Ricardo Segurado
Ricardo Segurado
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
1,,*,
Sevilla D Detera-Wadleigh
Sevilla D Detera-Wadleigh
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
3,,*,
Douglas F Levinson
Douglas F Levinson
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
5,,*,
Cathryn M Lewis
Cathryn M Lewis
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
6,,*,
Michael Gill
Michael Gill
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
2,,*,
John I Nurnberger, Jr
John I Nurnberger, Jr
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
8,9,
Nick Craddock
Nick Craddock
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
11,
J Raymond DePaulo
J Raymond DePaulo
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
12,
Miron Baron
Miron Baron
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
13,
Elliot S Gershon
Elliot S Gershon
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
15,
Jenny Ekholm
Jenny Ekholm
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
16,
Sven Cichon
Sven Cichon
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
18,
Gustavo Turecki
Gustavo Turecki
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
21,
Stephan Claes
Stephan Claes
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
19,20,
John R Kelsoe
John R Kelsoe
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
23,
Peter R Schofield
Peter R Schofield
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
24,
Renee F Badenhop
Renee F Badenhop
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
24,25,
J Morissette
J Morissette
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
27,
Hilary Coon
Hilary Coon
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
28,
Douglas Blackwood
Douglas Blackwood
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
29,
L Alison McInnes
L Alison McInnes
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
14,
Tatiana Foroud
Tatiana Foroud
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
8,9,
Howard J Edenberg
Howard J Edenberg
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
9,10,
Theodore Reich
Theodore Reich
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
30,
John P Rice
John P Rice
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
30,
Alison Goate
Alison Goate
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
30,
Melvin G McInnis
Melvin G McInnis
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
12,
Francis J McMahon
Francis J McMahon
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
3,
Judith A Badner
Judith A Badner
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
15,
Lynn R Goldin
Lynn R Goldin
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
4,
Phil Bennett
Phil Bennett
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
11,
Virginia L Willour
Virginia L Willour
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
12,
Peter P Zandi
Peter P Zandi
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
12,
Jianjun Liu
Jianjun Liu
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
13,
Conrad Gilliam
Conrad Gilliam
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
13,
Suh-Hang Juo
Suh-Hang Juo
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
13,
Wade H Berrettini
Wade H Berrettini
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
5,
Takeo Yoshikawa
Takeo Yoshikawa
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
31,
Leena Peltonen
Leena Peltonen
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
16,32,
Jouko Lönnqvist
Jouko Lönnqvist
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
17,
Markus M Nöthen
Markus M Nöthen
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
18,
Johannes Schumacher
Johannes Schumacher
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
34,
Christine Windemuth
Christine Windemuth
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
35,
Marcella Rietschel
Marcella Rietschel
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
37,
Peter Propping
Peter Propping
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
34,
Wolfgang Maier
Wolfgang Maier
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
36,
Martin Alda
Martin Alda
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
38,
Paul Grof
Paul Grof
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
39,
Guy A Rouleau
Guy A Rouleau
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
22,
Jurgen Del-Favero
Jurgen Del-Favero
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
20,
Christine Van Broeckhoven
Christine Van Broeckhoven
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
20,
Julien Mendlewicz
Julien Mendlewicz
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
40,
Rolf Adolfsson
Rolf Adolfsson
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
41,
M Anne Spence
M Anne Spence
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
42,
Hermann Luebbert
Hermann Luebbert
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
44,
Linda J Adams
Linda J Adams
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
24,
Jennifer A Donald
Jennifer A Donald
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
26,
Philip B Mitchell
Philip B Mitchell
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
25,
Nicholas Barden
Nicholas Barden
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
27,
Eric Shink
Eric Shink
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
27,
William Byerley
William Byerley
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
43,
Walter Muir
Walter Muir
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
29,
Peter M Visscher
Peter M Visscher
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
29,
Stuart Macgregor
Stuart Macgregor
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
29,
Hugh Gurling
Hugh Gurling
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
7,
Gursharan Kalsi
Gursharan Kalsi
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
7,
Andrew McQuillin
Andrew McQuillin
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
7,
Michael A Escamilla
Michael A Escamilla
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
45,
Victor I Reus
Victor I Reus
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
46,
Pedro Leon
Pedro Leon
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
47,48,
Nelson B Freimer
Nelson B Freimer
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
33,
Henrik Ewald
Henrik Ewald
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
49,
Torben A Kruse
Torben A Kruse
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
50,
Ole Mors
Ole Mors
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
49,
Uppala Radhakrishna
Uppala Radhakrishna
