

Abstract
One way to perform linkage-disequilibrium (LD) mapping of genetic traits is to use single markers. Since dense marker maps—such as single-nucleotide polymorphism and high-resolution microsatellite maps—are available, it is natural and practical to generalize single-marker LD mapping to high-resolution haplotype or multiple-marker LD mapping. This article investigates high-resolution LD-mapping methods, for complex diseases, based on haplotype maps or microsatellite marker maps. The objective is to explore test statistics that combine information from haplotype blocks or multiple markers. Based on two coding methods, genotype coding and haplotype coding, Hotelling’s T2 statistics TG and TH are proposed to test the association between a disease locus and two haplotype blocks or two markers. The validity of the two T2 statistics is proved by theoretical calculations. A statistic TC, an extension of the traditional χ2 method of comparing haplotype frequencies, is introduced by simply adding the χ2 test statistics of the two haplotype blocks together. The merit of the three methods is explored by calculation and comparison of power and of type I errors. In the presence of LD between the two blocks, the type I error of TC is higher than that of TH and TG, since TC ignores the correlation between the two blocks. For each of the three statistics, the power of using two haplotype blocks is higher than that of using only one haplotype block. By power comparison, we notice that TC has higher power than that of TH, and TH has higher power than that of TG. In the absence of LD between the two blocks, the power of TC is similar to that of TH and higher than that of TG. Hence, we advocate use of TH in the data analysis. In the presence of LD between the two blocks, TH takes into account the correlation between the two haplotype blocks and has a lower type I error and higher power than TG. Besides, the feasibility of the methods is shown by sample-size calculation.
Introduction
With the development of the Human Genome Project and of high-resolution microsatellite and early chromosomewide haplotype maps of the human genome, enormous amounts of genetic data on human chromosomes are becoming available. The opportunities for genomewide scans to map complex-disease genes are tremendous. However, it is not yet clear how to extract the most useful information for mapping complex-disease genes. To fully utilize the massive amount of genetic data for mapping complex-disease genes, novel mathematical and statistical methods are crucial. One urgent need is to explore statistical approaches of high-resolution haplotype or multiple-marker linkage disequilibrium (LD) mapping of complex diseases. One way to perform LD mapping of genetic traits is to use single markers. Since dense marker maps—such as single-nucleotide polymorphism (SNP) and high-resolution microsatellite maps—are available (Broman et al. 1998; The International SNP Map Work Group 2001; Kong et al. 2002), it is natural and practical to generalize single-marker LD mapping to high-resolution haplotype or multiple-marker LD mapping. With the recent discovery of haplotype block structures in the human genome and with the development of early chromosomewide haplotype maps, it is important to develop better statistical methods for analysis of data on SNPs, haplotype patterns, and related patterns of LD. The chromosomewide haplotype maps are expected to be key resources for mapping complex-disease genes. For example, a systematic case-control analysis of common haplotype variants in the human genome would reveal major causative genetic contributions to a disease.
For a case-control study, one can use a χ2 statistic to test the null hypothesis that the marker allele or haplotype frequencies are equal in the cases and controls on the basis of a multiple-allele marker (Olson and Wijsman 1984; Chapman and Wijsman 1998; Nielsen et al. 1998; Kaplan and Morris 2001). The method, however, can not be directly used for multiple markers or haplotype blocks, since the phase of a double heterozygote may be unknown (Ott 1999, p. 7). For multiple biallelic markers, such as SNPs, Xiong et al. (2002) proposed a Hotelling’s T2 statistic for LD mapping of qualitative traits for case-control studies, which can not be used for haplotype block data with multiple haplotypes. Hence, it is necessary to develop methods of genomic LD mapping of qualitative trait loci based on haplotype block data or multiple-marker data for case-control studies.
This paper investigates methods of high-resolution LD mapping for complex diseases based on haplotype maps or microsatellite marker maps. The objective is to explore test statistics that combine information from haplotype blocks or multiple markers. For uniformity of notation, we use “haplotype blocks” or simply “blocks” in our analysis, which can be changed to “multiallelic markers.” We are interested in developing statistical methods for efficient use of genomic patterns of LD to identify genetic variants that contribute to qualitative complex diseases on the basis of multiallelic markers or haplotype block data in a case-control study. Based on two coding methods, genotype coding and haplotype coding, we propose Hotelling’s T2 statistics to test the association between a disease locus and two haplotype blocks. The statistical property of the above T2 statistics will be investigated. An extension of traditional χ2 method of comparing haplotype frequencies is proposed by simply adding two χ2 test statistics of the two haplotype blocks together. The merit of the three methods will be explored by calculation and comparison of power and type I errors. Also, the feasibility of the methods is shown by calculation of sample sizes.
Methods
Test Statistics
Suppose that a disease locus D is flanked by two haplotype blocks H1 and H2, where H1 is a haplotype block on the left-hand side of D and H2 is a haplotype block on the right-hand side. Let us denote the haplotypes of block H1 by H11,…,H1l and the haplotypes of block H2 by H21,…,H2r, where l and r denote the number of observed haplotypes of blocks H1 and H2, respectively. Consider a case-control design with N cases from an affected population and M controls from a unaffected population. Let us define a coding vector for each case or control by one of the following two ways (Schaid 1996, p. 430).
Genotype coding
For the ith case, let Hap1i be his/her two haplotypes at block H1, and let Hap2i be his/her two haplotypes at block H2. Depending on the haplotypes Hap1i (or Hap2i), let us define an indicator vector X1i (or X2i) that contains exactly one component with value 1 and other components with value 0. That is, X1i=[x1i1,…,x1i(l-1),x1i12,…,x1i1l,…,x1i(l-1)l]τ, X2i=[x2i1,…,x2i(r-1),x2i12,…,x2i1r,…,x2i(r-1)r]τ, and
 |
where the indicator variables x1ij, x1ijk,j<k, x2is, and x2ist,s<t are defined by
 |
The dimension of X1i (or X2i) is l(l+1)/2-1 (or r(r+1)/2-1)—that is, the total number l(l+1)/2 (or r(r+1)/2) of genotypes of haplotype block H1 (or H2) minus 1 to remove the redundancy.
Haplotype coding
Define Xi=[z1i1,…,z1i(l-1),z2i1,…,z2i(r-1)]τ, where zuij is the number of haplotypes Huj for the ith case, i.e.,
 |
For the ith control, one may define a vector Yi in the same way. To illustrate the above coding methods, table1 gives an example of genotype and haplotype codings for a block H1 with three haplotypes. The coding method for block H2 is similar.
Table 1.
