

Abstract

Electrochemical nitrate reduction to ammonia (NO3RR) is promising to not only tackle environmental issues caused by nitrate but also produce ammonia at room temperatures. However, two critical challenges are the lack of effective electrocatalysts and the understanding of related reaction mechanisms. To overcome these challenges, we employed first-principles calculations to thoroughly study the performance and mechanisms of triple-atom catalysts (TACs) composed of transition metals (including 27 homonuclear TACs and 4 non-noble bimetallic TACs) anchored on N-doped carbon (NC). We found five promising candidates possessing not only thermodynamic and electrochemical stability, but also high activity and selectivity for ammonia production. Among them, non-noble homonuclear Ni3@NC TAC show high activity with low theoretical limiting potential of −0.31 VRHE. Surprisingly, bimetallic Co2Ni@NC, Co2Cu@NC, and Fe2Ni@NC TACs show ultrahigh activity with theoretical limiting potentials of 0.00 VRHE, without a potential determining step in the whole reaction pathways, representing the best theoretical activity been reported up to date. These promising candidates are facilitated by circumventing the limit of scaling relationships, a well-known obstacle for single-atom catalysts. This study indicates that designing suitable TACs can be a promising strategy for efficiently electro-catalyzing NO3RR and breaking the limit of the scaling relationship.
Keywords: ammonia synthesis, non-noble metal-based catalysts, In silico catalyst design, beyond single-atom catalysts, breaking scaling relationship
1. Introduction
Nitrate pollution arising from industrialization and agricultural activities poses significant risks to aquatic ecosystems and human health, contributing to diseases like cancer and blue baby syndrome.1−3 Consequently, effectively removing surplus nitrate ions from contaminated water is imperative for sustainable development. Recent findings4−10 suggest that electrocatalytic reduction of nitrate to ammonia (NO3RR) serves as a means to kill two birds with one stone.
Ammonia (NH3) is essential to nitrogen-based fertilizers and shows significant potential as a hydrogen-rich fuel for industry.3 Currently, NH3 is predominantly produced at a large scale using the energy-intensive Haber–Bosch (HB) process, which operates under high temperature conditions (400–600 °C) and pressures (150–350 atm).11,12 Moreover, the hydrogen required for the HB process usually originates from steam methane reforming (CH4 + H2O → CO + 3H2), resulting in the release of environmentally harmful CO and CO2. This reliance on fossil fuels underscores the need for more sustainable ammonia production methods. In view of this, electrochemical synthesis of ammonia from nitrate reduction (NO3RR) under ambient conditions (NO–3 + 9H+ + 8e– → NH3 + 3H2O) emerges as a promising approach. This is due to the ready availability of the nitrate anion in nature and its comparatively low N=O bond dissociation energy (2.1 eV).13,14 Besides, this strategy would simultaneously reduce nitrate pollutant. However, NO3RR is usually sluggish and complex with undesired byproducts, such as NO and NO26,9 Therefore, understanding the atomic-level mechanisms governing NO3RR pathways is crucial for designing new highly efficient electrocatalysts.15,16 Consequently, designing active, selective, stable, and cost-effective electrocatalysts for NO3RR continues to pose a significant challenge.17
Increasing amounts of research has focused on designing atomically dispersed metal catalysts supported by 2D substrates, initially from single atom catalysts (SACs),18−23 then to dual-atom catalysts (DACs),24,25 and even triple-atom catalysts (TACs).26−31 Compared with traditional metal-based catalysts, atomically dispersed metal catalysts require lower cost due to much higher utilization of metal atoms. Some of them demonstrate outstanding catalytic activity across a wide range of electrochemical reactions, such as the oxygen reduction reaction (ORR),23,32−36 the oxygen evolution reaction OER,33,36 the N2 reduction reaction (N2RR),37 and the CO2 reduction reaction (CO2RR).38−40 For NO3RR, SACs generally show limited activity due to the confinement of scaling relationship among intermediates.5,6 However, a few DACs are promising for NO3RR catalysis with high activity and selectivity. For example, Cr2 on expanded phthalocyanine (Cr2-Pc) is predicted theoretically to be a superior catalyst with a limiting potential of −0.02 V.8 Theoretical prediction and experimental validation show that the Cu dual-atom site with three nitrogen coordination is excellent for NO3RR with Faradaic efficiency (FE) of 97.4%.7 However, the development of NO3RR electrocatalysts is still in its infancy. Notably, there have been no reported TACs for NO3RR to NH3 to date, although some TACs are promising catalysts for other reactions, such as Fe3-TAC on graphdiyne for N2RR,29 Fe3-TAC on N-doped graphene for rapid CO electroreduction to propylene,41 and Cu3-TAC on the S-terminated MoSTe surface for CO2 electroreduction to CH4.42 Therefore, we are inspired to screen M3-TACs for NO3RR and to investigate the corresponding atomic-level mechanisms.
This study represents the inaugural comprehensive evaluation of the stability, selectivity, and electrocatalytic activity of M3-TACs on N-doped carbon (NC) for NO3RR. Employing in silico screening, we proposed both homonuclear and bimetallic M3-TACs for enhanced NO3RR toward NH3. Our computational findings revealed that non-noble bimetallic Co2Ni@NC, Co2Cu@NC, Fe2Ni@NC, Co2Fe@NC, and homonuclear Ni3@NC TACs, exhibit considerable promise as NO3RR electrocatalysts, characterized by low limiting potential and high selectivity. We investigated the potential of TACs to break the linear scaling relationship between reaction intermediates. We analyzed the electronic influence of secondary metal atoms in bimetallic Co2M′@NC TACs.
2. Computational Methods
We conducted all DFT calculations using VASP 5.4.4 software, applying a plane-wave cutoff energy of 500 eV and employing the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional within the generalized gradient approximation (GGA) framework, with spin polarization.43−46 van der Waals interactions were accounted for using the Grimme D3 method.47,48 Additionally, we utilized VASPsol to simulate the implicit solvation effect of water.49,50 Convergence thresholds were set at 0.04 eV/Å for force and 10–6 eV for energy and electronic structure.
N-doped carbon was modeled by a graphene p(5 × 5) supercell with a lattice constant of 12.28 Å, and a vacuum space of 20 Å was implemented to prevent interactions between adjacent periodic images. During structural relaxation, three carbon atoms in each TAC model are fixed to reduce the distortion and corresponding energy error (as shown in the relaxed structure file in Note S2 of the Supporting Information). Brillouin zone sampling followed the Monkhorst–Pack scheme and utilized 3 × 3 × 1 and 5 × 5 × 1 k-point grids, for structural optimization and single-point energy calculations, respectively. To compute the Gibbs free energy change (ΔG) for each step during nitrate reduction, we employed the computational hydrogen electrode (CHE) model, as follows51
| 1 |
Here, ΔE represents the electronic energy difference, ΔZPE stands for the zero-point energy, T denotes the temperature (298.15 K), and ΔS accounts for entropy corrections.
