 | 1,726 | 1,099 | 206 | 856 |
P<.005.
P<10-4.
The pattern for the NLSDs is similar. Among the six mutations for which a difference exists, four have higher frequency in CE, and two have higher frequency in EE. Among the three with differences that are statistically significant, two (CAN 693 and CF F508) are more common in CE, and one (BLM 2281) is more common in EE. The high frequency of carriers of BLM 2281 from Poland has been noted previously (Shahrabani-Gargir et al. 1998). In our sample, the highest frequency was observed in Galicia.
The similar geographic pattern for the LSD and NLSD mutations argues once again for the primary role of genetic drift or founder effects. Furthermore, for diseases with multiple mutations, there is no consistent geographic pattern. For GD, two mutations (1226G and 84GG) are more common centrally, whereas one (1604) is not; for TSD, one mutation (1277) is more common centrally, but the other two (1421 and G269) show the opposite trend. Similar discordant patterns also exist for CAN and CF.
In tables 4 and 5, the large unknown category approximately represents a mixture of CE and EE Ashkenazim comparable to that observed in the geographically defined sample. When the mutation frequencies in this group are compared with the CE and EE frequencies for the seven mutations with significant eastern-central differences, four are closer to CE, and three are closer to EE. Assuming that the unknown category is admixed between CE and EE, with proportion A from CE, we estimated A for each mutation by the formula A=(punk-pEE)/(pCE-pEE), where punk, pCE, and pEE are the observed mutation frequencies in the unknown, CE, and EE groups, respectively. Admixture estimates A for the seven loci range from 28% to 100%, with an average admixture estimate of 63% CE versus 37% EE. These figures are similar to those observed in the geographically defined sample, in which there are 53% from CE versus 47% from EE. By contrast, the mixed AJ group is much more characteristic of EE. In this group, seven of seven mutations are closer in frequency to EE than CE, and the admixture estimate A ranges from 0 to 41%, with an average estimate of 10% CE versus 90% EE. This finding is consistent with primarily EE Ashkenazi migrations to the countries included in this category.
A similar analysis of Germany reveals four mutation frequencies closer to CE versus three closer to EE. The average admixture estimate for this group is 68% CE versus 32% EE. Germany’s position intermediate between CE and EE likely represents its mix of longer-established central Ashkenazi founders, as well as more-recent immigrants from EE.
Only a few mutations are observed at any frequency in the Mediterranean or Middle Eastern Jews. The CF F508 mutation had a Mediterranean frequency of 0.0085, comparable to the frequencies found in Ashkenazim. Similarly, the 542 mutation is found in both Mediterranean and Mideast groups, whereas the 1282 mutation was restricted to Ashkenazim. These observations are consistent with earlier data from Israeli Jewish populations (Kerem et al. 1995). The only other mutation found outside Ashkenazim is the GD 1226G mutation, in the Mediterranean Jews. This observation is not surprising, given its high frequency and widespread distribution in Europeans in general, especially in Spain and Portugal (Lacerda et al. 1994). Other AJ mutations have also been found at very low frequency in non-Ashkenazi Jews (Peleg et al. 1994). These most likely represent migrations over the past 1,000 years, although earlier shared origins cannot be excluded.
Discussion
We have shown that no differences exist between LSDs and NLSDs in the AJ population with respect to (1) number and frequency of disease mutations, (2) coalescence times of the various mutations, and (3) the geographic distribution of mutation frequencies by country/region of origin in Europe. Thus, there is no evidence that special selective forces have operated on the LSDs, as compared with the broad array of other genetic diseases found in the AJ population.
In addition, we found highly disparate geographic distributions for mutations within single disease entities—in particular, GD, TSD, and CAN. Again, these data are more consistent with random founder effects than with systematic selection. In particular, three mutations appear to demonstrate impressive founder effects: BLM 2281, in Poland/Galicia; GD 84GG, in Hungary/Romania; and TSD 1421, in Lithuania/Russia.
