

Significance
Strategies for durable control of HIV are urgently needed. Preclinical models that recapitulate virological and immune interactions observed in humans enable evaluation of therapeutic interventions. Here, we administered a cocktail of anti-simian immunodeficiency virus (SIV) monoclonal antibodies to rhesus macaques spanning interruption of antiretroviral therapy (ATI) and demonstrated that combination SIV mAbs significantly delay SIV rebound following ATI. Antiviral activity was mediated primarily by a single, potent neutralizing mAb in the cocktail and increasing the number of lower potency mAbs in the cocktail to broaden epitope specificity did not augment delay in viral rebound. Our results establish SIV mAbs as a valuable research tool that mimics the effect of human bNAb interventions in a rigorous challenge model.
Keywords: HIV, SIV, bNAb, rhesus macaque
Abstract
HIV-1 envelope broadly neutralizing antibodies represent a promising component of HIV-1 cure strategies. To evaluate the therapeutic efficacy of combination monoclonal antibodies (mAbs) in a rigorous nonhuman primate model, we tested different combinations of simian immunodeficiency virus (SIV) neutralizing mAbs in SIVmac251-infected rhesus macaques. Antiretroviral therapy-suppressed animals received anti-SIV mAbs targeting multiple Env epitopes spanning analytical treatment interruption (ATI) in 3 groups (n = 7 each): i) no mAb; ii) 4-mAb combination; and iii) 2-mAb combination. Each mAb was administered at 15 mg/kg, and both mAb-treated groups received ITS103.01, a highly potent CD4-binding site targeting antibody. mAb treatment delayed viral rebound, lowered rebound viremia setpoint and viral diversity, and extended animal lifespan. Compared to controls, for which viremia rebounded 2 wk following ATI, mAb infusion delayed rebound for both groups (P = 0.0003). Animals that received the 4-mAb regimen rebounded 3 to 6 wk post-ATI while the 2-mAb regimen rebounded 5 to 22 wk post-ATI. Envelope escape mutations emerged in rebound virus of mAb-treated animals that abrogated neutralization by ITS103.01, the most potent in the cocktail. These data demonstrate in vivo antiviral activity of SIV mAbs in the context of ATI via immune pressure dominated by the most potent mAb and highlight their potential in adjunctive therapeutic studies.
HIV-1 broadly neutralizing monoclonal antibodies (bNAbs) are a promising class of clinical interventions for prevention of HIV-1, or as a component of HIV-1 treatment strategies. BNAbs target the HIV-1 envelope glycoprotein (Env) on the surface of HIV-1 virions, blocking viral entry into cells and preventing infection (1). Viral replication in people living with HIV (PLWH) can be suppressed by bNAb administration, an effect mediated by bNAb neutralization activity, in addition to Fc-mediated antibody effector functions (2–8). Multiple preclinical and clinical trials investigating bNAbs as therapeutics are planned or ongoing, with a major focus on bNAbs given either spanning analytical treatment interruption (ATI) or during antiretroviral therapy (ART) in combination with other agents to improve targeting of cellular reservoirs.
Numerous preclinical and clinical studies indicate that bNAbs can suppress replication of sensitive virus in vivo. Rhesus macaques infected with simian-HIV(SHIV) treated with bNAbs exhibit decreased viremia during chronic (9–13) or acute infection (10, 14–16). Subsequent clinical trials demonstrated similar bNAb-mediated antiviral effects. Specifically, 3BNC117 (17), VRC01 (18), and the combination of 3BNC117 and 10-1074 (2–4) suppressed viral replication when administered spanning ATI. BNAbs also suppress viral replication in people with chronic HIV-1, as seen with 3BNC117 (5, 6) and VRC01 (7), and in the context of combination treatments [e.g. PGDM1400, PGT121 and VRC07-523-LS (8); 3BNC117 and 10-1074 (3, 4)].
It has also been hypothesized that bNAb treatment is capable of exerting a “vaccinal effect”, whereby the endogenous adaptive immune response is enhanced following bNAb administration. In rhesus macaques, for example, bNAbs given either 3- or 14-d postinfection resulted in CD8-mediated control of infection in a subset of animals (15, 16), though no evidence of CD8 response modulation by bNAb treatment was observed. In chronically infected PLWH, increased endogenous neutralizing antibody titers were found following 3BNC117 infusion (19). T cell responses were also enhanced in PLWH administered bNAb at the time of ART initiation (3BNC117) (20), or at treatment interruption (3BNC117 and 10-1074) (21). It was hypothesized that this increase resulted from Fc–FcγR interactions between the bNAb and innate immune cells, and that the formation of bNAb-virus immune complexes exerted an immunomodulatory effect. However, augmented T cell immune responses have not been observed consistently in therapeutic bNAb trials, prompting further investigation (4, 22, 23).
While initial clinical studies investigating the therapeutic potential of HIV-1 bNAbs involved single monoclonal antibodies (5–7, 17, 18), single bNAb regimens selected for replication of viral isolates resistant to the bNAb. Similarly, in the context of bNAb immunoprophylaxis, the VRC01 bNAb used in the Antibody Mediated Prevention trials was only effective at preventing infection against sensitive viruses with an 80% inhibitory concentration less than 1 µg/mL (1). More recent and ongoing studies have therefore prioritized combination bNAbs for treatment and prevention both to address the challenge of HIV-1 env diversity and mitigate selection of escape mutants with monotherapy (2–4, 8). The exact bNAb combinations required for optimal antiviral effect, including the identity and number of antibodies, is an active area of investigation.
Here, we sought to evaluate the therapeutic efficacy of a combination of two or four anti-simian immunodeficiency virus (SIV) mAbs administered to SIV-infected rhesus macaques spanning ATI. Rhesus macaques are a valuable model for evaluating immunotherapeutic strategies due to their close genetic relatedness to humans and susceptibility to SIV, including development of simian AIDS. SIV was chosen here for its well-established, consistent, and vigorous viral replication kinetics in macaques, including following treatment interruption, providing a rigorous model system for evaluating therapeutic intervention efficacy. Recent isolation and extensive characterization of SIV Env-specific mAbs enable mAb prevention and cure studies in SIV infection models (24, 25). In a prior SIV study, monotherapy with the potent SIV mAb ITS103.01 spanning ATI delayed rebound by ~1 wk compared to controls (26). Viral rebound occurred in the presence of robust levels of ITS103.01, suggesting the possible development of resistance mutations. Here, we sought to build on this prior work and assess the impact of combination bNAb therapy on SIV infection using a panel of potent mAbs targeting multiple Env epitopes, theorized to reduce the development of escape mutations: ITS103.01, ITS102.01 [both CD4 binding site (CD4bs)], ITS09.01 [variable loop 2 (V2)], and ITS113.01 [membrane-proximal external region (MPER)]. These mAbs were chosen as the most potent anti-SIV mAbs available at the time of study initiation. ITS103.01 was administered in combination with one or three other mAbs spanning ATI to investigate differing numbers of bNAbs on virological and immunological parameters following treatment interruption.
Results
Study Design and Virologic Outcomes.
To identify optimal conditions for combination bNAb passive immunotherapy, we designed a macaque study comparing different numbers of anti-SIV mAbs with multiple antigen specificities and neutralization potencies administered over the period spanning ATI. Twenty-one rhesus macaques were infected intravenously with 200 TCID50 SIVmac251 followed by 18 wk of combination antiretroviral therapy (cART) initiated 1 wk postinfection (Fig. 1A). SIVmac251 was selected for being a diverse, polyclonal viral stock that may better resemble the diversity of HIV infection in humans than a single, clonal, viral preparation. Animals were assigned to 1 of 3 groups balanced for age, weight, sex, and peak viral load. MAbs were infused every 2 wk from weeks 15 to 29, for eight total doses. Daily cART was stopped at week 19. Animals were distributed into three treatment groups (n = 7/group); the control group received no mAbs while the two active arms received either a combination of 2 mAbs (ITS103.01 and ITS09.01) or 4 mAbs (ITS103.01, ITS09.01, ITS102.01, and ITS113.01) at 15 mg/kg of each antibody.
Fig. 1.

Study design and mAb therapeutic efficacy. (A) Schematic of study design. Animals were infected with SIVmac251 IV (day 0), and cART commenced on day 7 was continued until week 19. MAbs were administered to two active arms every 2 wk from week 15 to wk 29, as indicated by vertical black lines. Groups received either no mAbs, 4 mAbs (ITS103.01, ITS09.01, ITS102.01 and ITS113.01), or 2 mAbs (ITS103.01 and ITS09.01) (n = 7/group). (B) SIV strain neutralization sensitivity to the infused mAbs individually and in combination. IC50 values ± SD from 2 to 3 technical replicates are indicated. (C) Longitudinal plasma SIV RNA (pVL) for each animal by study group. Gray shading indicates period of ART treatment. (D) Weeks to pVL > 1,000 copies/mL. Statistical comparisons reflect log-rank (Mantel-Cox) test. (E) Setpoint pVL expressed as the geometric mean from weeks 8 to 30 post pVL > 1,000 copies/mL. (F) Peak plasma viral load within 4 wk of pVL > 1,000. Open symbols indicate animals with strongly delayed viral rebound. For (E) and (F) groups were compared using a Kruskal–Wallis test and Dunn’s posttest. Dots represent individual animals; horizontal bars, group geometric means. (G) Animal survival following ATI. Curves were compared using a log-rank (Mantel-Cox) test.