To circumvent the direct calculation of the energy of charged NO–3, we adopted gaseous HNO3 as a reference. The adsorption energy of NO–3 (ΔG*NO3) was determined as follows
| 2 |
In this equation, G*NO3, G*, 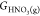 , and
, and 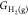 denote the intrinsic Gibbs free energies
of adsorbed NO–3, bare TAC, gas-phase HNO3, and gas-phase H2, respectively. Additionally, ΔGcorrect represents the correction for NO–3 adsorption energy, set to 0.392 eV.
denote the intrinsic Gibbs free energies
of adsorbed NO–3, bare TAC, gas-phase HNO3, and gas-phase H2, respectively. Additionally, ΔGcorrect represents the correction for NO–3 adsorption energy, set to 0.392 eV.
The total reaction of NO3RR can be expressed as NO3– + 9H+ + 8e– → NH3 + 3H2O. Note S1 shows the detailed formula of all elementary process observed during the search of minimum energy reaction pathways.
3. Results and Discussions
3.1. Structure and Stability of TACs
Figure 1a presents the configuration of studied M3-TACs. Such a TAC model is based on another theoretical research,41 and similar to some experimental results.26,52 Specifically, Fe3@NC52 and ZnCoFe@NC TAC with similar structures have been synthesized and their catalytic applications have been explored, suggesting the feasibility of experimentally synthesizing understudied TAC structures. Our work can bring fundamental insights into the future experimental synthesis of the proposed TACs. In this TAC model, metal trimers are located at the six carbon vacancies, and each metal trimer is anchored by six pyridine nitrogen atoms and one amino nitrogen atom in N-doped carbon, denoted as M3@NC TACs (M3-TACs for brevity). Each trimer consists of three metal atoms bonded together, designated as M#1 for the metal site in the upper left, M#2 for the metal site in the upper right, and M#3 for the metal site in the lower middle. We systematically considered the 27 homonuclear-metallic (Sc3, Ti3, V3, Cr3, Mn3, Fe3, Co3, Ni3, Cu3, Zn3, Y3, Zr3, Nb3, Mo3, Ru3, Rh3, Pd3, Ag3, Cd3, Hf3, Ta3, W3, Re3, Os3, Ir3, Pt3, and Au3) and 4 bimetallic trimers (Fe2Ni, Co2Fe, Co2Ni, and Co2Cu).
Figure 1.

(a) Schematics of studied TACs. (b) Four steps for screening promising TACs for NO3RR. (c) Stabilities of metal trimers in studied TACs.
In the hope of finding promising TACs for electrocatalytic reduction of nitrate to ammonia, we adopted a four-step screening to theoretically investigate the stability, selectivity, and activity of these TACs, as shown in Figure 1b.
To assess the thermodynamic and electrochemical stability of these TACs, we computed their formation energy (Eform) and dissolution potential (Udiss) as detailed below8,53
| 3 |
| 4 |
In the equation, ETAC stands for the total energy of the TAC, EN-G denotes the total energy of the nitrogen-doped carbon, and EM(i) represents the atomic energy of metal M(i) in its most stable bulk state. U0diss(M-bulk) represents the standard dissolution potential of the bulk metals, while n corresponds to the stoichiometric coefficient representing the number of electrons transferred during the dissolution. A negative value of Eform indicates that the TAC is thermodynamically stable and experimentally accessible, while a positive value of Udiss signifies the electrochemical stability of the TAC. Figure 1c provides a graphical representation of the formation energy and dissolution potential of the examined TACs. The values for both Eform and Udiss can be found in Tables S1 and S2. We find that Ru3, Ag3, W3, Re3, Os3, Ir3, and Au3-TACs have high formation energy, suggesting they will be difficult to synthesize. We find that Sc3, Ti3, V3, Cr3, Mn3, Zn3, Y3, Zr3, Nb3, Mo3, Cd3, Hf3, and Ta3-TACs have negative Eform but negative Udiss, suggesting that they can be synthesized but that they are not electrochemically stable. We find that seven homonuclear Fe3, Co3, Ni3, Cu3, Rh3, Pd3, and Pt3-TACs and all four bimetallic Fe2Ni, Co2Fe, Co2Ni, and Co2Cu-TACs have negative formation energies and positive dissolution potentials, suggesting they are synthetically feasible and electrochemically stable. Therefore, we considered only these 11 stable TACs (Fe3, Co3, Ni3, Cu3, Rh3, Pd3, Pt3, Fe2Ni, Co2Fe, Co2Ni, and Co2Cu-TACs) for the study of selectivity.
3.2. Selectivity of *NO3 and *H Adsorption on TACs
To kickstart the process of NO3RR, the first crucial step involves the absorption of the nitrate moiety. Figure 2a depicts the three different configurations of nitrate adsorption on the active site of TACs:
-
1
two oxygen atoms from the nitrate adsorbs on metal atoms M#1 or M#2 along with M#3 of the TAC;
-
2
two oxygen atoms from the nitrate adsorbs on metal atoms M#1 and M#2 of the TAC;
-
3
two oxygen atoms from the nitrate adsorbs on the metal atom M#3 atom of the TAC.
Figure 2.

Nitrate adsorption configurations on the triple-atom site and respective adsorption free energies: (a) three potential NO–3 configurations on TAC surfaces. (b) NO–3 adsorption free energies on TAC surfaces and their associated configurations. The background colors in (b) indicate the adsorption configurations of *NO3 on respective TACs.
The adsorption free energy of a nitrate and its corresponding binding configuration on TACs are presented in Figure 2b. Configuration (1) represents the most stable adsorption state on Co3, Cu3, Co2Fe, Co2Ni. Configuration (2) represents the most stable adsorption state on Co2Cu. Configuration (3) represents the most stable adsorption state on Fe3, Ni3, Rh3, Pd3, Pt3, Fe2Ni. For Pt3-TAC, the adsorption of *NO3 is very weak, with ΔG*NO3 of 0.66 eV. For the other 10 TACs, the adsorption of *NO3 is stronger, with ΔG*NO3 varying from −0.87 to 0.19 eV. None of studied TACs adsorb nitrate too strongly (ΔG*NO3< −3 eV), suggesting that these TACs would not be poisoned by nitrate during NO3RR. Values for the adsorption free energy of NO–3 for all possible configurations can be found in Table S3.
Before delving into the details of the NO3RR mechanism on TACs, the competitive adsorption between hydrogen and nitrate should be studied.8 If the binding of *H is stronger than that of NO–3, NO3RR would be hindered due to poisoning of the active sites by hydrogen adsorption. Moreover, weak adsorption of nitrate may lead to the desorption of NO–3 from active sites before being reduced to NH3. Therefore, NO3RR would be highly preferable on catalysts that binds nitrate selectively rather than hydrogen. Figure 3a shows the adsorption free energy of 3*H versus *NO3 on TACs.41 Catalysts above the dashed line prefer binding to *H while ones below the dashed line prefer *NO3. For Pt3-TAC, the adsorption of *NO3 is much weaker than 3*H, with ΔG*NO3 of 0.66 eV, indicating NO3RR is not likely to take place on Pt3-TAC. Thus, we considered only the other 10 TACs (Fe3, Co3, Ni3, Cu3, Rh3, Pd3, Fe2Ni, Co2Fe, Co2Ni, and Co2Cu-TACs) for studies of activity, excluding Pt3-TAC. However, Rh3-TAC and Pd3-TAC are located in or near the region selective for hydrogen adsorption, suggesting that the selectivity toward NO3RR is not very high; thus the FE on these two TACs would be lower compared with the other 8 TACs (Fe3, Co3, Ni3, Cu3, Fe2Ni, Co2Fe, Co2Ni, and Co2Cu-TACs).