Genotype and Haplotype Codings for a Block H1 with Three Haplotypes
|
Coding for Method |
||
| Hap1i | Genotype Coding | Haplotype Coding |
| H11H11 | 10000 | 20 |
| H12H12 | 01000 | 02 |
| H13H13 | 00000 | 00 |
| H11H12 | 00100 | 11 |
| H11H13 | 00010 | 10 |
| H12H13 | 00001 | 01 |
Let  and
and  be the mean vectors. Define a pooled-sample variance-covariance matrix by
be the mean vectors. Define a pooled-sample variance-covariance matrix by
 |
A Hotelling’s T2 statistic can be defined as
 |
(Hotelling 1931; Anderson 1984). Hereafter, we will denote the Hotelling’s T2 for haplotype coding as TH and the Hotelling’s T2 for genotype coding as TG. Assume that the sample sizes N and M are sufficiently large that the large-sample theory applies. Under the null hypothesis of no association, the statistic TH (or TG) is asymptotically distributed as central χ2 with l+r-2 (or [l(l+1)/2-1]+[r(r+1)/2-1]) df. Under the alternative hypothesis of association, TH (or TG) is asymptotically distributed as noncentral χ2. If only one haplotype block H1 is used in the analysis, the Hotelling’s T2 for haplotype coding will be denoted as TH1, and the Hotelling’s T2 for genotype coding will be denoted as TG1. Under the null hypothesis of no association, the statistic TH1 (or TG1) is asymptotically distributed as central χ2 with l-1 (or l(l+1)/2-1) df. Under the alternative hypothesis of association, TH1 (or TG1) is asymptotically distributed as noncentral χ2. Similarly, one may introduce test statistics TH2 (or TG2) if only one haplotype block H2 is used in the analysis.
If each of haplotype block Hu has only two haplotypes Hu1,Hu2,u=1,2, then the Hotelling’s T2 by haplotype coding described above coincides with the test statistic introduced by Xiong et al. (2002). To see this, notice that zui1=1+(zui1-1), where zui1-1 is equal to the indicator variable Xij (defined in Xiong et al. [2002], p. 1257). Hence, our method generalizes the method of using two biallelic markers in Xiong et al. (2002) to two haplotype blocks with multiple haplotypes.
In the definition above, we consider only two haplotype blocks H1 and H2. In practice, the test statistics TH and TG can be easily generalized to multiple haplotype blocks. To make the notation as simple as possible, we will focus on two haplotype blocks throughout the present article. In appendices A, B, and C, we will justify the use of the Hotelling’s T2 as an appropriate statistic to test association between the disease locus and the haplotype blocks by either the genotype coding method or the haplotype coding method. The basic idea is to show that the expectation of difference  is equal to 0 if there is no association between the disease locus and the haplotype blocks. Then one may construct a test statistic based on the difference vector
is equal to 0 if there is no association between the disease locus and the haplotype blocks. Then one may construct a test statistic based on the difference vector  , which leads to the Hotelling’s T2.
, which leads to the Hotelling’s T2.
Noncentrality Parameters
Let ΣA1=Cov{[z1i1,…,z1i(l-1)]|Aff} and ΣA2=Cov{[z2i1,…, z2i(r-1)]|Aff} be variance-covariance matrices of vectors [z1i1,…,z1i(l-1)]τ and [z2i1,…,z2i(r-1)]τ, respectively, in affected individuals. Similarly, let  z1i(l-1)]|Unaff} and
z1i(l-1)]|Unaff} and  z2i(r-1)]|Unaff} be variance-covariance matrices of column vectors [z1i1,…,z1i(l-1)]τ and [z2i1,…,z2i(r-1)]τ in controls. Let
z2i(r-1)]|Unaff} be variance-covariance matrices of column vectors [z1i1,…,z1i(l-1)]τ and [z2i1,…,z2i(r-1)]τ in controls. Let 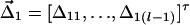 (or
(or  ) be the column vector of measures of LD between haplotype block H1 (or H2) and the disease locus D. Let αD be the average effect of gene substitution and
) be the column vector of measures of LD between haplotype block H1 (or H2) and the disease locus D. Let αD be the average effect of gene substitution and  , and let A be the disease prevalence in population and
, and let A be the disease prevalence in population and  (appendix A). Based on E(zuij|Aff) and E(zuij|Unaff), given in equation (A5) of appendix A, the noncentrality parameter λHu of Hotelling’s test statistic THu is given by
(appendix A). Based on E(zuij|Aff) and E(zuij|Unaff), given in equation (A5) of appendix A, the noncentrality parameter λHu of Hotelling’s test statistic THu is given by
 |
The elements of variance-covariance matrices ΣAu and  are calculated in appendix D. If the haplotype Hu has only two haplotypes—Hu1,Hu2 and N=M—then λHu = 4NΔ2u1(αD/A −
are calculated in appendix D. If the haplotype Hu has only two haplotypes—Hu1,Hu2 and N=M—then λHu = 4NΔ2u1(αD/A −  + Var(zui1|Unaff)]-1, where Var(zui1|Aff) and Var(zui1|Unaff) are given in equations (D1) and (D2) in appendix D.
+ Var(zui1|Unaff)]-1, where Var(zui1|Aff) and Var(zui1|Unaff) are given in equations (D1) and (D2) in appendix D.
Let ΣA=Cov{[z1i1,…,z1i(l-1),z2i1,…,z2i(r-1)]|Aff} be a variance-covariance matrix of column vector [z1i1,…,z1i(l-1),z2i1,…,z2i(r-1)]τ in affected individuals. Similarly, let  Unaff} be a variance-covariance matrix of column vector [z1i1,…,z1i(l-1),z2i1,…,z2i(r-1)]τ in controls. Let us denote
Unaff} be a variance-covariance matrix of column vector [z1i1,…,z1i(l-1),z2i1,…,z2i(r-1)]τ in controls. Let us denote
 |
Then the noncentrality parameter λH of Hotelling’s test statistic TH is given by
 |
The elements of variance-covariance matrices ΣA and  are calculated in appendices D and E. The noncentrality parameter λG (or λG1 or λG2) of TG (or TG1 or TG2) is given in appendix F.
are calculated in appendices D and E. The noncentrality parameter λG (or λG1 or λG2) of TG (or TG1 or TG2) is given in appendix F.