The neutralization capacity of each individual mAb and the 2- and 4-mAb combinations was measured against a panel of SIVmac251-derived env pseudovirus clones. This panel was chosen for its range in neutralization sensitivity to SIV mAbs and sequence diversity distributed across the SIVmac251 swarm (SI Appendix, Fig. S1) (24). While three of these pseudoviruses cluster phylogenetically, they differ in neutralization sensitivity (SIVmac251.6, SIVmac251.H9.15 (both tier 1), and SIVmac251.30 (tier 2)). The CD4bs-directed mAb, ITS103.01, was the most potent neutralizer of the four mAbs, with IC50s spanning 7-27 ng/mL (Fig. 1B). The other CD4bs-directed mAb, ITS102.01, and the MPER-directed mAb ITS113.01 neutralized most of the strains tested, albeit at lower potency. Full neutralization by the V2-directed mAb ITS09.01 was limited to the tier 1 viruses. Neutralization potency was equivalent between the 2- and 4-mAb combinations, which both neutralized with a marginally greater potency than the individual mAbs for 4/5 of the viruses tested.
The primary endpoint for mAb antiviral efficacy was time to plasma viremia rebound following ATI, which was monitored in all animals weekly until rebound, when sampling frequency was reduced to fortnightly, and then monthly. Prior to cART initiation, viremia peaked at ~106 copies/mL plasma, which was well suppressed by cART in most animals by week 5, with some animals continuing to display small “blips” in viremia (Fig. 1C). At week 15, the time of mAb treatment initiation, plasma viral loads were below the limit of detection in all animals. Following ATI, animals in the control group rebounded rapidly, with 7/7 animals in the control group developing plasma viral load >1,000 copies/mL 2 wk post-ATI (Fig. 1 C and D). Animals in the mAb-treated groups rebounded significantly later than the control animals: weeks 3 to 6 post-ATI for all animals in the 4-mAb group and 4/7 of the animals within the 2-mAb group. Viremia rebound was further delayed to 14, 18, and 22 wk post-ATI in three of the 2-mAb animals, resulting in a trend toward longer time to rebound in the 2-mAb group relative to the 4-mAb group (P = 0.09). For 11/14 of the mAb-treated animals, rebound occurred during mAb administration, prompting analyses assessing the impact of mAbs on viral replication. In general, mAb treatment reduced setpoint plasma viral load compared to controls (Fig. 1E; P = 0.04 and P = 0.06, 4- and 2-mAb groups, respectively). Strongly delayed rebound (14 to 22 wk post-ATI) corresponded with setpoint viremia in the bottom tertile. Peak viral load postrebound did not differ between groups (Fig. 1F). Overall, mAb treatment delayed viral rebound by up to 20 wk and decreased chronic infection virus replication postrebound.
The highly pathogenic nature of SIVmac251 enabled assessment of survival postrebound as an endpoint in this study. Control animals succumbed to simian AIDS-related disease sooner than those in the mAb-treated groups (Fig. 1G and SI Appendix, Fig. S2A). When assessed either relative to time following ATI or time following rebound, this difference was significant comparing the 4-mAb group with the untreated controls (P = 0.04, P = 0.04, respectively) and trended toward significance for the 2-mAb versus control animals (P = 0.05, P = 0.06, respectively). Survival time following ATI was negatively correlated with setpoint viral load (SI Appendix, Fig. S2B), implicating chronic virus replication in pathogenesis.
Monoclonal Antibody Pharmacokinetics.
To determine mAb in vivo persistence and its relationship to viremia suppression, plasma mAb concentrations were measured longitudinally using anti-idiotype antibodies specific for each mAb by ELISA. Levels were assessed preinfusion, 2 wk following the first infusion and continued fortnightly until mAb dropped below the limit of quantitation (Fig. 2). Geometric mean mAb concentrations 2 wk following administration were 153 µg/mL (ITS103.01), 180 µg/mL (ITS102.01), 189 µg/mL (ITS09.01), and 296 µg/mL (ITS113.01). Elevated assay background for ITS113.01 limited quantitative measurements below ~1 µg/mL for this mAb, although this background varied between animals. Following the final infusion at week 29, plasma mAb levels decayed logarithmically, achieving values below the assay limit of detection between weeks 40 to 56. Mean mAb half-life was 11.4 d for ITS103.01 and 14.2 d for each of ITS09.01, ITS102.01, and ITS113.01, consistent with prior half-life analyses of these mAbs (26, 27).
Fig. 2.
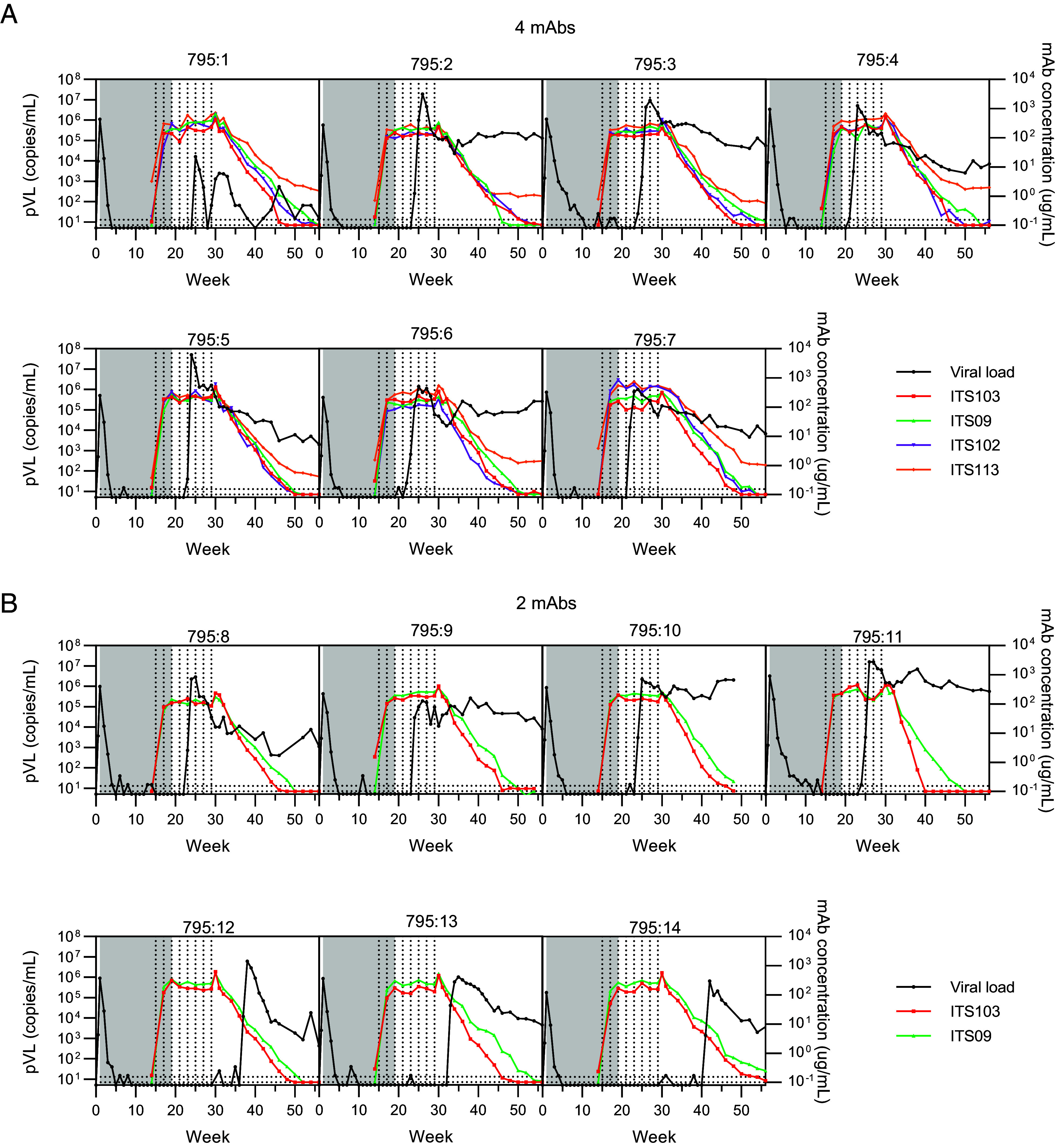
Antibody pharmacokinetics and individual plasma viral loads. Antibody levels in plasma were measured at baseline and from 1 wk post the first antibody infusion until week 56 by ELISA (colored lines). Longitudinal pVL for each animal are shown in black. The shaded area indicates the period of cART administration. Vertical dotted lines indicate mAb administrations. Horizontal dotted lines indicate lower limits of detection for the viral load assay (upper line) and mAb ELISA (lower line). Data are shown for the 4-mAb group (A) and 2-mAb group (B).