Figure 3.

Electrochemical activity of TACs in nitrate-to-ammonia conversion. (a) Comparison of H and NO–3 adsorption energies on TACs. (b) Limiting potentials for NO3RR to NH3 on studied metal trimers. Ni3-TAC exhibits notable homonuclear electrocatalytic activity, with a limiting potential of −0.31 V. Meanwhile, Co2Ni-TAC, Co2Cu-TAC, and Fe2Ni-TAC show superior activity without potential-determining steps. (c) Schematic of observed NO3RR to NH3 mechanism on TACs.
3.3. NO3RR Mechanism on TACs
Reducing NO–3 to NH3 electrochemically at ambient conditions involves eight electron transfers and nine proton transfers, with the surplus due to the anionic nature of NO–3. The complexity arises from various intermediates and byproducts such as NO, NO2, and N2, posing a challenge for investigation.6,8 Furthermore, diverse adsorption configurations of reaction intermediates have been reported, indicating multiple binding possibilities for each intermediate6,8
We assessed the NO3RR electrochemical performance
of the studied metal trimers based on the limiting potential (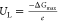 ), where ΔGmax represents the maximum free energy change among all elementary steps
that would be influenced by external potential. Although the overpotential
is obtained as zero for some TACs, the rate-limiting step could either
be the reaction barriers between each reaction intermediate or the
desorption of ammonia, as ammonia desorption is not spontaneous. This
evaluation culminated in the identification of the most favorable
candidates for NH3 synthesis on M3-TACs, as
detailed in Figure 3b. Here we are discussing reaction performance and mechanism at pH
= 7. Notably, NH3 synthesis was most prominently observed
on two homonuclear (Rh3 and Ni3) and four bimetallic
(Co2Ni, Co2Cu, Fe2Ni, and Co2Fe) TACs, showing significantly lower limiting potentials
compared to the others. Specifically, these six systems exhibit limiting
potentials of −0.04, −0.31, 0.00, 0.00, 0.00, and −0.22
V respectively. Conversely, the Pd3 (−1.06 V) and
Fe3 (−1.16 V) systems displayed unfavorable limiting
potentials for effective NO3RR electrochemical performance.
), where ΔGmax represents the maximum free energy change among all elementary steps
that would be influenced by external potential. Although the overpotential
is obtained as zero for some TACs, the rate-limiting step could either
be the reaction barriers between each reaction intermediate or the
desorption of ammonia, as ammonia desorption is not spontaneous. This
evaluation culminated in the identification of the most favorable
candidates for NH3 synthesis on M3-TACs, as
detailed in Figure 3b. Here we are discussing reaction performance and mechanism at pH
= 7. Notably, NH3 synthesis was most prominently observed
on two homonuclear (Rh3 and Ni3) and four bimetallic
(Co2Ni, Co2Cu, Fe2Ni, and Co2Fe) TACs, showing significantly lower limiting potentials
compared to the others. Specifically, these six systems exhibit limiting
potentials of −0.04, −0.31, 0.00, 0.00, 0.00, and −0.22
V respectively. Conversely, the Pd3 (−1.06 V) and
Fe3 (−1.16 V) systems displayed unfavorable limiting
potentials for effective NO3RR electrochemical performance.
Notably, although Rh3-TAC shows the lowest limiting potential among homonuclear TACs, we do not consider it as the best homonuclear NO3RR TAC due to both the low FE on Rh3-TAC, as discussed above in Section 3.2, and also because of the high cost and inappropriate oxidation state of precious Rh. The studied nitrogen-doped carbon is suitable to anchor metallic trimers with a total oxidation state of +6.41 However, the main oxidation state of a Rh atom is +3, suggesting that the Rh3 trimer may not fit into the structure. Co3-TAC is not regarded as a promising candidate for NO3RR, since its activity will be influenced by multiple nitrate adsorptions and preadsorption of H2O in the working environment, as shown in Table S5. In contrast, Ni3-TAC shows high selectivity and activity; its limiting potential will not be influenced by multiple nitrate adsorptions and preadsorption of *OH or H2O in the working environment; Ni is a nonprecious metal with a suitable oxidation state. Therefore, Ni3-TAC is regarded as the most promising homonuclear M3-TAC.
Compared to SACs or DACs, the synergy exhibited by metal trimers in TACs enables the accommodation of more intricate adsorption configurations and reaction pathways. Therefore, we conducted density functional theory (DFT) calculations to determine the Gibbs free energies for the elementary steps in nitrate reduction pathways toward NH3, aiming to explore the possible reaction pathways of NO3RR to NH3 on triple-atom sites. The NO3RR pathway is depicted by considering all plausible intermediates with the lowest total free energies. The observed NO3RR pathways on TACs are depicted in Figure 3c.
The initial step is the adsorption of a nitrate ion onto the surface.
Next is the protonation of the most accessible O atom accompanied by the breaking of a N–O bond, to form *NO2–*OH.
In steps 3 to 5, three protons react with O atoms to release two water molecules, leaving *NO on the surface.
Subsequently, in step 6, a proton attacks O atom in *NO accompanied by the breaking of the N–O bond, to form *N–*OH.
In the 4 subsequent protonation steps 7 to 10, a molecule of ammonia and an additional water molecule form on the surface.
For all elementary steps observed during the search for minimum energy reaction pathways, the formulas are shown in Note S1.
To delve more deeply into the reaction mechanism and to assess the efficacy of nitrate reduction to NH3 on the M3-TACs, we conducted computations to determine the free energy profiles for NO3RR. We established the reference state as the bare surface at 0.0 eV. The free energy profiles and corresponding DFT-optimized structures for homonuclear Ni3-TAC are presented in Figure 4. Likewise, for bimetallic Co2Ni-TAC, Co2Cu-TAC, and Fe2Ni-TAC, the free energy profiles and relevant DFT-optimized structures can be found in Figure 5. The free energy profiles and their associated DFT-optimized structures for the remaining TACs are available in Figures S2–S7.
Figure 4.

Reaction pathway (a) and corresponding optimized configurations (b) of NO3RR for Ni3-TAC.
Figure 5.

Reaction pathway of NO3RR for (a) Co2Ni-TAC, (b) Co2Cu-TAC, and (c) Fe2Ni-TAC. Corresponding optimized configurations of NO3RR for (d) Co2Ni-TAC, (e) Co2Cu-TAC, and (f) Fe2Ni-TAC.