The results for the TSD 1421 mutation are the most striking. We observed extreme and highly significant geographic variation, with a total absence from CE, a frequency of 0.7% in Russia, and a frequency of 1.1% in Lithuania. This eastern predominance and central absence is also reflected in the unknown and mixed AJ groups, in which the mutation frequency in the former (0.0009), a mostly CE group, is lower than in the latter (0.0041), a mostly EE group. This pattern is also remarkably similar to two other AJ mutations: delGAG, for DYT1 (Risch et al. 1995), and G197del, for FH (Meiner et al. 1991; Durst et al. 2001). On the basis of haplotype analysis, both of these mutations were dated to a coalescence time of 12 generations (range 8–20) consistent with more-recent founder events in the Lithuanian Jewish population (Risch et al. 1995; Durst et al. 2001). The extreme localization—both in time and space—observed for the TSD 1421 mutation, as compared with the primary TSD 1277 mutation, along with the other data reported above, provides compelling evidence for genetic drift and not selection as the explanation for the observed frequency of LSD mutations in the Ashkenazi population.
Appendix A
Table A1.
Diseases and Mutations Included in the Present Analysis[Note]
| Disease/Gene | Mutation(s) |
| Adenomatous polyposis coli (APC [*175100]) | I1307K |
| Bloom syndrome (BLM [#210900]) | 2281(−6del,+7ins) |
| Breast cancer: | |
| BRCA1 (*113705) | 185delAG |
| BRCA2 (*600185) | 6174delT |
| Canavan disease (CAN [*271900]) | 693(Y231X); 854(E285A) |
| Cystic fibrosis (CF [#219700]) | F508; W1282X |
| Cystinuria (CSNU [#220100]) | 808C→T |
| Deafness (NSRD1 [#220290]) due to connexin 26 (CX26 [#121011]) | 167delT; 35delG |
| Dysautonomia (DYS [#223900]) | IVS20 |
| Factor 11 deficiency (F11 [*264900]) | II (E117X); III (F283L) |
| Familial hypercholesterolemia (FH [#143890]) | G197del |
| Familial hyperinsulinism (HI [#256450]) | 3992(G→A) |
| Fanconi anemia type C (FACC [*227645]) | IVS4 |
| Gaucher disease (GD [#230800]) | 1226G(N370S); 84GG; 1604(R496H) |
| Glycogen storage disease VII (GLY7 [*232800]) | del5 |
| Mucolipidosis IV (MLIV [#252650]) | IVS3 |
| Niemann-Pick disease (NPD [#257200]) | L302P |
| Tay-Sachs disease (TSD [#272800]) | 1277(TATC); 1421(IVS12); G269 |
| Torsion dystonia (DYT1 [#128100]) | delGAG |
Note.— Derived from OMIM.
Electronic-Database Information
Accession numbers and the URL for data presented herein are as follows:
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for APC [MIM 175100], BLM [MIM 210900], BRCA1 [MIM 113705], BRCA2 [MIM 600185], CAN [MIM 271900], CF [MIM 219700], CSNU [MIM 220100], CX26 [MIM 121011], DYS [MIM 223900], DYT1 [MIM 128100], FACC [MIM 227645], FH [MIM 143890], F11 [MIM 264900], GD [MIM 230800], GLY7 [MIM 232800], HI [MIM 256450], MLIV [MIM 252650], NPD [MIM 257200], NSRD1 [MIM 220290], and TSD [MIM 272800])
References
- Bargal R, Avidan N, Olender T, Ben Asher E, Zeigler M, Raas-Rothschild A, Frumkin A, Ben-Yoseph O, Friedlander YA, Lancet D, Bach G (2001) Mucolipidosis type IV: novel MCOLN1 mutations in Jewish and non-Jewish patients and the frequency of the disease in the Ashkenazi Jewish population. Hum Mutat 17:397–402 [DOI] [PubMed] [Google Scholar]
- Bar-Sade RB, Kruglikova A, Modan B, Gak E, Hirsch-Yechezkel G, Theodor L, Novikov I, Gershoni-Baruch R, Risel S, Papa MZ, Ben-Baruch G, Friedman E (1998) The 185delAG BRCA1 mutation originated before the dispersion of Jews in the diaspora and is not limited to Ashkenazim. Hum Mol Genet 7:801–805 [DOI] [PubMed] [Google Scholar]