Since viral rebound occurred during the period of mAb administration in most mAb-treated animals, we sought to understand whether in vivo mAb concentrations were sufficient to achieve virus neutralization at the time of rebound. In most animals, mAb concentration at the time of rebound was greater than 100 µg/mL. The remaining 3/14 animals rebounded during the period of mAb decay, with ITS103.01 levels of 5, 19, or 99 µg/mL and ITS09.01 levels of 13, 37, or 146 µg/mL at the sampling 1 wk prior to plasma viremia >1,000 copies/mL. The IC80 of ITS103.01 ranges from 0.062 to 0.084 µg/mL for SIVmac251 isolates [Fig. 1B, (24)], thus ITS103.01 plasma levels were >1,000-fold greater than the IC80 and would be expected to retain neutralization activity against the challenge virus. Accordingly, insufficient mAb concentration is unlikely to explain the kinetics of viral rebound.
As the mAbs used here were native rhesus antibodies we did not expect the development of anti-drug antibodies (ADA) against the infused mAbs, despite repeated administration. We confirmed this by measuring antibody titers against the fragment antibody-binding (Fab) region of each individual mAb at baseline and following completion of all eight mAb infusions by ELISA (SI Appendix, Fig S3). Titers against each of the infused mAbs did not significantly increase post-mAb therapy, indicating limited evidence of ADA. We believe ADA response development beyond week 32 was unlikely given waning antigen levels, similar mAb PK observed among study animals, and prior in vivo experience with these mAbs in macaques (26), though we cannot exclude this possibility.
Endogenous Immune Responses Following MAb Administration.
Prior studies of HIV-1 mAb administration in both nonhuman primates (15, 16) and humans (19–21) suggest mAb passive transfer may enhance the development of endogenous HIV-1-specific immune responses, potentially conferring additional therapeutic benefit to passive immunization. Therefore, we assessed adaptive humoral and T cell immunity in SIV mAb recipient animals relative to controls. Env-specific binding antibody titers against gp140 and variable loops 1 and 2 (V1V2) were measured at week 70 (or the final blood collection prior to this time), when the infused mAbs were below detection limits in plasma and therefore unlikely to confound assay interpretation (Fig. 2). Gp140-binding antibody responses were greater in control animals compared to mAb-treated animals and positively correlated with setpoint viral load (Fig. 3A and SI Appendix, Fig. S4A), suggesting antigen burden contributed to response development. V1V2-specific titers did not differ between groups. In addition, we hypothesized that anti-CD4bs mAb infusion may limit development of immune responses targeting this site due to epitope occlusion and assessed CD4bs-directed responses by competition ELISA. Control animals mounted greater anti-CD4bs antibody responses than mAb-treated animals (Fig. 3B). These anti-CD4bs responses also positively correlated with setpoint viral load (SI Appendix, Fig. S4B), supporting antigen burden as driving the development of CD4bs-specific antibodies, although epitope blocking by the anti-CD4bs mAbs is also possible. Thus, SIV-specific binding antibody responses were not augmented by the mAb infusions.
Fig. 3.

Endogenous humoral responses following rebound. Endogenous plasma humoral responses were measured at week 70, or the final timepoint prior to necropsy for animals deceased prior to week 70. One animal in the 2-mAb group was excluded from analysis due to necropsy while infused mAb remained detectable. (A) Binding antibody responses to gp140 and V1V2 as assessed by ELISA. Data are expressed as area under the curve (AUC). (B) Serum antibody responses competing binding of the CD4bs-proximal mAb ITS103.01 measured by ELISA. Titers are expressed as the reciprocal serum titer required for 50% inhibition of ITS103.01 binding to SIVmac239 gp140 protein. (C) SIV pseudovirus neutralizing responses to SIVmac251-derived viral isolates of varying neutralization sensitivity. Virus neutralization reciprocal 50% inhibitory dilution (ID50) is shown. The dotted line indicates assay lower limit of detection. Each dot represents an individual animal; open symbols indicate animals with strongly delayed viral rebound. Horizontal bars indicate group medians. Significance was assessed using a Kruskal–Wallis test followed by a Dunn’s posttest.
In addition to binding antibody responses, we measured the impact of mAb infusion on endogenous neutralizing antibody titers in plasma for three SIVmac251-derived pseudovirus clones, spanning tier 1–tier 3 neutralization sensitivity. Neutralizing titers in all groups were high (~105 ID50) against the easy-to-neutralize tier 1 SIVmac251.6, moderate against the tier 2 SIVmac251.cs.41 [~103-4 ID50] and below the limit of detection in 15/20 measured animals for the difficult-to-neutralize tier 3 SIVmac239.cs.23 (Fig. 3C). Of note, these responses were assessed after mAb decay in plasma to limit the likelihood of mAb contributing to in vitro neutralization. MAb administration did not impact endogenous neutralization titers, as responses were comparable between the active and control groups. In addition, to explore whether the strong delay in viral rebound observed for three 2-mAb treated animals was linked to more potent adaptive immune responses, we compared their binding and neutralizing antibody responses to the other 2-mAb animals (Fig. 3). However, responses did not differ in this subgroup analysis, suggesting endogenous antibody responses were unlikely to contribute to these cases of prolonged virus control.
To explore potential augmentation of endogenous T cell responses, as previously reported following mAb administration in PLWH (20, 21), we assessed longitudinal SIV Gag and Env-specific T cell response magnitude by SIV peptide pool stimulation and intracellular cytokine staining for Th1, Th2, and T follicular helper (Tfh) markers (Fig. 4 and SI Appendix, Fig. S5). CD4 and CD8 T cell responses expressing Th1 cytokines were low at study week 14 (following 13 wk of cART-initiated 1-wk postinfection) and did not increase after mAb infusions during cART-suppressed infection (week 18) (Fig. 4 A and B). Just prior to mAb treatment (week 14), CD8 T cell responses against Env were marginally greater in animals assigned to the 4-mAb group (P = 0.03), but this difference was not observed at other study points. Following cART interruption, Gag- and Env-specific CD4 T cell responses increased at successive timepoints postrebound while robust CD8 responses were mounted 3 wk postrebound and sustained through 20 wk postrebound. Overall, anti-Gag T cell responses exceeded those against Env, and SIV-specific CD4 T cells produced predominantly Th1 cytokines. Neither Th1 CD4 nor CD8 T cell responses differed between control and mAb-treated groups. Responses by circulating Tfh-like cells were also of greater magnitude against Gag than Env, with detectable CD40L expression following Gag peptide stimulation during ART suppression, and increased responses following rebound (Fig. 4C). Gag-specific IL-21-producing Tfh responses were significantly greater in the 2-mAb group than controls at 20 wk postrebound (P = 0.02), potentially due to the lower viral loads in mAb-treated animals preserving T cell function, although this did not associate with delayed viral rebound. This increase was largely driven by IL-21 single-positive cells, as a similar increase was not observed for IL-21+CD40L+ Tfh (SI Appendix, Fig. S6). No other measured T cell responses differed between the study groups. Taken together, neither humoral nor cellular responses exhibited evidence of a vaccinal effect following mAb infusion.
Fig. 4.

Antigen-specific T cell responses. Gag- (Left) and Env- (Right) specific T cell responses were measured longitudinally in memory CD4, CD8, and Tfh cells by peptide pool stimulation and intracellular cytokine staining for PBMC. Boolean combinations of Th1 cytokines (IFNγ, TNF, and IL-2) and Th2 cytokines (IL-4 and IL-13) expression were summed for memory CD4 and memory CD8 T cells. CD40L (CD154) and IL-21 expression was assessed on Tfh. Each dot represents an individual animal. The Baseline timepoint samples were collected prior to SIV infection. The box plot reflects the interquartile range, and whiskers represent the minimum and maximum values. PR = postrebound. Significance was assessed using a Kruskal−Wallis test followed by a Dunn’s post hoc test, * indicates P < 0.05.
Envelope Sequence Changes Upon Viral Rebound.
To explore virus evolution during ART, and in the setting of mAb-mediated immune pressure, we performed two parallel sequencing methods to assess changes in env sequence at rebound. Deep sequencing of plasma SIV genomes was performed at peak rebound viral load from a subset of 12 animals (n = 3 to 5/group; ≤ 3 wk post-ATI) and generated an average of 871 sequences per animal. Single genome amplification (SGA) sequencing was also performed at rebound, as well as at peak viremia prior to ART commencement for all 21 animals, generating an average of 27 sequences per animal at each timepoint. Prior to ART, sequence diversity was similar across animals (Fig. 5A and SI Appendix, Fig. S7A), reflecting the diversity seen in the SIVmac251 stock (SI Appendix, Fig. S8). In contrast, rebound viremia was less diverse in mAb-treated animals (median pairwise diversity range = 0 to 0.002) compared to animals in the untreated group (median pairwise diversity range = 0.001 to 0.014) (Fig. 5B and SI Appendix, Fig. S7A). Similarly, SGA sequencing revealed a single lineage at rebound in mAb-treated animals versus a median of two lineages in untreated animals (range 1 to 3; P = 0.005 for mAb groups relative to control; SI Appendix, Fig. S7B). With the deep sequencing approach we identified an average of four lineages in the untreated animals versus 1.4 in the mAb groups (Fig. 5C), with a second, minor lineage in 3/8 of the mAb-treated animals. Correspondingly, while rebound sequences for both treated and untreated animals were dispersed among each other and the SIVmac251 swarm viral stock sequences, viral populations were monophyletic for animals treated with mAbs and polyphyletic for the untreated animals (SI Appendix, Figs. S8 and S9).