To investigate whether the adsorption of intermediates adheres to the scaling relationship, a pivotal challenge in developing high-performance single-atom electrocatalysts, we performed linear regression analysis for the adsorption free energies of intermediates in five protonation steps (step 1, step 3, step 4, step 5, and step 9) against ΔG*NO3, as depicted in Figure 6. These five steps were chosen because in these steps, intermediates on all studied TACs are the same, although the adsorption configurations differ in some steps. Intermediates in these five steps are *NO2 + *OH, *NO + *OH, *NO, *N + *OH, and *NH3, separately. Remarkably, for all these steps, the adsorption free energies of the intermediates demonstrate a weak linear correlation with ΔG*NO3, with R2 < 0.7, particularly in step 4 (*NO) with R2 = 0.33. This suggests that the design of TACs may serve as an effective approach to circumvent the scaling relationship among intermediates in NO3RR, which may facilitate the discovery of catalysts with ultrahigh activity.
Figure 6.

Adsorption free energies of intermediates in five protonation steps (step 1, step 3, step 4, step 5, and step 9) against ΔG*NO3.
To understand the advantage of TAC for NO3RR and the origin of breaking scaling relationship as discussed above, we analyzed the adsorption configurations of intermediates on all TACs along the NO3RR pathway. We found that for all these TACs, all three metal atoms participate in the adsorption of intermediates in many of the steps, although only one or two metal atoms contribute to the adsorption of *NO3 and *NH3. The collaborative function of metallic trimers enables the disruption of the scaling relationship that limits the efficacy of single-atom catalysts, in contrast to the conditions observed in SACs where intermediates can solely bind to a single active center. This suggests that the metallic trimers of TACs facilitate a smooth converting of nitrate to ammonia.
3.4. NO3RR Performance on Homonuclear TACs
The free energy profiles and corresponding DFT-optimized structures for Ni3-TAC are presented in Figure 4. Initially, two oxygen atoms in the nitrate moiety bind with Ni#3 to form *NO3, with a free energy change of 0.19 eV. The first step is the protonation of an O atom accompanied by breaking of a N–O bond in nitrate moiety, forming *NO2–*OH at −0.70 eV. In the optimized configuration of this step, *OH bonds with Ni#2 and Ni#3, while the N atom and an O atom in *NO2 bond with Ni#1 and Ni#3, respectively. The free energy of *OH + NO2 is −0.01 eV, indicating that the desorption of NO2 requires 0.69 eV, making it unfavorable. Next, a proton attacks the O in *NO2 that bonds with Ni#3, leading to breaking of an N–O bond to form *NO–2*OH at −1.00 eV. The free energy of 2*OH + NO is −0.27 eV, indicating the desorption of NO requires 0.73 eV, making it unfavorable. The two following downhill steps (*NO – 2*OH → *NO – *OH → *NO) are the protonation of two *OH to form two water molecules, leaving *NO bonding with three Ni atoms at −3.54 eV. Next is the protonation of O to form *N–*OH at −3.23 eV, with *OH bonding with Ni#3 and *N bonding with three Ni. Continuing, the sixth to eighth steps are consecutive protonation of N in intermediates (*N – *OH → *NH – *OH → *NH2 – *OH → *NH3 – *OH), with corresponding energy changes of −0.79, −0.37, and −0.59 eV, respectively. In the final step, a proton reacts with *OH exergonically to form a water molecule, leaving *NH3 at −5.66 eV. In the whole protonation process on Ni3-TAC, potential determining step is converting *NO to *N–*OH, requiring 0.31 eV.
For the Rh3-TAC case (as shown in Figure S2), the initial adsorption state of *NO3 and intermediates along the whole protonation pathway are the same as Ni3-TAC. In the whole protonation process on Rh3-TAC, the only endergonic step is converting *NH3–*OH to *NH3, requiring 0.04 eV. The desorption of *NO2 in *NO2–*OH and *NO in *NO–2*OH requires 1.48 and 1.35 eV, respectively, making them unfavorable. Although Rh3-TAC shows the lowest limiting potential among homonuclear TACs, we do not consider it as the best homonuclear NO3RR TAC due to the low FE on Rh3-TAC, as well as the high cost and inappropriate oxidation state of Rh, as discussed in 3.3.
Figure S3 presents the free energy profile and DFT-optimized structures for NO3RR for the Co3-TAC case without considering the influence of multiple nitrate adsorptions and preadsorption of H2O in the working environment, respectively. In the initial adsorption of the nitrate moiety, two oxygen atoms bind with Co#1 and Co#3, respectively, forming *NO3 at −0.12 eV. In the following five protonation steps (*NO3 → *NO2 – *OH → *NO – 2*OH → *NO – *OH → *NO → *N – *OH), intermediates in each step are the same as those on Ni3-TAC, with corresponding energy changes in each elementary step as −1.96, −0.77, −0.73, −1.01, and −0.97 eV, respectively. In the following step, the proton reacts with *OH to form a water molecule, leaving *N at −5.58 eV. Three subsequent steps are consecutive protonation of the nitrogen atom in intermediate steps to form an ammonia molecule (*N → *NH → *NH2 → *NH3), with corresponding energy changes of −0.31, 0.18, and −0.42 eV, respectively. In the whole protonation process on Co3-TAC, the only endergonic step is converting *NH to *NH2, requiring 0.18 eV. The desorption of *NO2 in *NO2–*OH and *NO in *NO–2*OH requires 1.37 and 1.70 eV, respectively, making them unfavorable.
For the Cu3-TAC case (as shown in Figure S4), intermediates in the first protonation step are also *NO2–*OH. Next is the protonation of *OH to form a water molecule, leaving *NO2 on the surface. In the following seven protonation steps, intermediates in each step are the same as those on Ni3-TAC. The potential determining step is converting *NO to form *N–*OH, with an energy change of 0.66 eV.
For the Pd3-TAC (as shown in Figure S5) and Fe3-TAC (as shown in Figure S6), intermediates along the whole protonation pathway are the same as those on Ni3-TAC. The potential determining step on Pd3-TAC is converting *NO to form *N–*OH, with an energy change of 1.06 eV. The potential determining step on Fe3-TAC (as shown in Figure S6) is converting *N–*OH to form *NH–*OH, with an energy change of 1.16 eV.
We compared the d-band centers and limiting potentials of Fe3, Ni3, and Cu3-TACs (as shown in Table S4), finding that the high activity of Ni3-TAC results from the medium energy level of its d-band center. For Fe3-TAC, its d-band center (−0.52 eV) is too high, and thus the adsorption of intermediates is too strong; for Cu3-TAC, the d-band center (−3.04 eV) is too low, and thus the adsorption of intermediates is too weak. Therefore, the NO3RR activity of Fe3 and Cu3 is low. By contrast, the d-band center of Ni3 (−1.48 eV) is located at medium energy level, leading to medium adsorption of intermediates and thus high activity.