For a case-control study using only one haplotype block H1, one may use a χ2 statistic to test the null hypothesis that the haplotype frequencies are equal in the cases and controls (Olson and Wijsman 1994; Chapman and Wijsman 1998; Kaplan and Morris 2001). Assume that N cases and N controls are sampled. Then the test statistic is given by  , where
, where  is the frequency of haplotype H1j in the cases, and
is the frequency of haplotype H1j in the cases, and  is the frequency of haplotype H1j in the controls. Using haplotype block H2, one may construct a similar test statistic
is the frequency of haplotype H1j in the controls. Using haplotype block H2, one may construct a similar test statistic  , where
, where  is the frequency of haplotype H2j in the cases, and
is the frequency of haplotype H2j in the cases, and  is the frequency of haplotype H2j in the controls. Using both haplotype blocks H1 and H2, one may construct a test statistic TC=TC1+TC2 by summing TC1 and TC2 together. If the two statistics TC1 and TC2 are independent, TC is asymptotically distributed as central χ2l+r-2, with l+r-2 df under the null hypothesis of no association. Under the alternative hypothesis, it is asymptotically distributed as noncentral χ2l+r-2(λC), where λC=λC1+λC2,
is the frequency of haplotype H2j in the controls. Using both haplotype blocks H1 and H2, one may construct a test statistic TC=TC1+TC2 by summing TC1 and TC2 together. If the two statistics TC1 and TC2 are independent, TC is asymptotically distributed as central χ2l+r-2, with l+r-2 df under the null hypothesis of no association. Under the alternative hypothesis, it is asymptotically distributed as noncentral χ2l+r-2(λC), where λC=λC1+λC2,
 |
and  +2P(H2j)]. To calculate λC1, one needs to notice that P(H1j|Aff)=E(z1ij|Aff)/2, and so the conditional expected frequencies P(H1j|Aff)=Δ1jαD/A+P(H1j) and
+2P(H2j)]. To calculate λC1, one needs to notice that P(H1j|Aff)=E(z1ij|Aff)/2, and so the conditional expected frequencies P(H1j|Aff)=Δ1jαD/A+P(H1j) and  (appendices A and C). However, the independence of TC1 and TC2 can be true only in the case that there is linkage equilibrium between the two blocks. Hence, TC may not be a valid test statistic unless one has strong evidence that the two blocks are in linkage equilibrium.
(appendices A and C). However, the independence of TC1 and TC2 can be true only in the case that there is linkage equilibrium between the two blocks. Hence, TC may not be a valid test statistic unless one has strong evidence that the two blocks are in linkage equilibrium.
Results
Type I Errors
To explore the performance of the test statistics, we calculate type I errors for statistics TC, TH, and TG for the four scenarios in table 2. We simulate 10,000 samples under an assumption of penetrance probabilities (fDD,fDd,fdd)=(0.05,0.05,0.05), which implies that the disease is not associated with the two haplotype blocks. Every sample contains 100 cases and 100 controls (N=M=100). For each sample, we calculate the empirical test statistics TC, TH, and TG. The type I error is calculated by dividing the count of those empirical test statistics, which are greater than or equal to the cut-off point at the significance level α=0.01, by 10,000. We repeat the above process a total of 100 times to get 101 type I errors for each of the test statistics TC, TH, and TG for the four models in table 2. On the basis of the 101 type I errors of each statistic, we calculate their mean, standard deviation (SD), minimum, and maximum, which are presented in table 2. For model I in table 2, a strong LD between the two blocks H1, l=2, and H2, r=2, is assumed (ΔH11H21=0.20); in this case, the type I error of TC (mean 0.026) is much greater than those of TH (mean 0.011) and TG (0.012). In model II in table 2, we assume that block H1 has two haplotypes and the block H2 has three haplotypes, and the measures of LD are ΔH11H21=0.15 and ΔH11H22=-0.075; in this case, the type I error of TC (mean 0.017) is the highest, and TG (mean 0.015) has higher type I error than TH (mean 0.012). In models III and IV of table 2, the block H2 has four haplotypes; in model III, the measures of LD are ΔH11H21=ΔH11H22=0.075 and ΔH11H23=ΔH11H24=-0.075, and in model IV, the two blocks are in linkage equilibrium; in these two cases, the type I errors of TG (mean 0.20) are the highest, which may be due to the large degree of freedom of TG. With LD (model III), TC (mean 0.16) has a slightly higher type I error than TH (mean 0.13); without LD (model VI), TH (mean 0.013) has a slightly higher type I error than TC (mean 0.010). Figure 1 shows the QQ plot for each statistic of TC, TH, and TG for the four models in table 2. Each of the QQ plots in figure 1 is drawn by comparing 10,000 sample statistic values with 10,000 related χ2-distribution values (X-axis). These QQ plots are consistent with the results of table 2. Moreover, it is evident that the type I error level of statistic TH is reasonable for N=M=100.
Table 2.
Type I Errors at Significance Level α=0.01 using Two Haplotype Blocks H1, l=2, P(H11)=P(H12)=0.50 and H2 with N=M=100
|
Type I Error |
|||||
| ModelandTest | Sizea | Mean | SD | Minimum | Maximum |
| I:b | |||||
| TG | 101 | .012 | .001 | .009 | .014 |
| TH | 101 | .011 | .001 | .008 | .014 |
| TC | 101 | .026 | .001 | .022 | .030 |
| II:c | |||||
| TG | 101 | .015 | .001 | .012 | .018 |
| TH | 101 | .012 | .001 | .010 | .015 |
| TC | 101 | .017 | .001 | .014 | .021 |
| III:d | |||||
| TG | 101 | .020 | .001 | .018 | .023 |
| TH | 101 | .013 | .001 | .011 | .015 |
| TC | 101 | .016 | .001 | .012 | .018 |
| IV:e | |||||
| TG | 101 | .020 | .001 | .016 | .023 |
| TH | 101 | .013 | .001 | .011 | .016 |
| TC | 101 | .010 | .001 | .007 | .012 |
Size is the total number of type I errors calculated for each statistic under a specific model.
In model I, r=2, P(H21)=P(H22)=0.50, and ΔH11H21=-ΔH11H22=0.20.
In model II, r=3, P(H21)=0.4,P(H22)=P(H23)=0.30, ΔH11H21=0.15, and ΔH11H22=ΔH11H23=-0.075.
In model III, r=4, P(H21)=P(H22)=P(H23)=P(H24)=0.25, ΔH11H21=P(H11H21)-P(H11)P(H21)=0.075, ΔH11H22=P(H11H22)-P(H11)P(H22)=0.075, ΔH11H23=P(H11H23)-P(H11)P(H23)=-0.075, and ΔH11H24=P(H11H24)-P(H11)P(H24)=-0.075.
In model IV, all parameters are the same as those of model III, except that ΔH11H21=ΔH11H22=ΔH11H23=ΔH11H24=0.
Figure 1.

QQ plot at significance level α=0.01 using two haplotype blocks H1, l=2, and H2. In graphs I.1, I.2, and I.3, all parameters are the same as those of model I in table 2. In graphs II.1, II.2, and II.3, all parameters are the same as those of model II in table 2. In graphs III.1, III.2, and III.3, all parameters are the same as those of model III in table 2. In graphs IV.1, IV.2, and IV.3, all parameters are the same as those of model IV in table 2.