Fig. 5.
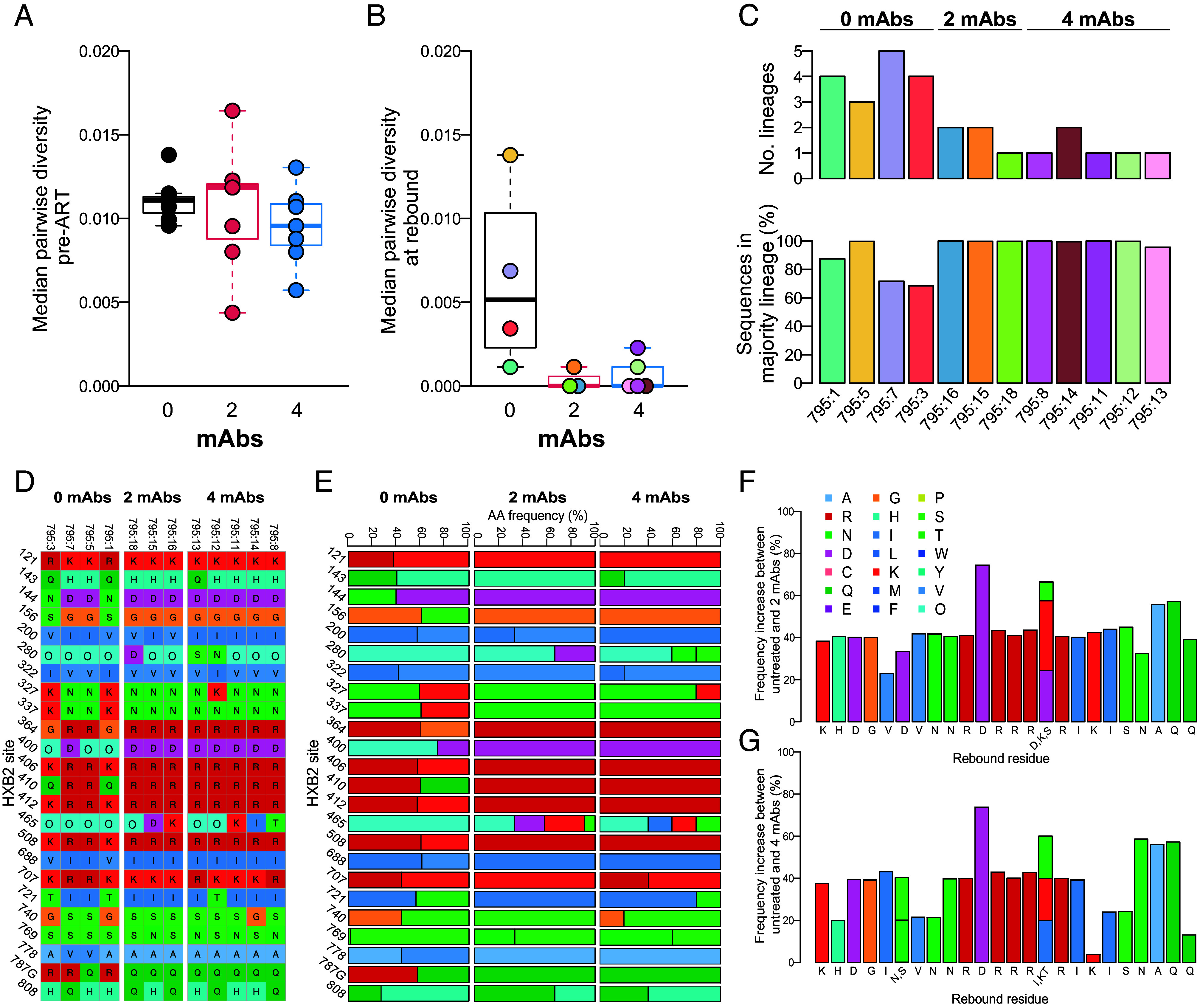
Impact of mAb treatment on SIV envelope sequence diversity and amino acid residue frequency. Boxplots of median pairwise envelope sequence diversity in untreated and treated animals at (A) pre-ART, measured by SGA, and (B) rebound, measured by deep sequencing in 12 of 21 animals. (C) The number of lineages and percentage of sequences found in the majority lineage in untreated and treated animals at rebound. (D–G) Env residues with differing frequencies between untreated and one or both of the treated groups in rebound virus. Amino acid identity corresponds to colors in legend at right; “O” indicates a PNGS. (D) Sequence alignment of consensus rebound sequences for animals treated with 0, 2, or 4 mAbs at sites identified to have shifted between untreated and treated animals. (E) Residue frequencies in PacBio rebound sequences. The increase in residue frequency compared to untreated animals in animals treated with (F) 2 mAbs and (G) 4 mAbs.
To investigate sites under selective pressure following mAb administration, we identified Env residues with variable amino acid frequencies between treated and untreated groups at rebound (18 Env sites were masked due to low information content). Twenty-four sites differed in residue frequency between the untreated group and one or both treated groups, as defined by a minimum of three SD of the mean residue frequency in untreated controls (Fig. 5 D and E). Three of these sites were potential N-linked glycosylation sites (PNGSs) (N280, D400N, and N465). The magnitude increase in residue frequency across the 24 sites ranged from 23 to 75% and 4 to 74% in animals treated with 2 and 4 mAbs, respectively (Fig. 5 F and G). We then assessed the residue frequency changes of the 24 sites identified using deep sequencing in the SGA sequences for which we had data from all animals at both pre-ART and rebound timepoints (SI Appendix, Fig. S10). Five of the 24 sites significantly varied between pre-ART and rebound in one or both mAb treatment groups (SI Appendix, Fig. S10 B–E; P < 0.05), supporting the deep sequencing findings and suggesting selective pressure at these key sites.
Exploring the mechanism by which these variants arose in the context of mAb therapy, two of the five identified sites under mAb selective pressure involved removal of a PNGS at either position 280 (within the D-loop) or 465 [immediately following variable loop 5 (V5)] (SI Appendix, Fig. S10B). In 12/14 mAb-treated animals, all rebound sequences had one of these two PNGSs removed; for the remaining two animals, these PNGSs were eliminated in 94% and 31% of sequences (Fig. 6A). The rebound sequences from mAb-treated animals did not cluster together in the phylogenetic tree (SI Appendix, Fig. S9), and the PNGS-removal arose and was selected independently in multiple lineages. Both PNGSs were removed in separate sequences within the same lineage in several animals (795:10, 795:17 & 795:21), supporting the independent development of these sequence variants.
Fig. 6.

Env residue hotspots observed following mAb therapy and decreased sensitivity of rebound virus to bNAb neutralization. (A) Proportion of recovered plasma viral sequences in each animal (rows) at the pre-ART baseline or peak rebound viremia timepoints containing removal of PNGSs at positions N280 or N465 (all HXB2 numbering) or containing either of these two mutations (“total”). (B) Proportion of sequences containing valine instead of alanine at position 778 for each animal (rows). (C) Neutralization sensitivity of pseudovirus cloned from the dominant rebound virus for each animal (columns) to the infused mAb (rows). Heat map displays the IC50 against each mAb.
Three additional sites displayed significant amino acid variance following mAb treatment. The most abundant change was in position 778 (located within the cytoplasmic domain). MAb treatment selected against V778, with all rebound isolates harboring A778, despite V778 representation in 13/14 animals at baseline (Fig. 6B and SI Appendix, Fig. S10C). V778 frequency at rebound within the control group ranged from 4 to 100% (median = 22%). The other two cases of potential mAb pressure — H143Q (variable loop 1) and I688V (transmembrane domain) — involved selection toward the consensus residue (H143 and I688, respectively) (SI Appendix, Fig. S10 D and E). The frequency of the nonconsensus residue in both cases was <40% at baseline in all animals, therefore it is possible that this change is driven by stochastic rebound of the higher frequency isolates containing the consensus residue.
To assess the impact of these amino acid changes on sensitivity to neutralization by the infused mAbs, we constructed pseudoviruses of the dominant rebound env plasma sequence for each animal. All rebound env sequences from control animals were highly sensitive to neutralization by ITS103.01 (Fig. 6C), mirroring the parent virus (Fig. 1B). In contrast, sequences cloned from mAb-treated animals were resistant to ITS103.01. Neutralization by the three other mAbs did not differ between study groups and was comparable to stock sensitivity. For the outlier mAb-treated animal (795.10) in which only 31% of rebound sequences lost a PNGS, we compared the neutralization sensitivity of two recovered sequences. The two sequences were identical except for the presence of a PNGS at position N280 in one of the sequences. Virus containing this PNGS was sensitive to neutralization by ITS103.01, but its removal rendered the virus resistant (SI Appendix, Table S1). Mapping the residues identified in the deep sequencing analysis as having an altered frequency on a tridimensional structure showed that mutations at the glycan sites 280 and 465 potentially interfere with the CD4bs (Fig. 7). Collectively, these data implicate the PNGSs N280 and N465 as being necessary for ITS103.01-mediated neutralization, which dominated the mAb selection pressure on rebound virus.