3.5. NO3RR Performance on Bimetallic TACs
In the hope of discovering superior NO3RR catalysts, we further examined four bimetallic TACs by replacing a metal atom in homonuclear metallic trimers with a secondary metal atom. All studied bimetallic TACs are composed of Fe, Co, Ni, or Cu, because they are nonprecious metals with suitable oxidation states. Among the homonuclear nonprecious TACs without considering the influence of multiple nitrate adsorptions and preadsorption of H2O in the working environment, Co3-TAC shows the lowest limiting potential as discussed above, but it will be influenced by multiple nitrate adsorptions and preadsorption of H2O in the working environment. Therefore, we investigated three bimetallic Co2M-TACs (Co2Fe, Co2Ni, and Co2Cu) to test whether the secondary metal atom can enhance the performance of Co-based TACs. In addition, we studied Fe2Ni-TAC to test the possibility of improving Fe-based TACs by introducing a secondary metal atom.
To find the favorable position of the secondary metal atom, we compared the energy of each bimetallic TAC with the secondary metal atom at M#1 and M#3 (Figure S1), respectively, since the atomic environment of M#1 and M#2 are the same. We found that the bimetallic TAC with the secondary metal atom at M#3 has a lower energy than the corresponding TAC with the secondary metal atom at M#1, suggesting that a secondary metal atom at M#3 is energetically preferable for bimetallic TACs. Therefore, we studied the stability, selectivity, and activity of TACs with the secondary metal atom at M#3. Since the stability and selectivity of these bimetallic TACs are favorable for NO3RR, as discussed above, we only analyze the activity below.
For Co2Ni-TAC (as shown in Figure 5a,d) and Co2Cu-TAC (as shown in Figure 5b,e), the reaction pathway is the same. The first step is protonation of the most accessible O atom in *NO3, forming *NO2–*OH with *NO2 bonding to Co#2 and *OH bonding with Co#1 and the secondary metal atom. In the second step, a proton attacks an O atom in *NO2, accompanied by the breaking of an N–O bond in *NO2 and an H–O bond in *OH to form a water molecule, leaving *NO–*O on the surface. Next is the protonation of *O to form *NO–*OH, the same intermediates as the third protonation step on Co3-TAC. For the following six protonation steps, the intermediates are the same as those on Co3-TAC. For Co2Ni-TAC, the desorption of *NO2 in *NO2–*OH and *NO in *NO–*O requires 1.30 and 1.88 eV, respectively, making them unfavorable. For Co2Cu-TAC, it requires 1.31 and 1.76 eV, respectively, making them unfavorable. Notably, the whole protonation pathway on Co2Ni-TAC and Co2Cu-TAC is downhill, suggesting that they are superior NO3RR catalysts with no potential determining step. Of course, in our current calculations, we do not consider activation barriers so kinetic factors are not considered. Thus, in realistic conditions, small overpotentials will be needed to drive the NO3RR on Co2Ni-TAC and Co2Cu-TAC.
For Fe2Ni-TAC (as shown in Figure 5c,f), intermediates in the whole reaction pathway are the same as those on Co3-TAC. The desorption of *NO2 in *NO2–*OH and *NO in *NO–*2OH require 1.40 and 2.23 eV, respectively, making them unfavorable. Notably, the whole protonation pathway on Fe2Ni-TAC is also downhill, just as for Co2Ni-TAC and Co2Cu-TAC discussed above, suggesting that they are superior NO3RR catalysts with no potential determining step. As discussed above, in realistic conditions, small overpotentials will be needed to drive the NO3RR on these three TACs.
For Co2Fe-TAC (as shown in Figure S7), intermediates in the whole reaction pathway are the same as those on Fe3-TAC. The desorption of *NO2 in *NO2–*OH and *NO in *NO–*2OH requires 1.21 and 1.55 eV, respectively, making them unfavorable. The potential determining step on Co2Fe-TAC is converting *N–*OH to form *NH–*OH, with an energy change of 0.22 eV.
The comparison of the electrochemical performance of the electrocatalysts and their potential-determining steps are listed in Table 1. Bimetallic Co2Ni@NC, Co2Cu@NC, and Fe2Ni@NC TACs show theoretical limiting potentials of 0.00 VRHE, representing the best theoretical activity been reported up to date.
Table 1. Comparison of Electrocatalytic Performance of NO3RR on Atomically Dispersed Metal Catalysts Supported by 2D Substrates.
| catalyst | potential-determining step | limiting potential (V) | reference |
|---|---|---|---|
| Co2Ni@NC | none | 0.00 | |
| Co2Cu@NC | none | 0.00 | |
| Fe2Ni@NC | none | 0.00 | this work |
| Co2Fe@NC | *N – *OH → *NH – *OH | –0.22 | |
| Ni3@NC | *NO → *N – *OH | –0.31 | |
| Ru/g-C3N4 | *NO → *NOH | –0.34 | (54) |
| Ti/g-CN | *NO → *NOH | –0.39 | (6) |
| Os–N4/C | *N → *NH | –0.42 | (55) |
| Pt–N4/C | *NO → *NOH | –0.48 | (56) |
| Cr2-Pc | *O2H3 → 2H2O | –0.02 | (8) |
| FeMo@g-CN | *NO → *NOH | –0.34 | (9) |
| Cu2@NC | *NO3 → *NO3H | –0.36 | (57) |
To understand the electronic impact of the secondary metal atom, we compared the projected density of states (PDOS) of related atomic orbitals on Co3-TAC, Co2Ni-TAC, and Co2Cu-TAC without and with adsorption of the *NH intermediate, as shown in Figure 7. We chose to analyze *NH based on the following considerations. Although the adsorption configuration of *NH is similar on three TACs, too strong adsorption of *NH on Co3-TAC contributes to the potential determining step, while relatively weak adsorption of *NH on Co2Ni-TAC and Co2Cu-TAC facilitates the smooth NO3RR without potential determining step. This indicates that compared with homonuclear Co3-TAC, the secondary Ni or Cu weakens the adsorption of *NH on Co-based TACs. By comparing the PDOS of d orbital of Co#2 in three TACs without *NH, as shown in Figure 7a–c, we found that the shape of the PDOS differs in the three conditions, suggesting that the secondary Ni and Cu influence the electron states of Co atoms. By comparing the PDOS of the d orbital of Co#2 and the p orbital of N in *NH for the three TACs (Figure 7d–f), we found that the N peaks overlap with the peaks of Co, indicating that *NH binds with Co atoms in the three TACs. By comparing the PDOS of the d orbital of the secondary Ni or Cu and the p orbital of N in *NH for bimetallic TACs (Figure 7e–f), we found that the peaks of N overlap with the peaks of the secondary atom, indicating that *NH binds to the secondary atoms in bimetallic TACs. Therefore, the secondary Ni and Cu atoms not only influence the electron states of Co atoms in bimetallic Co2Ni and Co2Cu TACs, but also bond with reaction intermediates.
Figure 7.

Electronic structures of Co3-TAC, Co2Ni-TAC, and Co2Cu-TAC. The partial density of states (PDOS) of Co3-TAC before (a) and after (d) the adsorption of *NH intermediate. PDOS of Co2Ni-TAC before (b) and after (e) the adsorption of *NH intermediate. PDOS of Co2Cu-TAC before (c) and after (f) the adsorption of *NH intermediate. The Fermi levels are indicated by the black dash lines.