- Bar-Sade RB, Theodor L, Gak E, Kruglikova A, Hirsch-Yechezkel G, Modan B, Kuperstein G, Seligsohn U, Rechavi G, Friedman E (1997) Could the 185delAG BRCA1 mutation be an ancient Jewish mutation? Eur J Hum Genet 5:413–416 [PubMed] [Google Scholar]
- Beutler E, Nguyen NF, Henneberger MW, Smolec JM, McPherson RA, West C, Gelbert T (1993) Gaucher disease: gene frequencies in the Ashkenazi Jewish population. Am J Hum Genet 52:85–88 [PMC free article] [PubMed] [Google Scholar]
- Blumenfeld A, Slaugenhaupt SA, Axelrod FB, Lucente DE, Maayan C, Liebert CB, Ozelius LJ, et al (1993) Localization of the gene for familial dysautonomia on chromosome 9 and definition of DNA markers for genetic diagnosis. Nat Genet 4:160–164 [DOI] [PubMed] [Google Scholar]
- Boas FE (2000) Linkage to Gaucher mutations in the Ashkenazi population: effect of drift on decay of linkage disequilibrium and evidence for heterozygote selection. Blood Cells Mol Dis 26:348–359 [DOI] [PubMed] [Google Scholar]
- Broide E, Zeigler M, Eckstein J, Bach G (1993) Screening for carriers of Tay-Sachs disease in the ultraorthodox Ashkenazi Jewish community in Israel. Am J Med Genet 47:213–215 [DOI] [PubMed] [Google Scholar]
- Caganna M, Eng CM, Desnick RJ, Schuchman EH (1994) Molecular population studies of Niemann-Pick disease type A. Am J Hum Genet Suppl 55:A147 [Google Scholar]
- Cavalli-Sforza LL (1979) The Ashkenazi gene pool: interpretations. In: Goodman RM, Motulsky AG (eds) Genetic diseases among Ashkenazi Jews. Raven Press, New York, pp 93–104 [Google Scholar]
- Chakravarti A, Chakraborty R (1978) Elevated frequency of Tay-Sachs disease among Ashkenazic Jews unlikely by genetic drift alone. Am J Hum Genet 30:256–261 [PMC free article] [PubMed] [Google Scholar]
- Chase GA, McKusick VA (1972) Controversy in human genetics: founder effect in Tay-Sachs disease. Am J Hum Genet 24:339–340 [PMC free article] [PubMed] [Google Scholar]
- Diamond JM (1994) Jewish lysosomes. Nature 368:291–292 [DOI] [PubMed] [Google Scholar]
- Diaz GA, Gelb BD, Risch N, Nygaard TG, Frisch A, Cohen IJ, Miranda CS, Amaral O, Maire I, Poenaru L, Caillaud C, Weizberg M, Mistry P, Desnick RJ (2000) Gaucher disease: the origins of the Ashkenazi N370S and 84GG acid β-glucosidase mutations. Am J Hum Genet 66:1821–1832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Edelmann L, Bajwa AM, Kornreich R, Desnick RJ (2002) Familial dysautonomia: detection of the IKBKAP IVS20+6T→C and R696P mutations and frequencies among Ashkenazi Jews. Am J Med Genet 110:253–257 [DOI] [PubMed] [Google Scholar]
- Dong J, Katz DR, Eng CM, Kornreich R, Desnick RJ (2001) Nonradioactive detection of the common connexin 26 167delT and 35delG mutations and frequencies among Ashkenazi Jews. Mol Genet Metab 73:160–163 [DOI] [PubMed] [Google Scholar]
- Drucker L, Shpilberg O, Neumann A, Shapira J, Stackievicz R, Beyth Y, Yarkoni S (2000) Adenomatous polyposis coli I1307 mutation in Jewish patients with different ethnicity: prevalence and phenotype. Cancer 88:755–760 [PubMed] [Google Scholar]