Fig. 7.

Structural position of Env residues under apparent selective pressure by SIV mAbs. SIV Env sites of interest identified through rebound virus sequence differences between mAb-treated and untreated animals were mapped on an SIVmac239 Env structure (PDB: 8DVD). Env region mediating CD4 binding is indicated by blue shading. Variable loops are colored as indicated, green shaded sites are within the V4 region.
MAb Suppression of Viral Replication In Vitro.
To measure viral susceptibility and capacity for escape from each mAb and mAb combinations, a viral resistance assay was performed in vitro (28). CD4+ T cells isolated from PBMC from two randomly selected control animals at rebound peak were incubated with the study mAbs and “feeder” CD4+ T cells from SIV-naïve animals over the course of 2 wk. MAb inhibition of viral replication was assessed longitudinally by monitoring SIV capsid (p27) concentration in culture supernatant. Control animals were chosen for this assay as their virus is expected to exhibit the greatest sequence similarity to the original viral stock, i.e. no immune pressure exerted by mAb treatment. The ITS09.01 and ITS113.01 mAbs did not suppress viral replication, indicating the presence of SIVmac251 isolates resistant to neutralization by these two antibodies (Fig. 8). ITS102.01 transiently suppressed replication in one of two animals, although virus quickly rebounded, suggesting selection for or development of virus containing resistance mutations. In contrast, ITS103.01 alone, as well as the 2- and 4-mAb combinations, suppressed viral replication for the duration of the assay, indicating that SIVmac251 virus in untreated animals is sensitive to ITS103.01-mediated neutralization.
Fig. 8.

In vitro viral replication sensitivity to SIV mAbs. Sensitivity of SIVmac251 replication in culture to SIV mAbs individually and in combination. CD4+ T cells were isolated from PBMC of two viremic rhesus macaques from the no-mAb group postrebound and cultured in the presence of the indicated mAb or the mAb combinations administered to study animals. CD4+ T cells from PBMC of uninfected rhesus macaques were added to the culture as feeder cells. Supernatant p27 protein levels were measured longitudinally by ELISA and normalized to the p27 concentration in the negative control mAb (100%). Dots represent the mean of three replicates, and error bars show SD.
Ex Vivo Detection of Cell Surface-Bound Monoclonal Antibody.
Reflecting upon the strong delay in rebound observed in three animals in the 2-mAb treated group, and considering prior evidence that ITS102.01 does not compete epitope binding of ITS103.01 (24), we hypothesized that the increased total mAb concentration in the 4-mAb condition reduced the ability of any individual mAb to interact with host cells, limiting effectiveness of the highly potent ITS103.01. We used anti-idiotype mAb-specific antibodies to stain surface Fc receptor-bound mAb on rhesus macaque PBMC incubated with either the 2-mAb or 4-mAb combination in vitro (SI Appendix, Fig. S11). Elevated levels of ITS103.01 and ITS09.01 (both present in the 2- and 4-mAb combinations) bound monocytes, and the classical monocyte subset in particular, in the context of 2 mAbs compared to 4 (SI Appendix, Fig. S12A). ITS103.01 preferentially bound monocytes expressing CD64 and CD32 (SI Appendix, Fig. S13A). The proportionate increase in surface ITS103.01 with monocyte CD64 expression, but not CD32, suggests that binding to classical monocytes was likely mediated by CD64 (FcγR1), the high-affinity IgG receptor (SI Appendix, Fig. S13B). No differences between the 2- and 4-mAb conditions were apparent in mAb binding to other monocyte subsets, NK cells, or dendritic cells in vitro (SI Appendix, Figs. S12A and S14). To measure cell-associated mAb following infusions in the SIV-infected animals, surface staining was performed on PBMC collected 24 h after the second mAb administration, while animals remained on suppressive cART (study week 17 + 1 d). Both ITS103.01 and ITS09.01 were detected on the surface of multiple leukocyte populations, with the most pronounced staining on monocytes (SI Appendix, Figs. S12B and S15). In contrast to the in vitro data, comparable mAb binding was observed between the 2- and 4-mAb treated animals for all cell types analyzed. The three animals in the 2-mAb group with a strongly delayed rebound did not substantially differ in mAb binding to other animals within the 2-mAb group. As binding was equivalent in vivo, it suggests levels of cell-bound mAb was unlikely to contribute to the delayed rebound in the 2-mAb group.
As mAb binding to cells is mediated by Fcγ receptors (FcγRs), we also sequenced the FcγR allotype of each animal to identify potential differences among the study groups, as well as within the 2-mAb group that may account for the divergent rebound phenotypes. FcγR allotypes were distributed evenly among the groups, and the three animals with strongly delayed rebound (795:19, 795:20, and 795:21) did not notably differ from other 2-mAb animals (SI Appendix, Table S2).
Discussion
We describe the use of combination SIV-specific mAb treatment during ATI in SIV-infected rhesus macaques as a therapeutic intervention to limit viral replication following discontinuation of cART. MAb treatment spanning ATI consisting of combinations of either 2 or 4 mAbs significantly delayed viral rebound relative to no mAb by 4 and 3 median weeks, respectively. However, rebound occurred in the presence of mAb at levels expected to be therapeutic (minimum ITS103.01 μg/mL > 150× IC50; ITS103.01 μg/mL > 3,000× IC50 for 12/14 animals). Both deep sequencing and SGA analysis of rebound plasma virus in treated animals revealed the emergence of env mutations that conferred neutralization resistance to the most potent infused mAb, ITS103.01, suggesting mAb-mediated immune pressure dominated by a single mAb. The primacy of ITS103.01 in limiting SIV replication in vivo was supported by ITS103.01 alone achieving suppression of in vitro SIV replication comparable to that of the 2- and 4-mAb cocktails. SIV replication in this model both before ART and following ATI was robust and consistent with plasma HIV-1 RNA levels in PLWH (29), supporting the use of SIV infection in rhesus macaques as a rigorous model for evaluating antibody-based interventions.
BNAb in vivo selection of neutralization-resistant HIV-1 env sequences has been observed previously, primarily in the setting of monotherapy (5–7, 17, 18). Here, rebound virus in all SIV mAb-treated animals contained mutations that removed a PNGS at either position 280 (in the D loop) or 465 (V5), which are located proximal to the CD4bs and conferred resistance to the highly potent, CD4bs-directed mAb, ITS103.01. Mutations at equivalent positions in HIV-1 env arose in ART-untreated viremic PLWH following VRC01 administration (7), although due to the viral sequence diversity between SIV and SHIV, mutations at these positions in HIV-1 did not consistently involve mutation of a PNGS. Other studies administering VRC01 either during acute infection or at ATI did not observe evidence of selection mutations (30, 31). D-loop and V5 mutations have been described as conferring resistance to VRC01-class HIV-1 bNAbs in in vitro assays (32, 33), and in naturally resistant strains (34). The removal of either PNGS did not confer resistance to neutralization by ITS102.01 to the same extent as ITS103.01. These two CD4bs-directed antibodies have differing modes of recognition (24), suggesting that glycans at N280 or N465 are likely less critical for ITS102.01-mediated neutralization. Additionally, the mAb regimens selected against a valine at position 778 within the cytoplasmic domain (A778V). While it is unlikely bNAbs are directly contacting this site, changes in the cytoplasmic domain sequence may impact Env structure (35, 36), Env incorporation into virions (37), and viral replication capacity (38, 39). There is some evidence that residue 778 coevolves with residues in the gp41 and V3 domains (40); an alanine at position 778 (or the absence of a valine) may confer a replication advantage to SIV under pressure from the infused mAbs. It is unclear whether these mutations would be retained in the absence of mAb selection pressure.
Multiple lines of evidence indicate that ITS103.01 was the primary driver of viral suppression in this study. First, SIV env pseudoviruses generated from rebound virus isolates that arose in the mAb-treated animals acquired neutralization resistance to ITS103.01, but not to the other mAbs. Second, the viral resistance assay, using replication-competent, biologically relevant intact virus derived from study animals, only demonstrated suppression of replication by ITS103.01. And third, ITS103.01 was the most potent SIV-neutralizing mAb within the cocktails (24). While we expect most of the antiviral activity was a result of ITS103.01-mediated neutralization (as has been previously demonstrated for in vivo prophylactic efficacy (12, 41, 42)), we do not have direct evidence of the efficacy of this mAb alone in vivo against SIVmac251. Functions other than neutralization may also be contributing – ITS103.01 exhibits potent antibody-dependent cellular cytotoxicity, exceeding that of the other mAbs in the cocktail (43), which may also mediate antiviral activity in vivo. In a previous study in NHPs infected with SIVmac239, ITS103.01 monotherapy administered spanning ATI delayed rebound by a mean ~1.3 wk (span 0 to 4 wk) (26), shorter than the 1 to 20 wk delay (mean 5.8 wk for all mAb-treated animals) observed in the current study. Likely sources for this difference include the use of different SIV strains (SIVmac251 clones are more sensitive to mAb neutralization than SIVmac239) and more limited viral reservoir seeding due to earlier initiation of cART (1 versus 5 wk). Inclusion of additional mAbs may have contributed to the longer delay, but this is less probable. While the other mAbs neutralized SIVmac251 pseudovirus variants, they are less potent than ITS103.01 and were unable to inhibit replication of the SIVmac251 swarm in vitro, suggesting that potent neutralization is required to block SIVmac251. The mAbs for this study were chosen, in part, due to the lack of other, more potent alternatives. These results therefore highlight the need for using combinations of highly potent bNAbs in SIV/HIV-1 therapeutic studies. Additional SIV bNAb isolation efforts have identified ITS99 and ITS104, which potently neutralize predominately tier 1 viruses, but are less effective against tier 2 or 3 (24). Others, like ITS90.03, neutralize tier 3 isolates like SIVmac239, and have some potency against SIVmac251 isolates, but are less effective against other strains (44). Continued efforts isolating potent antibodies will better enable preclinical evaluation of combination mAb therapy approaches in SIV infection models with the goal of informing optimal HIV-1 intervention strategies using combination mAbs to combat HIV-1 diversity.