Both the secondary metal elements and Co bind with *NH intermediates (as shown in the adsorption configuration in Figure 5d,e, as well as the PDOS in Figure 7e,f). Compared with Co3-TAC (ΔG*NH = −5.89 eV in Figure S3a), we can find that the Ni (ΔG*NH = −5.33 eV of Co2Ni-TAC in Figure 5a) or Cu replacement (ΔG*NH = −4.86 eV of Co2Cu-TAC in Figure 5b) weakens the binding strength between Co and *NH. Besides, the weakening effect is also indicated by the PDOS. The p orbital of the N atom forms strong bonding with the d orbital of the Co atom in Co3-TAC around −4.5 eV below Fermi level (shown in Figure 7d), while only weak peaks appear in Co2Ni-TAC (shown in Figure 7e) and Co2Cu-TAC (shown in Figure 7f).
4. Conclusions
In summary, Quantum Mechanics calculations were utilized to evaluate the potential of TACs supported on nitrogen-doped carbon for nitrate reduction to ammonia. By studying the reaction pathways, we found five highly active nonprecious candidates with significantly low limiting potentials. Four of them are bimetallic TACs: Co2Ni (0.00 V), Co2Cu (0.00 V), Fe2Ni (0.00 V), and Co2Fe (−0.22 V). And the other one is homonuclear Ni3-TAC (−0.31 V). We predicted that these catalysts show high selectivity for converting NO–3 to NH3, due to stronger adsorption of nitrate than hydrogen, and high energy barriers for releasing byproducts (NO2 and NO). By analyzing the density of states, we find that in bimetallic Co2Ni and Co2Cu-TACs, the secondary Ni and Cu atoms not only bond with intermediates, but also influence the electron states of Co atoms. We find that the collaboration among three active sites can disrupt the scaling relationship among intermediates in NO3RR, resulting in catalysts boasting remarkably high activity. This heralds a new era in the development of efficient electrocatalysts for nitrate reduction and ammonia synthesis based on the triple-atom platform.
Acknowledgments
G.H.C. acknowledges financial support by the General Research Fund (grant no. 17309620) and Research Grants Council (RGC: T23-713/22-R). G.H.C. and W.A.G. acknowledge support from the Hong Kong Quantum AI Lab, AIR@InnoHK of the Hong Kong Government. W.A.G. thanks the U.S. National Science Foundation (CBET- 2311117) for support.
Data Availability Statement
Data will be available on request.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsami.4c17726.
Formula of all elementary process observed during the search of minimum energy reaction pathways. During structural relaxation, three carbon atoms in each TAC model are fixed to reduce the distortion and corresponding energy error (PDF)
Author Contributions
⊥ X.Z. and M.T. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Gruber N.; Galloway J. N. An Earth-System Perspective of the Global Nitrogen Cycle. Nature 2008, 451 (7176), 293–296. 10.1038/nature06592. [DOI] [PubMed] [Google Scholar]
- Canfield D. E.; Glazer A. N.; Falkowski P. G. The Evolution and Future of Earth’s Nitrogen Cycle. Science 2010, 330 (6001), 192–196. 10.1126/science.1186120. [DOI] [PubMed] [Google Scholar]
- Chen J. G.; Crooks R. M.; Seefeldt L. C.; Bren K. L.; Bullock R. M.; Darensbourg M. Y.; Holland P. L.; Hoffman B.; Janik M. J.; Jones A. K.; Kanatzidis M. G.; King P.; Lancaster K. M.; Lymar S. V.; Pfromm P.; Schneider W. F.; Schrock R. R. Beyond Fossil Fuel–Driven Nitrogen Transformations. Science 2018, 360 (6391), eaar6611 10.1126/science.aar6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher D. P.; Gewirth A. A. Nitrate Reduction Pathways on Cu Single Crystal Surfaces: Effect of Oxide and Cl. Nano Energy 2016, 29, 457–465. 10.1016/j.nanoen.2016.06.024. [DOI] [Google Scholar]
- Gao X.; Tse E. C. M. Unraveling the Performance Descriptors for Designing Single-Atom Catalysts on Defective MXenes for Exclusive Nitrate-To-Ammonia Electrocatalytic Upcycling. Small 2024, 20 (11), 2306311. 10.1002/smll.202306311. [DOI] [PubMed] [Google Scholar]
- Niu H.; Zhang Z.; Wang X.; Wan X.; Shao C.; Guo Y. Theoretical Insights into the Mechanism of Selective Nitrate-to-Ammonia Electroreduction on Single-Atom Catalysts. Adv. Funct. Mater. 2021, 31 (11), 2008533. 10.1002/adfm.202008533. [DOI] [Google Scholar]
- Zhao T.; Chen K.; Xu X.; Li X.; Zhao X.; Cai Q.; Chu K.; Zhao J. Homonuclear Dual-Atom Catalysts Embedded on N-Doped Graphene for Highly Efficient Nitrate Reduction to Ammonia: From Theoretical Prediction to Experimental Validation. Appl. Catal., B 2023, 339, 123156. 10.1016/j.apcatb.2023.123156. [DOI] [Google Scholar]
- Rehman F.; Kwon S.; Musgrave C. B.; Tamtaji M.; Goddard W. A.; Luo Z. High-Throughput Screening to Predict Highly Active Dual-Atom Catalysts for Electrocatalytic Reduction of Nitrate to Ammonia. Nano Energy 2022, 103, 107866. 10.1016/j.nanoen.2022.107866. [DOI] [Google Scholar]
- Shu Z.; Chen H.; Liu X.; Jia H.; Yan H.; Cai Y. High-Throughput Screening of Heterogeneous Transition Metal Dual-Atom Catalysts by Synergistic Effect for Nitrate Reduction to Ammonia. Adv. Funct. Mater. 2023, 33 (32), 2301493. 10.1002/adfm.202301493. [DOI] [Google Scholar]
- Luo F.; Guo L. Bimetallic Synergistic Catalysts Based on Two-Dimensional Carbon-Rich Conjugated Frameworks for Nitrate Electrocatalytic Reduction to Ammonia: Catalyst Screening and Mechanism Insights. Nanotechnology 2024, 35 (12), 125201. 10.1088/1361-6528/ad1649. [DOI] [PubMed] [Google Scholar]
- Haber F.; Le Rossignol R. Über Die Technische Darstellung von Ammoniak Aus Den Elementen. Z. Elektrochem. Angew. Phys. Chem. 1913, 19 (2), 53–72. 10.1002/bbpc.19130190201. [DOI] [Google Scholar]