- Durst R, Colombo R, Shpitzen S, Avi LB, Friedlander Y, Wexler R, Raal FJ, Marais DA, Defesche JC, Mandelshtam MY, Kotze MJ, Leitersdorf E, Meiner V (2001) Recent origin and spread of a common Lithuanian mutation, G197del LDLR, causing familial hypercholesterolemia: positive selection is not always necessary to account for disease incidence among Ashkenazi Jews. Am J Hum Genet 68:1172–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelmann L, Dong J, Desnick RJ, Kornreich R (2002) Carrier screening for mucolipidosis type IV in the American Ashkenazi Jewish population. Am J Hum Genet 70:1023–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis NA, Ciocci S, Proytcheva M, Lennon D, Groden J, German J (1998) The Ashkenazic Jewish Bloom syndrome mutation blmAsh is present in non-Jewish Americans of Spanish ancestry. Am J Hum Genet 63:1685–1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraikor AL (1977) Tay Sachs disease: genetic drift among Ashkenazim Jews. Soc Biol 24:117–134 [DOI] [PubMed] [Google Scholar]
- Goldstein DB, Reich ED, Bradman N, Usher S, Seligsohn U, Peretz H (1999) Age estimates of two common mutations causing factor XI deficiency: recent genetic drift is not necessary for elevated disease incidence among Ashkenazi Jews. Am J Hum Genet 64:1071–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman RM (1979) Genetic disorders among the Jewish people. Johns Hopkins University Press, Baltimore [Google Scholar]
- Goodman RM, Motulsky AG (eds) (1979) Genetic diseases among Ashkenazi Jews. Raven Press, New York [Google Scholar]
- Grebner EE, Tomczak J (1991) Distribution of three α-chain β hexosaminidase A mutations among Tay-Sachs carriers. Am J Hum Genet 48:604–607 [PMC free article] [PubMed] [Google Scholar]
- Jorde LB (1992) Genetic diseases in the Ashkenazi population: evolutionary considerations. In: Bonne-Tamir B, Adam A (eds) Genetic diversity among Jews. Oxford University Press, New York, pp 305–318 [Google Scholar]
- Kerem E, Kalman YM, Yahav Y, Shoshani T, Abeliovich D, Szeinberg A, Rivlin J, et al (1995) Highly variable incidence of cystic fibrosis and different mutation distribution among different Jewish ethnic groups in Israel. Hum Genet 96:193–197 [DOI] [PubMed] [Google Scholar]
- Kronn D, Oddoux C, Phillips J, Ostrer H (1995) Prevalence of Canavan disease heterozygotes in the New York metropolitan Ashkenazi Jewish population. Am J Hum Genet 57:1250–1252 [PMC free article] [PubMed] [Google Scholar]
- Lacerda L, Amaral O, Pinto R, Oliveira P, Aerts J, Sa Miranda MC (1994) Gaucher disease: N370S glucocerebrosidase gene frequency in the Portuguese population. Clin Genet 45:298–300 [DOI] [PubMed] [Google Scholar]
- Laken SJ, Petersen GM, Gruber SB, Oddoux C, Ostrer H, Giardiello FM, Hamilton SR, Hampel H, Markowitz A, Klimstra D, Jhanwar S, Winawer S, Offit K, Luce MC, Kinzler KW, Vogelstein B (1997) Familial colorectal cancer in Ashkenazim due to a hypermutable tract in APC. Nat Genet 17:79–83 [DOI] [PubMed] [Google Scholar]
- Matalon R, Kaul R, Aloya M, Jin M, Stoiloff S, Nallasivam P, Balamurugan K, Michals K, Levy PS (1995) Canavan-disease–carrier rate of 1/36 among Ashkenazi Jews. Pediatr Res 37 Part 2:A150 [Google Scholar]
- Meiner V, Landsberger D, Berkman N, Reshef A, Segal P, Seftel HC, van der Westhuyzen DR, Jeenah MS, Coetzee GA, Leitersdorf E (1991) A common Lithuanian mutation causing familial hypercholesterolemia in Ashkenazi Jews. Am J Hum Genet 49:443–449 [PMC free article] [PubMed] [Google Scholar]
- Morell RJ, Kim HJ, Hood LJ, Goforth L, Friderici K, Fisher R, Van Camp G, Berlin CI, Oddoux C, Ostrer H, Keats B, Friedman TB (1998) Mutations in the connexin 26 gene (GJB2) among Ashkenazi Jews with nonsyndromic recessive deafness. N Engl J Med 339:1500–1505 [DOI] [PubMed] [Google Scholar]