Power Calculation and Comparison
To calculate the noncentrality parameters, we assume a deterministic population genetic model. Assume that a single disease mutation was introduced into the population T generations ago, with a frequency PD. First, we consider only one haplotype block Hu, u=1,2. At the initial generation of the occurrence of the mutation, the haplotype frequencies P(Hu1D)(0)=PD and P(HujD)(0)=0,j=2,…, l, if u=1, or j=2,…, r,if u=2. Moreover, P(Hu1d)(0)=P(Hu1)-PD and P(Hujd)(0)=P(Huj),j=2,…,l if u=1, or j=2,…,r if u=2. Let θu be the recombination fraction between haplotype block Hu and disease locus D,u=1,2. Given a map distance λu between haplotype block Hu and disease locus D, the recombination fraction θu can be calculated by Haldane’s map function θu=[1-exp(-2λu)]/2, under the assumption of no interference. At generation T, the haplotype frequencies can be approximately calculated by P(HujD)(T)=P(HujD)(0)e-Tθu+PDP(Huj)(1-e-Tθu) and P(Hujd)(T)=P(Hujd)(0)e-Tθu+PdP(Huj)(1-e-Tθu),j=1,…,l, if u=1, or j=2,…,r, if u=2. Second, we consider both haplotype blocks H1 and H2. At the initial generation of the occurrence of mutation, the haplotype frequencies P(H11DH21)(0)=PD and P(H1jDH2s)(0)=0,j=1,…,l,s=1,…,r, and (j,s)≠(1,1). That is, the disease-susceptibility allele D was carried by haplotype H11H21 at the initial generation of mutation. The other initial haplotype frequencies are P(H11dH21)(0)=P(H11H21)-PD and P(H1jdH2s)(0)=P(H1jH2s),j=1,…,l,s=1,…,r and (j,s)≠(1,1).
At generation T, the haplotype frequencies can be approximately calculated by P(H1jDH2s)(T)=ΔjDs(0)e-T(θ1+θ2) + P(H1j)Δ2s(0)e-Tθ2 + P(H2s)Δ1j(0)e-Tθ1+ P(H1j)PDP(H2s) and P(H1jdH2s)(T)=P(H1jH2s)-P(H1jDH2s)(T),j=1,…, l, s=1,…, r, where ΔjDs(0) = P(H1jDH2s)(0) − P(H1j)Δ2s(0) − P(H2s)Δ1j(0) − P(H1j)PDP(H2s) is the measure of initial LD at the three loci for haplotypes H1j and H2s, Δ1j(0)=P(H1jD)(0)-P(H1j)PD is the measure of initial LD between haplotype H1j and disease locus D, and Δ2s(0)=P(DH2s)(0)-PDP(H2s) is the measure of initial LD between haplotype H2s and disease locus D (Akey et al. 2001).
To make a power comparison, we consider four genetic models: heterogeneous recessive, heterogeneous dominant, additive, and multiplicative. First, we consider optimistic penetrance probabilities and genotype relative risks given in table 3 (Nielson et al. 1998). For less optimistic models, with lower penetrance probabilities and genotype relative risks, we consider the four models in table 4. For each model in table 4, the population disease prevalence is ∼0.05 and the sib recurrence risk is ∼0.06 (Iles 2002). We assume that the distance between the two haplotype blocks is 4 cM. The block H1 is located at position 0 cM, and the block H2 is located at position 4 cM. Since the disease locus D is usually unknown, we assume that it is located in the interval between H1 and H2. Given the location of disease locus D, the map distance λu between Hu and D can be used to calculate the recombination fraction θu by Haldane’s map function, u=1,2, λ1+λ2=4 cM. To calculate the power, we first partition the interval of 4 cM between block H1 and H2 to be 100 subintervals with 101 end-points. Given that the disease locus D is located at an end-point, we may perform power calculation at this locus. We assume that the haplotype H1 has two haplotypes H11 and H12 with equal frequencies, PD=0.10, N=M=100, and T=50 for the four models in table 3. For the four models in table 4, PD=0.30, N=M=500. For each genetic model in table 4, figures 2, 3, and 4 show power curves of TC,TH,TG,TC2,TH2, and TG2 for r=2,3,4 haplotypes of block H2, respectively. The related parameters, such as measures of LD between block H1 and block H2, are given in the legend of each figure. First, it is clear from these three figures that the power of using two haplotype blocks is generally higher than that of using one block. When the disease locus D is far from block H2, the power of using two haplotype blocks is significantly higher. When the disease locus D is close to block H2, the power of using two haplotype blocks is similar to that of using only one block H2. Second, the power of TC is generally higher than or similar to that of TH, and the power of TH is higher than or similar to that of TG. This may be due to the lack of consideration of correlation between the two blocks by TC (see the type I error comparison in table 2). Third, the power of TC2 is similar to that of TH2 and higher than that of TG2.
Table 3.
First Set of Parameters of Simulated Genetic Models
| Model Type | fDD | fDd | fdd |
| Heterogeneous recessive | 1.00 | .05 | .05 |
| Heterogeneous dominant | 1.00 | .95 | .05 |
| Additive | 1.00 | .50 | .0 |
| Multiplicative | .81 | .045 | .0025 |
Table 4.
Second Set of Parameters of Simulated Genetic Models
| Model Type | fDD | fDd | fdd |
| Heterogeneous recessivea | .16 | .04 | .04 |
| Heterogeneous dominantb | .08 | .08 | .02 |
| Additivec | .108 | .0675 | .027 |
| Multiplicatived | .12 | .06 | .03 |
fDD=4fDd=4fdd.
fDD=fDd=4fdd.
fDD=4fdd, fDd=(fDD+fdd)/2.
fDD=4fdd, fDd=2fdd.
Figure 2.

Power curves of TC, TH, TG, TC2, TH2, and TG2 at significance level α=0.01, using two haplotype blocks H1, l=2, and H2, r=2, when P(H11)=P(H12)=P(H21)=P(H22)=0.50, ΔH11H21=P(H11H21)-P(H11)P(H21)=0.075, ΔH11H22=P(H11H22)-P(H11)P(H22)=-0.075, PD=0.30, N=M=500, T=50, for the four genetic models in table 4.
Figure 3.

Power curves of TC, TH, TG, TC2, TH2, and TG2 at significance level α=0.01, using two haplotype blocks H1, l=2, and H2, r=3, when P(H11)=P(H12)=0.5, P(H21)=0.4, P(H22)=P(H23)=0.30, ΔH11H21=P(H11H21)-P(H11)P(H21)=0.075, ΔH11H22=P(H11H22)-P(H11)P(H22)=-0.0375, ΔH11H23=P(H11H23)-P(H11)P(H23)=-0.0375, PD=0.30,N=M=500,T=50, for the four genetic models in table 4.
Figure 4.

Power curves of TC, TH, TG, TC2, TH2, and TG2 at significance level α=0.01 using two haplotype blocks H1, l=2, and H2, r=4, when P(H11)=P(H12)=0.5,P(H21)=P(H22)=P(H23)=P(H24)=0.25, ΔH11H21=P(H11H21)-P(H11)P(H21)=0.075, ΔH11H22=P(H11H22)-P(H11)P(H22)=0.075, ΔH11H23=P(H11H23)-P(H11)P(H23)=-0.075, ΔH11H24=P(H11H24)-P(H11)P(H24)=-0.075, PD=0.30, N=M=500, T=50, for the four genetic models in table 4.