In this study, the combination of one highly potent bNAb with other less potent mAbs effectively acted as monotherapy, albeit with different total amounts of infused mAb (30 versus 60 mg/kg -in the 2- and 4-mAb groups). These results may therefore not be generalizable to cocktails of potent bNAbs; however, there is potential for similar situations to occur in PLWH administered a bNAb cocktail, in which their virus is resistant to a subset of the included mAbs. Here, we saw that in three animals from the 2-mAb group there was a strong delay in time to viral rebound posttreatment interruption, not observed in any animals in the 4-mAb group. While the difference in time to rebound between these active arms was not statistically significant (P = 0.09), we investigated potential viral and immune mechanisms that could explain the increased delay. None of the assessed parameters, including mAb pharmacokinetics, viral sequence diversity, and adaptive immune responses consistently differed between the study groups. Increasing the number of mAbs did impact the in vitro binding of individual mAbs to cell surface Fc receptors, as less ITS103.01 coated monocytes in the context of a 4-mAb cocktail relative to 2-mAb. This was likely due to increased competition for the high affinity FcγR1 CD64. We did not observe a similar reduction on ex vivo PBMC from study animals postinfusion, and thus a role for FcγR competition is unclear. Another possible explanation is epitope competition between mAbs for Env binding when some mAbs bind but do not neutralize, especially given two of the mAbs used, ITS103.01 and ITS102.01, target a similar epitope. In previous results, we have shown that ITS103.01 competes binding of ITS102.01 while ITS102.01 does not compete binding of ITS103.01 (24), therefore we did not expect ITS102.01 would directly compete and reduce the effectiveness of ITS103.01. However, in vitro assays may not fully recapitulate mAb interactions occurring in vivo. Thus, the addition of ITS102.01 in the 4-mAb group may have reduced the potency of ITS103.01 relative to the 2-mAb combination. Further studies are required to assess the implications of incorporating additional mAbs in combination approaches when the potency of one mAb greatly exceeds that of the other(s).
Previous studies of passive HIV-1 mAb infusion in PLWH (19–21) and infected macaques (15, 16) have observed a vaccinal effect in which recipients exhibit increased virus-specific adaptive immunity, while others have reported no differences (4, 22, 23). We assessed endogenous immune responses following mAb treatment in this study and found no evidence of augmented SIV-specific binding antibody, neutralizing antibody, or T cell responses. In performing these assays, we took great care to ensure there was no contamination from infused antibodies, selecting time points well past antibody washout. These results add to the literature that a “vaccinal effect” arising from monoclonal antibody administration is not universal. Further research into factors contributing to the elicitation of a vaccinal effect, such as antibody characteristics (e.g. specificity, subclass), endogenous virus sensitivity, as well as host immunological features, are warranted.
Combination mAb interventions will be required for HIV-1 therapeutic and prevention strategies to effectively target the diversity of HIV-1 in the human population. Here, we report the efficacy of combination anti-SIV mAb passive immunization during ATI in the setting of a rigorous SIV infection model characterized by consistent and robust viral replication. Extending the ~5-wk delay in viral rebound that was observed in most treated animals will likely require incorporation of additional mAb(s) with neutralization potency more comparable to that of ITS103.01, which appeared to mediate the antiviral activity in this study. The prolonged 12- to 20-wk delay in viral rebound observed only in the 2-mAb group highlights the potential for reduced efficacy with larger bNAb cocktails with variable neutralization potency, which may warrant consideration when optimizing bNAb cocktails aimed at maximizing neutralization breadth. The relatively rapid and consistent development of env mutations conferring neutralization resistance to passively administered mAbs poses a considerable challenge to mAb-based therapeutic strategies, supporting incorporation of complementary therapeutic interventions.
Methods
A full, detailed description of the methods is given in SI Appendix.
Study Design and Procedures.
Twenty-one research naïve Indian-origin rhesus macaques were intravenously challenged with SIVmac251 virus. ART commenced 1 wk following infection, continuing until week 19. Beginning at week 15, and continuing every 2 wk until week 29, animals received intravenous infusions of either a combination of 2 SIV mAbs (ITS103.01-LS and ITS09.01-LS) or 4 SIV mAbs (ITS103.01-LS, ITS09.01-LS, ITS102.01-LS, and ITS113.01-LS) at 15 mg/kg each mAb. Plasma SIV RNA levels were measured as described previously (45). Blood was collected weekly.
Antibody Pharmacokinetics.
Infused antibody concentrations were measured longitudinally in plasma by ELISA. ELISA plates were coated with an anti-idiotype antibody specific for the mAb of interest. Following blocking, dilutions of non-heat inactivated plasma or monoclonal antibody used as a positive control were added to the plate as the primary antibody. Binding was detected with anti-NHP HRP-conjugated antibody. Reactions were developed using SureBlue TMB 1-Component Microwell Peroxidase Substrate (KPL) and reactions stopped with 1 N sulfuric acid. Absorbance was measured at 450 nm. Concentrations were interpolated from the positive control curve.
ADA.
Development of polyclonal antibody responses specific for the infused SIV mAbs were measured in plasma by ELISA using plates coated with the Fab of each SIV mAb. ELISA conditions were as described above. Data are presented as AUC of the dilution series.
Endogenous Env-Specific Binding Antibody Immune Responses.
Anti-Env antibody titers were measured in plasma by ELISA as described above using either SIVmac239 gp140 or V1V2 antigens. Data are displayed as the AUC of a ½-log dilution series. Responses directed against the CD4bs of SIV Env were measured by competition ELISA with ITS103 Fab as follows. ELISA plates were coated with SIVmac239 gp140. Biotinylated ITS103 Fab protein was added to wells followed by plasma serial twofold dilutions. ITS103 Fab binding was detected using high-sensitivity Streptavidin-HRP (Pierce). Data are expressed as the plasma dilution achieving 50% inhibition of Fab binding.
SIV Neutralization Assays.
SIV Env pseudotyped virus was produced as described previously (25). SIV gp160-encoding plasmids for the clones SIVmac251.H9.15 and SIVmac251.30 (46); SIVmac251.6 and SIVmac239.cs.23 (47); and SIVmac251.cs.41 (48) were kindly provided by David Montefiori. SIV env genes obtained from sequencing plasma of infected study animals were synthesized in plasmid DNA expression vectors. Virus neutralization was measured using infection of TZM-bl target cells by pseudotyped virus as previously described (49). Titers were calculated as either the 50% inhibitory concentration (IC50) for mAbs or the reciprocal plasma dilution (ID50) of plasma that achieved a 50% reduction of infection relative to virus alone.
Intracellular Cytokine Staining (ICS).
Cryopreserved PBMC were thawed and rested overnight. Cells were stimulated with SIVmac239 Gag and Env peptide pools (NIH HIV Reagent Program). Negative controls were cultured in 0.5% DMSO, the peptide pool diluent and diluent concentration present in peptide-stimulated conditions. Cytokine staining was performed as described (50). Samples were acquired on a BD FACSymphony flow cytometer. Gating schematic is shown in SI Appendix, Fig. S5.
Deep Sequencing of SIV env.
SIV env genetic material in macaque plasma and the SIVmac251 challenge stock was derived following methods described by Westfall and colleagues (51). Pacific Biosciences and SGA sequences were codon-aligned to a SIVmac239 env reference sequence (Genbank id: M33262) and translated. A maximum-likelihood phylogenetic tree was constructed for stock and rebound env sequences. Pairwise diversity was calculated on alignments for each animal under a HIVw model (52). The number of clusters (i.e., sequences descending from a stock lineage) in each rebound nucleotide alignment was determined using the Gap Procedure (53). Changes in amino acid frequencies between treated and untreated animals were determined by aligning sequences from each group separately. Residue frequencies were computed for each site. Significant associations between sites were determined on phylogenies and alignments for each treated group using a Bayesian graphical model that analyzes the joint distribution of substitution events between sites (54). N-linked glycosylation sites were predicted for each rebound alignment of PacBio sequences by identifying NXT and NXS patterns (55). Env sites of interest were mapped on a SIVmac239 Env structure (PDB: 8DVD) using homology with HIV-1 Env for the variable loops and using the structure PDB: 6TYB to determine the mCD4 binding sites.
SGA Analysis.
SGA sequencing of the env gene was performed as described previously (56).