- Kandemir T.; Schuster M. E.; Senyshyn A.; Behrens M.; Schlögl R. The Haber–Bosch Process Revisited: On the Real Structure and Stability of “Ammonia Iron” under Working Conditions. Angew. Chem., Int. Ed. 2013, 52 (48), 12723–12726. 10.1002/anie.201305812. [DOI] [PubMed] [Google Scholar]
- Hirakawa H.; Hashimoto M.; Shiraishi Y.; Hirai T. Selective Nitrate-to-Ammonia Transformation on Surface Defects of Titanium Dioxide Photocatalysts. ACS Catal. 2017, 7 (5), 3713–3720. 10.1021/acscatal.7b00611. [DOI] [Google Scholar]
- Rosca V.; Duca M.; de Groot M. T.; Koper M. T. M. Nitrogen Cycle Electrocatalysis. Chem. Rev. 2009, 109 (6), 2209–2244. 10.1021/cr8003696. [DOI] [PubMed] [Google Scholar]
- Zheng X.; Yan Y.; Li X.; Liu Y.; Yao Y. Theoretical Insights into Dissociative-Associative Mechanism for Enhanced Electrochemical Nitrate Reduction to Ammonia. J. Hazard. Mater. 2023, 446, 130679. 10.1016/j.jhazmat.2022.130679. [DOI] [PubMed] [Google Scholar]
- Yang L.; Feng S.; Zhu W. Achieving Reaction Pathway Separation for Electrochemical Nitrate Fixation on Triatomic Catalysts: A New Mechanism. J. Hazard. Mater. 2023, 441, 129972. 10.1016/j.jhazmat.2022.129972. [DOI] [Google Scholar]
- Fang L.; Wang S.; Song C.; Yang X.; Li Y.; Liu H. Enhanced Nitrate Reduction Reaction via Efficient Intermediate Nitrite Conversion on Tunable CuxNiy/NC Electrocatalysts. J. Hazard. Mater. 2022, 421, 126628. 10.1016/j.jhazmat.2021.126628. [DOI] [PubMed] [Google Scholar]
- Wang A.; Li J.; Zhang T. Heterogeneous Single-Atom Catalysis. Nat. Rev. Chem 2018, 2 (6), 65–81. 10.1038/s41570-018-0010-1. [DOI] [Google Scholar]
- Mitchell S.; Pérez-Ramírez J. Single Atom Catalysis: A Decade of Stunning Progress and the Promise for a Bright Future. Nat. Commun. 2020, 11 (1), 4302. 10.1038/s41467-020-18182-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C.; Fu S.; Shi Q.; Du D.; Lin Y. Single-Atom Electrocatalysts. Angew. Chem., Int. Ed. 2017, 56 (45), 13944–13960. 10.1002/anie.201703864. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Wang D.; Li Y. Rational Design of Single-Atom Site Electrocatalysts: From Theoretical Understandings to Practical Applications. Adv. Mater. 2021, 33 (34), 2008151. 10.1002/adma.202008151. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Guan J. Single-Atom Catalysts for Electrocatalytic Applications. Adv. Funct. Mater. 2020, 30 (31), 2000768. 10.1002/adfm.202000768. [DOI] [Google Scholar]
- Tamtaji M.; Kim M. G.; Wang J.; Galligan P. R.; Zhu H.; Hung F.-F.; Xu Z.; Zhu Y.; Luo Z.; Goddard W. A.; Chen G. A High-Entropy Single-Atom Catalyst Toward Oxygen Reduction Reaction in Acidic and Alkaline Conditions. Advanced Science 2024, 11 (26), 2309883. 10.1002/advs.202309883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R.; Wang D. Superiority of Dual-Atom Catalysts in Electrocatalysis: One Step Further Than Single-Atom Catalysts. Adv. Energy Mater. 2022, 12 (9), 2103564. 10.1002/aenm.202103564. [DOI] [Google Scholar]
- Tamtaji M.; Kim M. G.; Li Z.; Cai S.; Wang J.; Galligan P. R.; Hung F.-F.; Guo H.; Chen S.; Luo Z.; Wu W.; Goddard W. A.; Chen G. High-Throughput Screening of Dual Atom Catalysts for Oxygen Reduction and Evolution Reactions and Rechargeable Zinc-Air Battery. Nano Energy 2024, 126, 109634. 10.1016/j.nanoen.2024.109634. [DOI] [Google Scholar]
- Lin X.; Li Q.; Hu Y.; Jin Z.; Reddy K. M.; Li K.; Lin X.; Ci L.; Qiu H.-J. Revealing Atomic Configuration and Synergistic Interaction of Single-Atom-Based Zn-Co-Fe Trimetallic Sites for Enhancing Oxygen Reduction and Evolution Reactions. Small 2023, 19 (30), 2300612. 10.1002/smll.202300612. [DOI] [PubMed] [Google Scholar]
- Ji S.; Chen Y.; Fu Q.; Chen Y.; Dong J.; Chen W.; Li Z.; Wang Y.; Gu L.; He W.; Chen C.; Peng Q.; Huang Y.; Duan X.; Wang D.; Draxl C.; Li Y. Confined Pyrolysis within Metal–Organic Frameworks To Form Uniform Ru3 Clusters for Efficient Oxidation of Alcohols. J. Am. Chem. Soc. 2017, 139 (29), 9795–9798. 10.1021/jacs.7b05018. [DOI] [PubMed] [Google Scholar]
- Xiao J.; Liu Z.; Wang X.; Li F.; Zhao Z. Homonuclear Multi-Atom Catalysts for CO2 Electroreduction: A Comparison Density Functional Theory Study with Their Single-Atom Counterparts. J. Mater. Chem. A 2023, 11 (46), 25662–25670. 10.1039/D3TA05498E. [DOI] [Google Scholar]
- Chen Z. W.; Chen L. X.; Jiang M.; Chen D.; Wang Z. L.; Yao X.; Singh C. V.; Jiang Q. A Triple Atom Catalyst with Ultrahigh Loading Potential for Nitrogen Electrochemical Reduction. J. Mater. Chem. A 2020, 8 (30), 15086–15093. 10.1039/D0TA04919K. [DOI] [Google Scholar]
- Chen Z. W.; Chen L. X.; Yang C. C.; Jiang Q. Atomic (Single, Double, and Triple Atoms) Catalysis: Frontiers, Opportunities, and Challenges. J. Mater. Chem. A 2019, 7 (8), 3492–3515. 10.1039/C8TA11416A. [DOI] [Google Scholar]
- Cai G.; Lv H.; Zhang G.; Liu D.; Zhang J.; Zhu J.; Xu J.; Kong X.; Jin S.; Wu X.; Ji H. A Volcano Correlation between Catalytic Activity for Sulfur Reduction Reaction and Fe Atom Count in Metal Center. J. Am. Chem. Soc. 2024, 146, 13055. 10.1021/jacs.3c14312. [DOI] [PubMed] [Google Scholar]
- Shang H.; Zhou X.; Dong J.; Li A.; Zhao X.; Liu Q.; Lin Y.; Pei J.; Li Z.; Jiang Z.; Zhou D.; Zheng L.; Wang Y.; Zhou J.; Yang Z.; Cao R.; Sarangi R.; Sun T.; Yang X.; Zheng X.; Yan W.; Zhuang Z.; Li J.; Chen W.; Wang D.; Zhang J.; Li Y. Engineering Unsymmetrically Coordinated Cu-S1N3 Single Atom Sites with Enhanced Oxygen Reduction Activity. Nat. Commun. 2020, 11 (1), 3049. 10.1038/s41467-020-16848-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng L.; Shang L.; Zhang T.; Waterhouse G. I. N. Recent Advances in the Development of Single-Atom Catalysts for Oxygen Electrocatalysis and Zinc–Air Batteries. Adv. Energy Mater. 2020, 10 (48), 2003018. 10.1002/aenm.202003018. [DOI] [Google Scholar]