To explore the effect of the degree of LD on the test statistics, figure 5 plots power curves under an assumption of linkage equilibrium between the two blocks H1 and H2 for four models in table 4. From the four graphs of figure 5, the power of TH is similar to or slightly higher than that of TC, except for heterogeneous recessive and multiplicative models, in which the power of TH is slightly lower than that of TC. In all graphs of figure 5, the power of TC and TH is higher than that of TG. Figure 6 plots power curves for different mutation ages of the disease allele D for four models in table 4. For the four models in table 4, the power is very high for a disease mutation of T=30, high for T=40, and relatively high for T=50 generations old. Figure 7 plots power curves of TH for different disease frequencies PD for the four models in table 4. For recessive disease model in table 4, a disease with frequency PD⩾0.30 would have high power if the haplotype block is close to the disease locus. For the other three models in table 4, a disease with frequency PD⩾0.20 would have high power if the haplotype block is close to the disease locus (fig. 7).
Figure 5.

Power curves of TC, TH, and TG at significance level α=0.01 using two haplotype blocks H1, l=2, and H2, r=4, when P(H11)=P(H12)=0.5,P(H21)=P(H22)=P(H23)=P(H24)=0.25, ΔH11H21=P(H11H21)-P(H11)P(H21)=0.0, ΔH11H22=P(H11H22)-P(H11)P(H22)=0.0, ΔH11H23=P(H11H23)-P(H11)P(H23)=0.0, ΔH11H24=P(H11H24)-P(H11)P(H24)=0.0, PD=0.30, N=M=500, T=50, for the four genetic models in table 4.
Figure 6.

Power curves of TH for different mutation ages at significance level α=0.01, using two haplotype blocks H1, l=2, and H2, r=4, when P(H11)=P(H12)=0.5, P(H21)=P(H22)=P(H23)=P(H24)=0.25, ΔH11H21=P(H11H21)-P(H11)P(H21)=0.075, ΔH11H22=P(H11H22)-P(H11)P(H22)=0.075, ΔH11H23=P(H11H23)-P(H11)P(H23)=-0.075, ΔH11H24=P(H11H24)-P(H11)P(H24)=-0.075, PD=0.30, N=M=500, for the four genetic models in table 4.
Figure 7.

Power curves of TH for different disease frequency at significance level α=0.01, using two haplotype blocks H1, l=2, and H2, r=4, when P(H11)=P(H12)=0.5, P(H21)=P(H22)=P(H23)=P(H24)=0.25, ΔH11H21=P(H11H21)-P(H11)P(H21)=0.075, ΔH11H22=P(H11H22)-P(H11)P(H22)=0.075, ΔH11H23=P(H11H23)-P(H11)P(H23)=-0.075, ΔH11H24=P(H11H24)-P(H11)P(H24)=-0.075, T=50, N=M=500, for the four genetic models in table 4.
Corresponding to the six figures for the less optimistic models in table 4, we provide six figures for the optimistic models in table 3 on our Web site. The power of the heterogeneous recessive model in table 3 is low (figs. 1, 2, and 3 on our Web site). In contrast, the power of the heterogeneous recessive model in table 4 is reasonably high (figs. 2, 3, and 4). In the absence of LD, the power of TH is similar to or slightly higher than that of TC for the four models in table 3 (fig. 4 on our Web site). For recessive disease model in table 3, the power is low even for very young disease mutation (T=10) (fig. 5 on our Web site). For the recessive disease model in table 3, a disease with frequency PD⩾0.15 would have high power if the haplotype block is close to the disease locus. For the other three models in table 3, a disease with frequency PD⩾0.10 would have high power (fig. 6 on our Web site).
Sample Size
Table 5 gives sample size required for the four genetic models in table 3 at significance level .01 and 80% power using two haplotype blocks H1, l=2, and H2, r=4. Except for heterozygous recessive disease with low disease-allele frequency PD=0.05, the sample sizes required are <400 and are feasible in practice. For most cases, the sample sizes required are <100. Table 6 gives the sample sizes required for the four genetic models in table 4 at significance level 0.01 and 80% power, using two haplotype blocks H1, l=2, and H2, r=4. Compared with the sample sizes in table 5 for the four models in table 3, the sample sizes in table 6 for the four models in table 4 are much greater. For the recessive disease model in table 4, the sample sizes required for low frequency (PD⩽0.10) are >5,000, and so it may not be realistic to recruit enough patients for such disease studies. For all dominant disease models and recessive disease models with high disease frequency (PD=0.20 or 0.30), the sample sizes required are <1,000 and are feasible in practice. For the additive and multiplicative disease models in table 4, the sample sizes required are <1,000, except for low–disease-frequency cases (PD=0.05) or old disease mutations (T⩾50).
Table 5.
Sample Sizes Required for the Four Genetic Models in Table 3, at Significance Level 0.01 and 80% Power, Using Two Haplotype Blocks H1, l=2, and H2, r=4[Note]
|
Required Sample Size |
|||||||||||||||
|
T=20 |
T=30 |
T=40 |
T=50 |
T=60 |
|||||||||||
| Model Typeand PD | TC | TH | TG | TC | TH | TG | TC | TH | TG | TC | TH | TG | TC | TH | TG |
| Heterogeneous recessive: | |||||||||||||||
| .05 | 2,296 | 3,005 | 2,145 | 2,534 | 3,297 | 2,455 | 2,798 | 3,619 | 2,806 | 3,088 | 3,976 | 3,202 | 3,409 | 4,369 | 3,650 |
| .10 | 193 | 279 | 241 | 213 | 303 | 271 | 235 | 329 | 303 | 258 | 358 | 339 | 285 | 390 | 379 |
| .15 | 57 | 85 | 82 | 62 | 92 | 92 | 68 | 99 | 102 | 75 | 107 | 113 | 83 | 116 | 125 |
| Heterogeneous dominant: | |||||||||||||||
| .05 | 45 | 47 | 60 | 50 | 53 | 67 | 55 | 59 | 75 | 60 | 65 | 84 | 66 | 73 | 94 |
| .10 | 26 | 24 | 29 | 29 | 27 | 33 | 32 | 30 | 38 | 35 | 34 | 43 | 39 | 38 | 48 |
| .15 | 19 | 15 | 17 | 21 | 17 | 21 | 23 | 20 | 24 | 26 | 23 | 28 | 28 | 26 | 32 |
| Additive: | |||||||||||||||
| .05 | 22 | 18 | 21 | 25 | 21 | 24 | 27 | 24 | 28 | 30 | 27 | 32 | 33 | 31 | 37 |
| .10 | 20 | 16 | 18 | 22 | 18 | 21 | 24 | 21 | 24 | 27 | 24 | 28 | 29 | 27 | 32 |
| .15 | 18 | 13 | 15 | 20 | 16 | 18 | 22 | 18 | 21 | 24 | 20 | 24 | 26 | 23 | 28 |
| Multiplicative: | |||||||||||||||
| .05 | 29 | 34 | 47 | 32 | 38 | 51 | 35 | 41 | 56 | 38 | 46 | 62 | 42 | 50 | 68 |
| .10 | 15 | 17 | 23 | 17 | 19 | 25 | 19 | 21 | 28 | 21 | 23 | 31 | 23 | 25 | 34 |
| .15 | 12 | 11 | 15 | 13 | 13 | 17 | 14 | 14 | 19 | 15 | 16 | 22 | 17 | 18 | 24 |
Note.— Data shown are for P(H11)=P(H12)=0.5, P(H21)=P(H22)=P(H23)=P(H24)=0.25, ΔH11H21=P(H11H21)-P(H11)P(H21)=0.075, ΔH11H22=P(H11H22)-P(H11)P(H22)=0.075, ΔH11H23=P(H11H23)-P(H11)P(H23)=-0.075, ΔH11H24=P(H11H24)-P(H11)P(H24)=-0.075, and θ1=θ2=0.005.