Viral Resistance Assay.
CD4+ T cells were negatively selected from cryopreserved PBMC samples obtained from viremic SIV-infected rhesus macaques. Cells were activated overnight using a T Cell Activation/Expansion Kit (Miltenyi Biotec, Cat# 130-092-919). Additionally, rhesus CD4+ T cells from 3 SIV-naïve donors were similarly activated separately. After ~18 h, activation beads were removed, and all cell cultures were counted and combined into a single bulk culture. The bulk culture was maintained at a density of 106 cells/ml for 1 to 3 d until supernatant SIV p27 levels exceeded 100 pg/mL. Cells were then transferred to a 96-well culture plate. SIV mAbs were diluted and added to each respective well, including a negative control anti-influenza mAb, 9114 (57). Half of the supernatant was removed 3 times each week for analysis and replaced with fresh media and antibody. Additionally, activated CD4 feeder T cells were added to each well once a week to sustain the conditions necessary for further virus growth. Supernatant samples were analyzed for SIV-1 p27 antigen by capture ELISA (ABL, Cat# 5450) per the manufacturer’s instructions.
Cell Surface-Bound Monoclonal Antibody Detection.
Cryopreserved PBMC were thawed, washed, and then stained with a viability stain. Direct ex vivo detection of membrane-bound SIV mAb was measured by surface staining with anti-idiotype antibodies specific for each mAb fluorescently labeled with Alexa Fluor 647 using an NHS Ester kit (Thermo Fisher). Concurrently, cells were stained with fluorescently labeled antibodies specific for cell surface lineage markers. For in vitro cell labeling with mAbs, cells from mAb-naïve animals were incubated with SIV mAbs during viability dye staining followed by surface anti-idiotype staining described above. Cells were resuspended in 0.5% paraformaldehyde and staining was measured on a BD FACS Symphony. Gating schematic and anti-idiotype staining are shown in SI Appendix, Fig S11.
Fcγ Receptor Allotype Sequencing.
FcγR allotypes were sequenced as previously described (58). FcγR allotypes were identified based upon published sequences (59).
Statistical Analysis.
Virological and immunological comparisons were made between the three study groups using the two-tailed Kruskal−Wallis test adjusting for the multiple comparisons, or a two-tailed Mann–Whitney test when comparing only between the two mAb-treated groups. Nonparametric pair-wise comparisons between time points were made using a two-tailed Wilcoxon matched-pairs signed rank test. Survival analysis was performed using a log-rank (Mantel-Cox) test and correlates were assessed by nonparametric Spearman correlation. Statistical significance was preset at an alpha level of 0.05.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Acknowledgments
We would like to thank Rebecca Lynch for discussions regarding HIV-1 envmutations. We also thank the Vaccine Research Center (VRC) Nonhuman Primate Immunogenicity Core for superb technical support, especially from Evan Lamb, Samantha J. Provost, Dillon R. Flebbe, Josue Marquez, and Anna Mychalowych. We are grateful to the VRC Vaccine Production Program for SIV mAb production and to the veterinary staff and animal support staff at the Vaccine Research Center and BIOQUAL, Inc., especially Dr. Ruth Woodward, Dr. Diana Scorpio, and John N. Graves. The Quantitative Molecular Diagnostics Core of the AIDS and Cancer Virus Program, Frederick National Laboratory, provided expert assistance with plasma viral load determinations. This work was supported by a gift from Gilead Sciences, Inc., with additional support from federal funds from the National Cancer Institute, NIH, under Contract No. 75N91019D00024/HHSN261201500003I, and a cooperative agreement (W81XWH-18-2-0040) between the Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc., and the U.S. Department of Defense. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. Material has been reviewed by the Walter Reed Army Institute of Research. There is no objection to its presentation and/or publication. The opinions or assertions contained herein are the private views of the author, and are not to be construed as official, or as reflecting true views of the Department of the Army or the Department of Defense or H.J.F.
Author contributions
H.A.D.K., M. Roederer, and D.B. designed research; H.A.D.K., D.B., C.M.F., K.M.M., H.R.S., S.A., P.K., M.L., P.P., E.S.-B., K.S., E.M., and J.-P.T. performed research; H.A.D.K., E.L., S.A., H.B., R.M., S.H.H., A.P., K.E.F., J.D.L., B.F.K., M. Rolland, M. Roederer, and D.L.B. analyzed data; E.M. and J.-P.T. animal study coordination and execution; and H.A.D.K., M. Roederer, and D.L.B. wrote the paper.
Competing interests
This work was supported by a gift from Gilead Sciences, Inc.
Footnotes
This article is a PNAS Direct Submission M.C. is a guest editor invited by the Editorial Board.
Contributor Information
Mario Roederer, Email: marior@mail.nih.gov.
Diane L. Bolton, Email: dbolton@hivresearch.org.
Data, Materials, and Software Availability
The datasets generated during and/or analyzed during the current study are available as Dataset S1. All other data are included in the manuscript and/or supporting information.
Supporting Information
References
- 1.Corey L., et al. , Two randomized trials of neutralizing antibodies to prevent HIV-1 acquisition. N. Engl. J. Med. 384, 1003–1014 (2021), 10.1056/NEJMoa2031738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gaebler C., et al. , Prolonged viral suppression with anti-HIV-1 antibody therapy. Nature 606, 368–374 (2022), 10.1038/s41586-022-04597-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mendoza P., et al. , Combination therapy with anti-HIV-1 antibodies maintains viral suppression. Nature 561, 479–484 (2018), 10.1038/s41586-018-0531-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sneller M. C., et al. , Combination anti-HIV antibodies provide sustained virological suppression. Nature 606, 375–381 (2022), 10.1038/s41586-022-04797-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caskey M., et al. , Viraemia suppressed in HIV-1-infected humans by broadly neutralizing antibody 3BNC117. Nature 522, 487–491 (2015), 10.1038/nature14411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caskey M., et al. , Antibody 10–1074 suppresses viremia in HIV-1-infected individuals. Nat. Med. 23, 185–191 (2017), 10.1038/nm.4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lynch R. M., et al. , Virologic effects of broadly neutralizing antibody VRC01 administration during chronic HIV-1 infection. Sci. Transl. Med. 7, 319ra206 (2015), 10.1126/scitranslmed.aad5752. [DOI] [PubMed] [Google Scholar]
- 8.Julg B., et al. , Safety and antiviral activity of triple combination broadly neutralizing monoclonal antibody therapy against HIV-1: A phase 1 clinical trial. Nat. Med. 28, 1288–1296 (2022), 10.1038/s41591-022-01815-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barouch D. H., et al. , Therapeutic efficacy of potent neutralizing HIV-1-specific monoclonal antibodies in SHIV-infected rhesus monkeys. Nature 503, 224–228 (2013), 10.1038/nature12744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shingai M., et al. , Antibody-mediated immunotherapy of macaques chronically infected with SHIV suppresses viraemia. Nature 503, 277–280 (2013), 10.1038/nature12746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Asokan M., et al. , Fc-mediated effector function contributes to the in vivo antiviral effect of an HIV neutralizing antibody. Proc. Natl. Acad. Sci. U.S.A. 117, 18754–18763 (2020), 10.1073/pnas.2008236117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parsons M. S., et al. , Fc-dependent functions are redundant to efficacy of anti-HIV antibody PGT121 in macaques. J. Clin. Invest. 129, 182–191 (2019), 10.1172/jci122466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Julg B., et al. , Virological control by the CD4-binding site antibody N6 in simian-human immunodeficiency virus-infected rhesus monkeys. J. Virol. 91, e00498 (2017), 10.1128/jvi.00498-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bolton D. L., et al. , Human immunodeficiency virus type 1 monoclonal antibodies suppress acute simian-human immunodeficiency virus viremia and limit seeding of cell-associated viral reservoirs. J. Virol. 90, 1321–1332 (2016), 10.1128/jvi.02454-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nishimura Y., et al. , Early antibody therapy can induce long-lasting immunity to SHIV. Nature 543, 559–563 (2017), 10.1038/nature21435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nishimura Y., et al. , Immunotherapy during the acute SHIV infection of macaques confers long-term suppression of viremia. J. Exp. Med. 218, e20201214 (2021), 10.1084/jem.20201214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scheid J. F., et al. , HIV-1 antibody 3BNC117 suppresses viral rebound in humans during treatment interruption. Nature 535, 556–560 (2016), 10.1038/nature18929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bar K. J., et al. , Effect of HIV antibody VRC01 on viral rebound after treatment interruption. N. Engl. J. Med. 375, 2037–2050 (2016), 10.1056/NEJMoa1608243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schoofs T., et al. , HIV-1 therapy with monoclonal antibody 3BNC117 elicits host immune responses against HIV-1. Science 352, 997–1001 (2016), 10.1126/science.aaf0972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosás-Umbert M., et al. , Administration of broadly neutralizing anti-HIV-1 antibodies at ART initiation maintains long-term CD8(+) T cell immunity. Nat. Commun. 