- Motulsky AG (1979) Possible selective effects of urbanization on Ashkenazi Jewish populations. In: Goodman RM, Motulsky AG (eds) Genetic disease among Ashkenazi Jews. Raven Press, New York, pp 301–312 [Google Scholar]
- ——— (1995) Jewish diseases and origins. Nat Genet 9:99–101 [DOI] [PubMed] [Google Scholar]
- Myrianthopoulos NC, Aronson SM (1966) Population dynamics of Tay-Sachs disease. I. Reproductive fitness and selection. Am J Hum Genet 18:313–327 [PMC free article] [PubMed] [Google Scholar]
- Nestorowicz A, Wilson BA, Schoor KP, Inoue H, Glaser B, Landau H, Stanley CA, Thornton PS, Clement JP IV, Bryan J, Aguilar-Bryan L, Permutt MA (1996) Mutations in the sulfonylurea receptor gene are associated with familial hyperinsulinism in Ashkenazi Jews. Hum Mol Genet 5:1813–1822 [DOI] [PubMed] [Google Scholar]
- Neuhausen SL, Godwin AK, Gershoni-Baruch R, Schubert E, Garber J, Stoppa-Lyonnet D, Olah E, et al (1998) Haplotype and phenotype analysis of nine recurrent BRCA2 mutations in 111 families: results of an international study. Am J Hum Genet 62:1381–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhausen SL, Mazoyer S, Friedman L, Stratton M, Offit K, Califo A, Tomlinson G, Cannon-Albright L, Bishop T, Kelsell D, Solomon E, Weber B, Couch F, Struewing J, Tonin P, Durocher F, Narod S, Skolnick MH, Lenoir G, Serova O, Ponder B, Stoppa-Lyonnet D, Easton D, King MC, Goldgar DE (1996) Haplotype and phenotype analysis of six recurrent BRCA1 mutations in 61 families: results of an international study. Am J Hum Genet 58:271–280 [PMC free article] [PubMed] [Google Scholar]
- Oddoux C, Struewing JP, Clayton CM, Neuhausen S, Brody LC, Kaback M, Haas B, Norton L, Borgen P, Jhanwar S, Goldgar D, Ostrer H, Offit K (1997) The carrier frequency of the BRCA2 6174delT mutation among Ashkenazi Jewish individuals is approximately 1%. Nat Genet 14:188–190 [DOI] [PubMed] [Google Scholar]
- Paw BH, Tieu PT, Kaback MM, Lim J, Neufeld EF (1990) Frequency of three Hex A mutant alleles among Jewish and non-Jewish carriers identified in a Tay-Sachs screening program. Am J Hum Genet 47:698–705 [PMC free article] [PubMed] [Google Scholar]
- Peleg L, Karpati M, Gazit R, Raas-Rothschild A, Goldman B (1994) Mutations of the hexosaminidase A gene in Ashkenazi and non-Ashkenazi Jews. Biochem Med Metab Biol 52:22–26 [DOI] [PubMed] [Google Scholar]
- Peleg L, Pesso R, Goldman B, Dotan K, Omer M, Friedman E, Berkenstadt M, Reznik-Wolf H, Barkai G (2002) Bloom syndrome and Fanconi’s anemia: rate and ethnic origin of mutation carriers in Israel. Isr Med Assoc J 4:95–97 [PubMed] [Google Scholar]
- Peretz H, Mulai A, Usher S, Zivelin A, Segal A, Weisman Z, Mittelman M, Lupo H, Lanir N, Brenner B, Shpilberg O, Seligsohn U (1997) The two common mutations causing factor XI deficiency in Jews stem from distinct founders: one of ancient Middle Eastern origin and another of more recent European origin. Blood 90:2654–2659 [PubMed] [Google Scholar]
- Petersen GM, Rotter JI, Cantor RM, Field LL, Greenwald S, Lim JS, Roy C, Schoenfeld V, Lowden JA, Kaback MM (1983) The Tay-Sachs disease gene in North American Jewish populations: geographic variations and origin. Am J Hum Genet 35:1258–1269 [PMC free article] [PubMed] [Google Scholar]
- Pras E, Raben N, Golomb E, Arber N, Aksentijevich I, Schapiro JM, Aarel D, et al (1995) Mutations in the SLC3A1 transporter gene in cystinuria. Am J Hum Genet 56:1297–1303 [PMC free article] [PubMed] [Google Scholar]
- Rao DC, Morton NE (1973) Large deviations in the distribution of rare genes. Am J Hum Genet 25:594–597 [PMC free article] [PubMed] [Google Scholar]