Table 6.
Sample Sizes Required for the Four Genetic Models in Table 4 at Significance Level 0.01 and 80% Power Using Two Haplotype Blocks H1, l=2, and H2, r=4[Note]
|
Required Sample Size |
||||||||||||
|
T=30 |
T=40 |
T=50 |
T=60 |
|||||||||
| Model Typeand PD | TC | TH | TG | TC | TH | TG | TC | TH | TG | TC | TH | TGc |
| Heterogeneous recessive: | ||||||||||||
| .05 | 94,373 | 117,340 | 78,996 | 104,275 | 129,519 | 91,520 | 115,217 | 142,977 | 105,797 | 127,309 | 157,850 | 122,031 |
| .10 | 6233 | 7965 | 5707 | 6883 | 8763 | 6555 | 7601 | 9644 | 7517 | 8394 | 10618 | 8607 |
| .20 | 477 | 657 | 550 | 525 | 716 | 620 | 579 | 782 | 698 | 639 | 854 | 785 |
| .30 | 125 | 183 | 173 | 138 | 198 | 192 | 152 | 215 | 214 | 167 | 233 | 238 |
| Heterogeneous dominant: | ||||||||||||
| .05 | 469 | 576 | 771 | 517 | 635 | 851 | 570 | 699 | 939 | 629 | 770 | 1,036 |
| .10 | 196 | 233 | 302 | 216 | 257 | 335 | 238 | 284 | 370 | 262 | 313 | 410 |
| .20 | 109 | 121 | 145 | 120 | 134 | 162 | 132 | 149 | 181 | 145 | 165 | 202 |
| .30 | 93 | 96 | 104 | 102 | 107 | 117 | 112 | 119 | 133 | 123 | 133 | 150 |
| Additive: | ||||||||||||
| .05 | 1,296 | 1,607 | 2,188 | 1,430 | 1,771 | 2,412 | 1,578 | 1,952 | 2,658 | 1,741 | 2,151 | 2,931 |
| .10 | 421 | 520 | 703 | 464 | 572 | 774 | 512 | 630 | 853 | 564 | 694 | 940 |
| .20 | 162 | 195 | 260 | 178 | 215 | 287 | 196 | 237 | 317 | 216 | 261 | 350 |
| .30 | 102 | 119 | 157 | 112 | 132 | 174 | 123 | 145 | 192 | 136 | 160 | 212 |
| Multiplicative: | ||||||||||||
| .05 | 2,397 | 2,979 | 4,070 | 2,646 | 3,284 | 4,487 | 2,921 | 3,620 | 4,946 | 3,224 | 3,992 | 5,454 |
| .10 | 669 | 836 | 1,141 | 737 | 919 | 1,256 | 813 | 1,012 | 1,382 | 897 | 1,114 | 1,522 |
| .20 | 203 | 254 | 347 | 224 | 279 | 382 | 246 | 307 | 419 | 271 | 337 | 460 |
| .30 | 107 | 133 | 182 | 118 | 146 | 200 | 130 | 160 | 219 | 143 | 176 | 241 |
Note.— All other parameters except the penetrance probabilities are the same as those in table 5.
For the sample sizes given in tables 5 and 6, we perform an empirical power calculation by 10,000 replicates. The results for TH are pretty consistent with the theoretical value of 0.80.
Discussion
The objective of this paper is to explore methods for high-resolution haplotype or multiple-marker genome-association studies of complex diseases by case-control designs. We investigated test statistics that combine information from haplotype blocks or multiple markers. We introduced two Hotelling’s T2 statistics TG and TH to test association between a disease locus and two haplotype blocks on the basis of two coding methods, genotype coding and haplotype coding. By theoretical analysis, we showed that they are valid test statistics. Ignoring the correlation between the two blocks, one may use an extension sum statistic, TC, of two traditional χ2 test statistics, TC1 and TC2, for comparing haplotype frequencies in cases and controls. For each of the three statistics, the power of using two haplotype blocks is higher than that of using only one haplotype block. By power comparison, we notice that TC has higher power than TH, and TH has higher power than TG.
In the absence of LD between the two blocks, the power of TC is similar to that of TH and is higher than that of TG. In the presence of LD between the two blocks, the type I error of TC is higher than those of TH and TG. Hence, we advocate to use TH in the data analysis. In the presence of LD between the two blocks, TH takes into account of the correlation between the two haplotype blocks and has the lowest type I error and a higher power than TG. On the one hand, TG has the lowest power, although it takes into account the correlation between the two haplotype blocks. On the other hand, the type I error of TG gets bigger as the number of haplotypes increases, which may be due to the large degree of freedom. Therefore, TG is less favorable than TH.
Several empirical studies showed that the haplotypes have block structures in human genome, and each haplotype block has limited diversity (Daly et al. 2001; Goldstein 2001; Patil et al. 2001; Reich et al. 2001; Rioux et al. 2001; Stephens et al. 2001; Gabriel et al. 2002). The haplotype blocks are punctuated by apparent sites of recombination or hot-spot areas. Within a haplotype block, there are only a few (2–4) haplotypes, and LD decays only gradually with distance. Within the hot-spot areas, however, there may have been several recombination events, and thus LD decays rapidly with distance. The recombination events are clustered to be hot spots. These patterns of LD are very relevant to genomewide association studies for mapping complex-disease genes. However, the general properties of haplotype structure in human genome are not fully understood. It is necessary to characterize patterns of LD in the human genome, and to investigate approaches of high resolution LD mapping of complex traits based on haplotype block data.
The test statistics, such as TH and TG, that are based on multiallelic markers or haplotype blocks can usually lead to a large number of df. However, when haplotype block data are used, the df would not be very large if one took into account the recent discovery of haplotype structure in human genome. Although a haplotype block may enclose many SNPs, it takes only a few SNPs to uniquely identify each of the haplotypes in the block. This implies that the number of df when haplotype block data are used may be even less than that when multiple SNP markers are used in an analysis. Moreover, haplotype block data already take into account the haplotype structure and potentially are more powerful.