- Wan C.; Duan X.; Huang Y. Molecular Design of Single-Atom Catalysts for Oxygen Reduction Reaction. Adv. Energy Mater. 2020, 10 (14), 1903815. 10.1002/aenm.201903815. [DOI] [Google Scholar]
- Zhang J.; Yang H.; Liu B. Coordination Engineering of Single-Atom Catalysts for the Oxygen Reduction Reaction: A Review. Adv. Energy Mater. 2021, 11 (3), 2002473. 10.1002/aenm.202002473. [DOI] [Google Scholar]
- Li Y.; Feng Y.; Zheng D.; Zhao X.; Zhou Y.; Fu X.; Chen X. High-Performance Screening of Carbon-Nitride Single-Atom Catalysts for Oxygen Electrode Reaction in Rechargeable Metal–Air Batteries. Chem. Eng. J. 2023, 476, 146753. 10.1016/j.cej.2023.146753. [DOI] [Google Scholar]
- Pang Y.; Su C.; Xu L.; Shao Z. When Nitrogen Reduction Meets Single-Atom Catalysts. Prog. Mater. Sci. 2023, 132, 101044. 10.1016/j.pmatsci.2022.101044. [DOI] [Google Scholar]
- Li M.; Wang H.; Luo W.; Sherrell P. C.; Chen J.; Yang J. Heterogeneous Single-Atom Catalysts for Electrochemical CO2 Reduction Reaction. Adv. Mater. 2020, 32 (34), 2001848. 10.1002/adma.202001848. [DOI] [PubMed] [Google Scholar]
- Wang T.; Zhao Q.; Fu Y.; Lei C.; Yang B.; Li Z.; Lei L.; Wu G.; Hou Y. Carbon-Rich Nonprecious Metal Single Atom Electrocatalysts for CO2 Reduction and Hydrogen Evolution. Small Methods 2019, 3 (10), 1900210. 10.1002/smtd.201900210. [DOI] [Google Scholar]
- Wang Y.; Liu Y.; Liu W.; Wu J.; Li Q.; Feng Q.; Chen Z.; Xiong X.; Wang D.; Lei Y. Regulating the Coordination Structure of Metal Single Atoms for Efficient Electrocatalytic CO2 Reduction. Energy Environ. Sci. 2020, 13 (12), 4609–4624. 10.1039/D0EE02833A. [DOI] [Google Scholar]
- Tamtaji M.; Kwon S.; Musgrave C. B. I. I. I.; Goddard W. A. I. I. I.; Chen G. Reaction Mechanism of Rapid CO Electroreduction to Propylene and Cyclopropane (C3+) over Triple Atom Catalysts. ACS Appl. Mater. Interfaces 2024, 16, 50567. 10.1021/acsami.4c06257. [DOI] [PubMed] [Google Scholar]
- Yang H.; Zou W.; Zhang C.; Du A. Ab Initio Studies of Electrocatalytic CO2 Reduction for Small Cu Cluster Supported on Polar Substrates. ACS Appl. Mater. Interfaces 2024, 16 (26), 33688–33695. 10.1021/acsami.4c07445. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77 (18), 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Furthmüller J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6 (1), 15–50. 10.1016/0927-0256(96)00008-0. [DOI] [Google Scholar]
- Kresse G.; Furthmüller J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54 (16), 11169–11186. 10.1103/PhysRevB.54.11169. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Joubert D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59 (3), 1758–1775. 10.1103/PhysRevB.59.1758. [DOI] [Google Scholar]
- Grimme S. Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27 (15), 1787–1799. 10.1002/jcc.20495. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132 (15), 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Mathew K.; Sundararaman R.; Letchworth-Weaver K.; Arias T. A.; Hennig R. G. Implicit Solvation Model for Density-Functional Study of Nanocrystal Surfaces and Reaction Pathways. J. Chem. Phys. 2014, 140 (8), 084106. 10.1063/1.4865107. [DOI] [PubMed] [Google Scholar]
- Mathew K.; Kolluru V. S. C.; Mula S.; Steinmann S. N.; Hennig R. G. Implicit Self-Consistent Electrolyte Model in Plane-Wave Density-Functional Theory. J. Chem. Phys. 2019, 151 (23), 234101. 10.1063/1.5132354. [DOI] [PubMed] [Google Scholar]
- Nørskov J. K.; Rossmeisl J.; Logadottir A.; Lindqvist L.; Kitchin J. R.; Bligaard T.; Jónsson H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108 (46), 17886–17892. 10.1021/jp047349j. [DOI] [PubMed] [Google Scholar]
- Cai G.; Lv H.; Zhang G.; Liu D.; Zhang J.; Zhu J.; Xu J.; Kong X.; Jin S.; Wu X.; Ji H. A Volcano Correlation between Catalytic Activity for Sulfur Reduction Reaction and Fe Atom Count in Metal Center. J. Am. Chem. Soc. 2024, 146 (19), 13055–13065. 10.1021/jacs.3c14312. [DOI] [PubMed] [Google Scholar]
- Fang C.; Zhou J.; Zhang L.; Wan W.; Ding Y.; Sun X. Synergy of Dual-Atom Catalysts Deviated from the Scaling Relationship for Oxygen Evolution Reaction. Nat. Commun. 2023, 14 (1), 4449. 10.1038/s41467-023-40177-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv L.; Shen Y.; Liu J.; Meng X.; Gao X.; Zhou M.; Zhang Y.; Gong D.; Zheng Y.; Zhou Z. Computational Screening of High Activity and Selectivity TM/g-C3N4 Single-Atom Catalysts for Electrocatalytic Reduction of Nitrates to Ammonia. J. Phys. Chem. Lett. 2021, 12 (45), 11143–11150. 10.1021/acs.jpclett.1c03005. [DOI] [PubMed] [Google Scholar]
- Wang S.; Gao H.; Li L.; Hui K. S.; Dinh D. A.; Wu S.; Kumar S.; Chen F.; Shao Z.; Hui K. N. High-Throughput Identification of Highly Active and Selective Single-Atom Catalysts for Electrochemical Ammonia Synthesis through Nitrate Reduction. Nano Energy 2022, 100, 107517. 10.1016/j.nanoen.2022.107517. [DOI] [Google Scholar]
- Wang Y.; Shao M. Theoretical Screening of Transition Metal–N4-Doped Graphene for Electroreduction of Nitrate. ACS Catal. 2022, 12 (9), 5407–5415. 10.1021/acscatal.2c00307. [DOI] [Google Scholar]
- Lv P.; Wu D.; He B.; Li X.; Zhu R.; Tang G.; Lu Z.; Ma D.; Jia Y. An Efficient Screening Strategy towards Multifunctional Catalysts for the Simultaneous Electroreduction of NO3–, NO2– and NO to NH3. J. Mater. Chem. A 2022, 10 (17), 9707–9716. 10.1039/D2TA00192F. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be available on request.