13, 6473 (2022), 10.1038/s41467-022-34171-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Niessl J., et al. , Combination anti-HIV-1 antibody therapy is associated with increased virus-specific T cell immunity. Nat. Med. 26, 222–227 (2020), 10.1038/s41591-019-0747-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gruell H., et al. , Effect of 3BNC117 and romidepsin on the HIV-1 reservoir in people taking suppressive antiretroviral therapy (ROADMAP): A randomised, open-label, phase 2A trial. Lancet Microbe 3, e203–e214 (2022), 10.1016/s2666-5247(21)00239-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gunst J. D., et al. , Impact of a TLR9 agonist and broadly neutralizing antibodies on HIV-1 persistence: The randomized phase 2a TITAN trial. Nat. Med. 29, 2547–2558 (2023), 10.1038/s41591-023-02547-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Welles H. C., et al. , Broad coverage of neutralization-resistant SIV strains by second-generation SIV-specific antibodies targeting the region involved in binding CD4. PLoS Pathog. 18, e1010574 (2022), 10.1371/journal.ppat.1010574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mason R. D., et al. , Targeted isolation of antibodies directed against major sites of SIV Env vulnerability. PLoS Pathog. 12, e1005537 (2016), 10.1371/journal.ppat.1005537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iwamoto N., et al. , Blocking α4β7 integrin binding to SIV does not improve virologic control. Science 365, 1033–1036 (2019), 10.1126/science.aaw7765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dashti A., et al. , AZD5582 plus SIV-specific antibodies reduce lymph node viral reservoirs in antiretroviral therapy-suppressed macaques. Nat. Med. 29, 2535–2546 (2023), 10.1038/s41591-023-02570-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pegu A., et al. , Potent anti-viral activity of a trispecific HIV neutralizing antibody in SHIV-infected monkeys. Cell Rep. 38, 110199 (2022), 10.1016/j.celrep.2021.110199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sneller M. C., et al. , Kinetics of plasma HIV rebound in the era of modern antiretroviral therapy. J. Infect. Dis. 222, 1655–1659 (2020), 10.1093/infdis/jiaa270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crowell T. A., et al. , Safety and efficacy of VRC01 broadly neutralising antibodies in adults with acutely treated HIV (RV397): A phase 2, randomised, double-blind, placebo-controlled trial. Lancet HIV 6, e297–e306 (2019), 10.1016/S2352-3018(19)30053-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cale E. M., et al. , Neutralizing antibody VRC01 failed to select for HIV-1 mutations upon viral rebound. J. Clin. Invest. 130, 3299–3304 (2020), 10.1172/JCI134395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dingens A. S., Arenz D., Weight H., Overbaugh J., Bloom J. D., An antigenic atlas of HIV-1 escape from broadly neutralizing antibodies distinguishes functional and structural epitopes. Immunity 50, 520–532.e523 (2019), 10.1016/j.immuni.2018.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Otsuka Y., et al. , Diverse pathways of escape from all well-characterized VRC01-class broadly neutralizing HIV-1 antibodies. PLoS Pathog. 14, e1007238 (2018), 10.1371/journal.ppat.1007238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lynch R. M., et al. , HIV-1 fitness cost associated with escape from the VRC01 class of CD4 binding site neutralizing antibodies. J. Virol. 89, 4201–4213 (2015), 10.1128/jvi.03608-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Piai A., et al. , Structural basis of transmembrane coupling of the HIV-1 envelope glycoprotein. Nat. Commun. 11, 2317 (2020), 10.1038/s41467-020-16165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen J., et al. , Effect of the cytoplasmic domain on antigenic characteristics of HIV-1 envelope glycoprotein. Science 349, 191–195 (2015), 10.1126/science.aaa9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Postler T. S., Desrosiers R. C., The tale of the long tail: The cytoplasmic domain of HIV-1 gp41. J. Virol. 87, 2–15 (2013), 10.1128/jvi.02053-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Santos da Silva E., et al. , The envelope cytoplasmic tail of HIV-1 subtype C contributes to poor replication capacity through low viral infectivity and cell-to-cell transmission. PLoS ONE 11, e0161596 (2016), 10.1371/journal.pone.0161596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Snetkov X., et al. , A conserved tryptophan in the envelope cytoplasmic tail regulates HIV-1 assembly and spread. Viruses 14, 129 (2022), 10.3390/v14010129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Travers S. A. A., Tully D. C., McCormack G. P., Fares M. A., A study of the coevolutionary patterns operating within the env gene of the HIV-1 group M subtypes. Mol. Biol. Evol. 24, 2787–2801 (2007), 10.1093/molbev/msm213. [DOI] [PubMed] [Google Scholar]
- 41.Moldt B., et al. , A nonfucosylated variant of the anti-HIV-1 monoclonal antibody b12 has enhanced FcγRIIIa-mediated antiviral activity in vitro but does not improve protection against mucosal SHIV challenge in macaques. J. Virol. 86, 6189–6196 (2012), 10.1128/jvi.00491-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hangartner L., et al. , Effector function does not contribute to protection from virus challenge by a highly potent HIV broadly neutralizing antibody in nonhuman primates. Sci. Transl. Med. 13, eabe3349 (2021), 10.1126/scitranslmed.abe3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grunst M. W., et al. , Antibody-dependent cellular cytotoxicity, infected cell binding and neutralization by antibodies to the SIV envelope glycoprotein. PLoS Pathog. 19, e1011407 (2023), 10.1371/journal.ppat.1011407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gorman J., et al. , Isolation and structure of an antibody that fully neutralizes isolate SIVmac239 reveals functional similarity of SIV and HIV glycan shields. Immunity 51, 724–734.e724 (2019), 10.1016/j.immuni.2019.09.007. [DOI] [PubMed] [Google Scholar]
- 45.Li H., et al. , Envelope residue 375 substitutions in simian-human immunodeficiency viruses enhance CD4 binding and replication in rhesus macaques. Proc. Natl. Acad. Sci. U.S.A. 113, E3413–E3422 (2016), 10.1073/pnas.1606636113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schell J. B., et al. , Significant protection against high-dose simian immunodeficiency virus challenge conferred by a new prime-boost vaccine regimen. J. Virol. 85, 5764–5772 (2011), 10.1128/jvi.00342-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fairman J., et al. , Enhanced in vivo immunogenicity of SIV vaccine candidates with cationic liposome-DNA complexes in a rhesus macaque pilot study. Hum. Vaccin. 5, 141–150 (2009), 10.4161/hv.5.3.6589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yeh W. W., et al. , Partial protection of simian immunodeficiency virus (SIV)-infected rhesus monkeys against superinfection with a heterologous SIV isolate. J. Virol. 83, 2686–2696 (2009), 10.1128/jvi.02237-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Montefiori D. C., Evaluating neutralizing antibodies against HIV, SIV, and SHIV in luciferase reporter gene assays. Curr. Protoc. Immunol. 64, 12.11.11–12.11.17 (2004), 10.1002/0471142735.im1211s64. [DOI] [PubMed] [Google Scholar]
- 50.Donaldson M. M., Kao S. F., Foulds K. E., OMIP-052: An 18-color panel for measuring Th1, Th2, Th17, and Tfh responses in rhesus macaques. Cytometry A 95, 261–263 (2019), 10.1002/cyto.a.23670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Westfall D. H., et al. , Optimized SMRT-UMI protocol produces highly accurate sequence datasets from diverse populations-Application to HIV-1 quasispecies. Virus Evol. 10, veae019 (2024). 10.1093/ve/veae019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schliep K. P., Phangorn: Phylogenetic analysis in R. Bioinformatics 27, 592–593 (2011), 10.1093/bioinformatics/btq706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vrbik I., Stephens D. A., Roger M., Brenner B. G., The Gap Procedure: For the identification of phylogenetic clusters in HIV-1 sequence data. BMC Bioinformatics 16, 355 (2015), 10.1186/s12859-015-0791-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Poon A. F., Lewis F. I., Frost S. D., Kosakovsky Pond S. L., Spidermonkey: Rapid detection of co-evolving sites using Bayesian graphical models. Bioinformatics 24, 1949–1950 (2008), 10.1093/bioinformatics/btn313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang M., et al. , Tracking global patterns of N-linked glycosylation site variation in highly variable viral glycoproteins: HIV, SIV, and HCV envelopes and influenza hemagglutinin. Glycobiology 14, 1229–1246 (2004), 10.1093/glycob/cwh106. [DOI] [PubMed] [Google Scholar]
- 56.Breed M. W., et al. , Loss of a tyrosine-dependent trafficking motif in the simian immunodeficiency virus envelope cytoplasmic tail spares mucosal CD4 cells but does not prevent disease progression. J. Virol. 87, 1528–1543 (2013), 10.1128/jvi.01928-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dreyfus C., et al. , Highly conserved protective epitopes on influenza B viruses. Science 337, 1343–1348 (2012), 10.1126/science.1222908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nguyen D. C., Scinicariello F., Attanasio R., Characterization and allelic polymorphisms of rhesus macaque (Macaca mulatta) IgG Fc receptor genes. Immunogenetics 63, 351–362 (2011), 10.1007/s00251-011-0514-z. [DOI] [PubMed] [Google Scholar]
- 59.Chan Y. N., et al. , IgG binding characteristics of rhesus macaque FcγR. J. Immunol. 197, 2936–2947 (2016), 10.4049/jimmunol.1502252. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available as Dataset S1. All other data are included in the manuscript and/or supporting information.