In our analysis, only two haplotype blocks are discussed. One could generalize the method to use multiple haplotype blocks in the analysis. One interesting topic is to study the merit of a generalized TH that uses multiple haplotype blocks, instead of the current version of TH, which uses only two haplotype blocks. Moreover, the methods can be generalized to analyze pedigree data, including sib pairs (Cordell and Clayton 2002). Other issues, such as population-stratification effects and methods of combining population and pedigree data, are exciting research topics (Ardlie et al. 2000; Rannala and Reeve 2001). If the data contain individuals with missing genotypes within the haplotype blocks or with genotyping errors, some potential problems can arise in actual data analysis. The effect of uncertainty in the haplotype block’s start and stop positions is unclear. More investigations will be necessary to cope with these challenges.
Acknowledgments
We thank two reviewers for very detailed and thoughtful critiques, which made the paper more clear. R.F. was supported partially by a research fellowship from the Alexander von Humboldt Foundation, Germany, and an International Research Travel Assistance Grant from Texas A&M University. M.K. was supported by grant KN 370/1-1 (Project D1 of FOR 423) from the Deutsche Forschungsgemeinschaft.
Appendix A
Suppose that the disease locus has two alleles D and d, D being the allele for disease susceptibility and d being normal. Assume that the disease-susceptibility allele D has population frequency PD, and normal allele d has population frequency Pd. Let fDD, fDd=fdD, and fdd be the probabilities that an individual with genotypes DD, Dd, and dd is affected with the disease, respectively. Since allele D is disease susceptible, one may assume fDD⩾fDd⩾fdd. Let  and
and  . Denote the disease prevalence in the population by A=fDDP2D+2fDdPDPd+fddP2d, and
. Denote the disease prevalence in the population by A=fDDP2D+2fDdPDPd+fddP2d, and  . As in quantitative genetics, let us introduce some notation. Let a=fDD-(fDD+fdd)/2,d=fDd-(fDD+fdd)/2, δD=2d, and αD=a+(Pd-PD)d. In terms of quantitative genetics, αD is the average effect of gene substitution, and δD is the dominant deviation (Falconer and Mackay 1996). Similarly, denote
. As in quantitative genetics, let us introduce some notation. Let a=fDD-(fDD+fdd)/2,d=fDd-(fDD+fdd)/2, δD=2d, and αD=a+(Pd-PD)d. In terms of quantitative genetics, αD is the average effect of gene substitution, and δD is the dominant deviation (Falconer and Mackay 1996). Similarly, denote 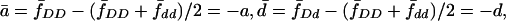
 , and
, and  . Denote the measures of LD between haplotype H1j of the first haplotype block H1 and the disease locus D by Δ1j=P(H1jD)-P(H1j)PD,j=1,…,l, and the measures of LD between haplotype H2s of the second haplotype block H2 and the disease locus D by Δ2s=P(DH2s)-P(H2s)PD,s=1,…,r. For u=1,2, the frequencies of heterozygous genotype HujHuk,j≠k, in affected and unaffected individuals are calculated in appendix B as
. Denote the measures of LD between haplotype H1j of the first haplotype block H1 and the disease locus D by Δ1j=P(H1jD)-P(H1j)PD,j=1,…,l, and the measures of LD between haplotype H2s of the second haplotype block H2 and the disease locus D by Δ2s=P(DH2s)-P(H2s)PD,s=1,…,r. For u=1,2, the frequencies of heterozygous genotype HujHuk,j≠k, in affected and unaffected individuals are calculated in appendix B as
The frequencies of homozygous genotype HujHuj in affected and unaffected individuals are calculated in appendix B as
Under the null hypothesis of no association between the haplotype blocks Hu,u=1,2 and the disease locus D—that is, Δuj=0 for all j, equations (A1), (A2), (A3) and (A4), imply the expectation  for genotype coding method. In appendix C, we show
for genotype coding method. In appendix C, we show
Hence, we have  which implies the expectation
which implies the expectation  for the haplotype coding method, under the null hypothesis of no association between the haplotype blocks Hu,u=1,2 and the disease locus D.
for the haplotype coding method, under the null hypothesis of no association between the haplotype blocks Hu,u=1,2 and the disease locus D.
Appendix B
Notice that P(HujD)=Δuj+P(Huj)PD,P(Hujd)=-Δuj+P(Huj)Pd, P(HukD)=Δuk+P(Huk)PD, and P(Hukd)=-Δuk+P(Huk)Pd for u=1,2. Using the expression αD=(fDD-fdd)/2+(Pd-PD)[fDd-(fDD+fdd)/2]=PDfDD+PdfDd-PDfDd-Pdfdd, the frequency of genotype HujHuk,j≠k, in affected can be calculated as
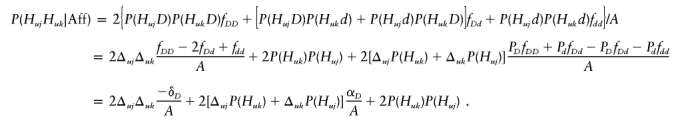 |
Similarly, the frequency of genotype HujHuj in affected can be calculated as
 |
Appendix C
Notice that  and
and  . From equations (A1) and (A3), the expectation of numbers of haplotypes Huj in affected is equal to
. From equations (A1) and (A3), the expectation of numbers of haplotypes Huj in affected is equal to
 |
Similarly, one may show that the expectation of numbers of haplotypes Huj in unaffected is equal to 
Appendix D
Using the notations of 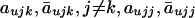 in equations (A1), (A2), (A3), and (A4), we calculate the variance-covariance matrices ΣA1 and
in equations (A1), (A2), (A3), and (A4), we calculate the variance-covariance matrices ΣA1 and  . First, we calculate the variance of the number of haplotypes Huj in affected by equations (A3) and (A5)
. First, we calculate the variance of the number of haplotypes Huj in affected by equations (A3) and (A5)
 |
Similarly, the variance of the number of haplotypes Huj in controls is
By use of equations (A1) and (A5), the covariance between the number of haplotypes Huj and the number of haplotypes Huk, j≠k in affected individuals is
 |
Similarly, the covariance between the number of haplotypes Huj and the number of haplotypes Huk, j≠k in controls is
Appendix E
To calculate the covariance between z1ij,z2is, denote for j≠k,s≠t
 |
For j=1,…,l-1 and s=1,…,r-1, the covariance
 |
Similarly, for j=1,…,l-1 and s=1,…,r-1, the covariance
 |
where  and
and  are expected genotype frequencies in controls like those defined in equation (E1) for cases.
are expected genotype frequencies in controls like those defined in equation (E1) for cases.
Appendix F
To calculate the noncentrality parameter λG, we notice first that the expectation
 |
where  is equal to
is equal to  , and
, and  is equal to
is equal to  .
.