- Risch N, deLeon D, Ozelius L, Kramer P, Almasy L, Singer B, Fahn S, Breakefield X, Bressman S (1995) Genetic analysis of idiopathic torsion dystonia in Ashkenazi Jews and their recent descent from a small founder population. Nat Genet 9:152–159 [DOI] [PubMed] [Google Scholar]
- Roa BB, Boyd AA, Volcik K, Richards CS (1997) Ashkenazi Jewish population frequencies for common mutations in BRCA1 and BRCA2. Nat Genet 14:185–187 [DOI] [PubMed] [Google Scholar]
- Rotter JI, Diamond JM (1987) What maintains the frequencies of human genetic diseases? Nature 329:289–290 [DOI] [PubMed] [Google Scholar]
- Rozen P, Shomrat R, Strul H, Naiman T, Karminsky N, Legum C, Orr-Urtreger A (1999) Prevalence of the I1307K APC gene variant in Israeli Jews of differing ethnic origin and risk for colorectal cancer. Gastroenterology 116:54–57 [DOI] [PubMed] [Google Scholar]
- Shahrabani-Gargir L, Shomrat R, Yaron Y, Orr-Urtreger A, Groden J, Legum C (1998) High frequency of a common Bloom syndrome Ashkenazi mutation among Jews of Polish origin. Genet Test 2:293–296 [DOI] [PubMed] [Google Scholar]
- Sherman JB, Raben N, Nicastri C, Argov Z, Nakajima H, Adams EM, Eng CM, Cowan TM, Plotz PH (1994) Common mutations in the phosphofructokinase-M gene in Ashkenazi Jewish patients with glycogenesis VII—and their population frequency. Am J Hum Genet 55:305–313 [PMC free article] [PubMed] [Google Scholar]
- Shpilberg O, Peretz H, Zivelin A, Yatuv R, Chetrit A, Kulka T, Stern C, Weiss E, Seligsohn U (1995) One of the two common mutations causing factor XI deficiency in Ashkenazi Jews (type II) is also prevalent in Iraqi Jews, who represent the ancient gene pool of Jews. Blood 85:429–432 [PubMed] [Google Scholar]
- Slaugenhaupt SA, Acierno JS Jr, Helbling LA, Bove C, Goldin E, Bach G, Schiffmann R, Gusela JF (1999) Mapping of the mucolipidosis type IV gene to chromosome 19p and definition of founder haplotypes. Am J Hum Genet 65:773–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struewing JP, Hartge P, Wacholder S, Baker SM, Berlin M, McAdams M, Timmerman MM, Brody C, Tucker MA (1997) The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N Engl J Med 336:1401–1408 [DOI] [PubMed] [Google Scholar]
- Triggs-Raine BL, Feigenbaum AS, Natowicz M, Skomorowski MA, Schuster SM, Clarke JT, Mahuran DJ, Kolodny EH, Gravel RA (1990) Screening for carriers of Tay-Sachs disease among Ashkenazi Jews: a comparison of DNA-based and enzyme-based tests. N Engl J Med 323:6–12 [DOI] [PubMed] [Google Scholar]
- Verlander PC, Kaporis A, Liu Q, Zhang Q, Seligsohn U, Auerbach AD (1995) Carrier frequency of the IVS+4 A→T mutation of the Fanconi anemia gene FAC in the Ashkenazi Jewish population. Blood 86:4034–4038 [PubMed] [Google Scholar]
- Wagener D, Cavalli-Sforza LL, Barakat R (1978) Ethnic variation of genetic disease: roles of drift for recessive lethal genes. Am J Hum Genet 30:262–270 [PMC free article] [PubMed] [Google Scholar]
- Woodage T, King SM, Wacholder S, Hartge P, Struewing JP, McAdams M, Laken SJ, Tucker MA (1998) The APC I1307K allele and cancer risk in a community-based study of Ashkenazi Jews. Nat Genet 20:62–65 [DOI] [PubMed] [Google Scholar]
- Yokoyama S (1979) Role of genetic drift in the high frequency of Tay-Sachs disease among Ashkenazic Jews. Ann Hum Genet 43:133–136 [DOI] [PubMed] [Google Scholar]
- Zlotogora J, Zieglier M, Bach G (1988) Selection in favor of lysosomal storage disorders? Am J Hum Genet 42:271–273 [PMC free article] [PubMed] [Google Scholar]