Let ΣG be the variance-covariance matrix of genotype coding Xi. Then its elements can be calculated by Var(xuij|Aff)=aujj-a2ujj, where j=1,…,l-1 if u=1 and j=1,…,r-1 if u=2, Var(xuijk|Aff)=aujk-a2ujk,j≠k, Cov(xuij,xui(j+k)|Aff)=-aujjau(j+k)(j+k) if k⩾1, Cov(xuij,xuimk|Aff)=-aujjaumk for m≠k, Cov(xuijk,xuist|Aff)=-aujkaust for j≠k and s≠t.
Using the notation in equations (A1), (A3), and (E1), the covariances between x1ij,x1ijk and x2is,x2ist are given by
 |
Similarly, we may calculate the variance-covariance matrix  for the controls. Then the noncentrality parameter λG of TG is given by
for the controls. Then the noncentrality parameter λG of TG is given by
 |
Using the variance-covariance matrices ΣG1 and  of the genotype coding vector X1i in affected and unaffected individuals, one may calculate the noncentrality parameter λG1 similarly.
of the genotype coding vector X1i in affected and unaffected individuals, one may calculate the noncentrality parameter λG1 similarly.
Electronic-Database Information
Accession numbers and URLs for data presented herein are as follows:
- R.F.'s Web site, http://stat.tamu.edu/~rfan/paper.html/case_control_Figs_supplement.pdf and http://stat.tamu.edu/~rfan/paper.html/case_control_powsim.pdf (for supplementary information)
References
- Akey J, Jin L, Xiong MM (2001) Haplotype vs. single marker linkage disequilibrium tests: what do we gain? Eur J Hum Genet 9:291–300 [DOI] [PubMed] [Google Scholar]
- Anderson TW (1984) An introduction to multivariate statistical analysis, 2nd edition. John Wiley and Sons, New York [Google Scholar]
- Ardlie KG, Lunetta KL, Seielstad M (2002) Testing for population subdivision and association in four case-control studies. Am J Hum Genet 71:304–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman KW, Murray JC, Sheffied VC, White RL, Weber JL (1998) Comprehensive human genetic map: individual and sex-specific variation in recombination. Am J Hum Genet 63:861–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman NH, Wijsman EM (1998) Genome screens using linkage disequilibrium tests: optimal marker characteristics and feasibility. Am J Hum Genet 63:1872–1885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordell HJ, Clayton DG (2002) A unified stepwise regression procedure for evaluating the relative effects of polymorphisms within a gene using case/control or family data: application to HLA in type 1 diabetes. Am J Hum Genet 70:124–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly MJ, Rioux JD, Schaffner SF, Hudson TJ, Lander ES (2001) High-resolution haplotype structure in the human genome. Nat Genet 29:229–232 [DOI] [PubMed] [Google Scholar]
- Falconer DS, Mackay TFC (1996) Introduction to quantitative genetics, 4th edition. Longman, London [Google Scholar]
- Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly MJ, Altshuler D (2002) The structure of haplotype blocks in the human genome. Science 296:2225–2229 [DOI] [PubMed] [Google Scholar]
- Goldstein GB (2001) Islands of LD. Nat Genet 29:109–111 [DOI] [PubMed] [Google Scholar]
- Hotelling H (1931) The generalization of student’s ratio. Ann Math Stat 2:360–378 [Google Scholar]
- Iles MM (2002) Effect of mode of inheritance when calculating the power of a transmission/disequilibrium test study. Hum Hered 53:153–157 [DOI] [PubMed] [Google Scholar]
- The International SNP Map Working Group (2001) A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409:928–933 [DOI] [PubMed] [Google Scholar]
- Kaplan N, Morris R (2001) Issues concerning association studies for fine mapping a susceptibility gene for a complex disease. Genet Epidemiol 20:432–457 [DOI] [PubMed] [Google Scholar]
- Kong A, Gudbjartsson DF, Sainz J, Jonsdottir GM, Gudjonsson SA, Richardsson B, Sigurdardottir S, Barnard J, Hallbeck B, Masson G, Shlien A, Palsson ST, Frigge ML, Thorgeirsson TE, Gulcher JR, Stefansson K (2002) A high resolution recombination map of the human genome. Nat Genet 31:241–247 [DOI] [PubMed] [Google Scholar]
- Nielsen DM, Ehm MG, Weir BS (1998) Detecting marker-disease association by testing for Hardy-Weinberg disequilibrium at a marker locus. Am J Hum Genet 63:1531–1540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson JM, Wijsman EM (1994) Design and sample size considerations in the detection of linkage disequilibrium with a marker locus. Am J Hum Genet 55:574–580 [PMC free article] [PubMed] [Google Scholar]
- Ott J (1999) Analysis of human genetic linkage, 3rd edition. Johns Hopkins University Press, Baltimore and London [Google Scholar]
- Patil NP, Berno AJ, Hinds DA, Barrett WA, Doshi JM, Hacker CR, Kautzer CR, Lee DH, Marjoribanks C, McDonough DP, Nguyen BTN, Norris MC, Sheehan JB, Shen N, Stern D, Stokowski RP, Thomas DJ, Trulson MO, Vyas KR, Frazer KA, Fodor SPA, Cox DR (2001) Blocks of limited haplotype diversity revealed by high-resolution scanning of human chromosome 21. Science 294:1719–1723 [DOI] [PubMed] [Google Scholar]
- Rannala B, Reeve JP (2001) High-resolution multipoint linkage-disequilibrium mapping in the context of a human genome sequence. Am J Hum Genet 69:159–178 (erratum 69:172) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich DE, Cargill M, Bolk S, Ireland J, Sabett RC, Richter DJ, Lavery T, Kouyounmjian R, Farhadian SF, Ward R, Lander ES (2001) Linkage disequilibrium in the human genome. Nature 411:199–204 [DOI] [PubMed] [Google Scholar]
- Rioux JD, Daly MJ, Silverberg MS, Lindblad K, Steinhart H, Cohen Z, Delmonte T, et al (2001) Genetic variation in the 5q31 cytokine gene cluster confers susceptibility to Crohn disease. Nat Genet 29:223–228 [DOI] [PubMed] [Google Scholar]
- Schaid DJ (1996) General score tests for associations of genetic markers with disease using cases and their parents. Genet Epidemiol 13:423–449 [DOI] [PubMed] [Google Scholar]
- Schaid DJ, Rowland C (1998) Use of parents, sibs, and unrelated controls for detection of associations between genetic markers and disease. Am J Hum Genet 63:1492–1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens JC, Schneider JA, Tanguay DA, Choi J, Acharya T, Stanley SE, Jiang R, et al (2001) Haplotype variation and linkage disequilibrium in 313 human genes. Science 293:489–493 [DOI] [PubMed] [Google Scholar]
- Xiong MM, Zhao J, Boerwinkle E (2002) Generalized T2 test for genome association studies. Am J Hum Genet 70:1257–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]