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
51,
Jean-Louis Blouin
Jean-Louis Blouin
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
51,
Stylianos E Antonarakis
Stylianos E Antonarakis
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
51,
Nurten Akarsu
Nurten Akarsu
1Neuropsychiatric Genetics Unit, Department of Genetics, Trinity College, and 2Trinity Centre for Health Sciences, Department of Psychiatry, St. James Hospital, Dublin; 3Mood and Anxiety Disorders Program, National Institute of Mental Health/National Institutes of Health, and 4Genetic Epidemiology Branch, National Cancer Institute, National Institutes of Health, Bethesda; 5Center for Neurobiology and Behavior, Department of Psychiatry, University of Pennsylvania, Philadelphia; 6Division of Genetics and Development, Guy’s, King’s & St Thomas’ School of Medicine, and 7Molecular Psychiatry Laboratory, Windeyer Institute for Medical Science, Department of Psychiatry and Behavioral Sciences, Royal Free and University College Medical School, University College London, London; Departments of 8Psychiatry, 9Medical and Molecular Genetics, and 10Biochemistry and Molecular Biology, Indiana University, Indianapolis; 11Molecular Psychiatry Group, Division of Neuroscience, University of Birmingham, Queen Elizabeth Psychiatric Hospital, Edgbaston, Birmingham, United Kingdom; 12Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore; 13Division of Psychiatric Genetics, Department of Medical Genetics, Columbia University, New York State Psychiatric Institute, and 14Departments of Psychiatry and Human Genetics, Mount Sinai School of Medicine, New York; 15Department of Psychiatry, University of Chicago, Chicago; 16Department of Molecular Medicine, National Public Health Institute, and Department of Medical Genetics, University of Helsinki, and 17Department of Mental Health, National Public Health Institute, Helsinki; Departments of 18Medical Genetics and 19Psychiatry, University of Antwerp, and 20Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerp, Belgium; 21Department of Psychiatry and 22Centre for Research in Neuroscience, The Montreal General Hospital Research Institute, McGill University, Montreal; 23Laboratory of Psychiatric Genomics, Department of Psychiatry, University of California San Diego (UCSD), La Jolla; 24Garvan Institute of Medical Research, 25School of Psychiatry, University of New South Wales, and Mood Disorders Unit, Prince of Wales Hospital, and 26School of Biological Science, Macquarie University, Sydney; 27Neuroscience Centre Hospitalier de l'Université Laval Research Center and Laval University, Quebec; 28University of Utah, Salt Lake City; 29Psychiatry and Biological Sciences, University of Edinburgh, Edinburgh; 30Department of Psychiatry, Washington University School of Medicine, St. Louis; 31Laboratory for Molecular Psychiatry, RIKEN (Institute of Physical and Chemical Research) Brain Science Institute, Saitama, Japan; Departments of 32Human Genetics and 33Psychiatry, University of California, Los Angeles; 34Institute of Human Genetics, 35Institute for Medical Biometry, Informatics, and Epidemiology, and 36Department of Psychiatry, University of Bonn, Bonn, Germany; 37Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Mannheim, Germany; 38Department of Psychiatry, Dalhousie University, Halifax, Canada; 39Department of Psychiatry, University of Ottawa, Ottawa, Canada; 40Department of Psychiatry, Erasme Hospital, University of Brussels, Brussels; 41Department of Clinical Sciences Division of Psychiatry University of Umea, Umea, Sweden; Departments of 42Pediatrics and 43Psychiatry, University of California, Irvine; 44Biofrontera Pharmaceuticals, Leverkusen, Germany; 45Department of Psychiatry, University of Texas Health Science Center, San Antonio; 46Department of Psychiatry, University of California, San Francisco; 47Centro de Biología Molecular y Celular and 48Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 49Institute for Basic Psychiatric Research, Aarhus University Hospital, Risskov, Denmark; 50Department of Clinical Biochemistry and Genetics, Odense University Hospital, Odense, Denmark; 51Department of Medical Genetics, University of Geneva Medical School and University Hospitals, Geneva, Switzerland; and 52Department of Pediatrics, Hacettepe University, Ankara, Turkey
52


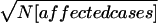 , the square root of the number of genotyped affected cases), which simulation studies demonstrated will increase power when the genetic effect of a locus is similar across samples. Thus, we considered our primary analyses to be the weighted analyses of models 1 and 2 (defined below).
, the square root of the number of genotyped affected cases), which simulation studies demonstrated will increase power when the genetic effect of a locus is similar across samples. Thus, we considered our primary analyses to be the weighted analyses of models 1 and 2 (defined below).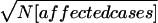 , divided by the mean of this value over all studies), as discussed in the first article in this series (Levinson et al. 2003 [in this issue]). Two pointwise P values were determined by permutation (5,000 permutations per analysis): PAvgRnk and Pord, as defined in appendix A.
, divided by the mean of this value over all studies), as discussed in the first article in this series (Levinson et al. 2003 [in this issue]). Two pointwise P values were determined by permutation (5,000 permutations per analysis): PAvgRnk and Pord, as defined in appendix A.
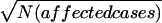 for each study; and the overall place of each bin in ascending order of average ranks (the lowest/best average rank is first-place). Average ranks with significant PAvgRnk values are highlighted above the columns (black for PAvgRnk<.01, gray for PAvgRnk<.05). Tied ranks sometimes resulted in uneven numbers of bins in some groupings, particularly for lower ranks when there were many zero or negative scores. Marshfield (“Mfd”) locations are shown for the marker at the distal boundary of each bin. Bin boundaries were selected at ∼30-cM spacing on the Généthon map; mean bin width is 29.1 cM on the Marshfield map. Peds = number of pedigrees; Aff = number of genotyped affected cases. See table 1 for the references associated with each study.
for each study; and the overall place of each bin in ascending order of average ranks (the lowest/best average rank is first-place). Average ranks with significant PAvgRnk values are highlighted above the columns (black for PAvgRnk<.01, gray for PAvgRnk<.05). Tied ranks sometimes resulted in uneven numbers of bins in some groupings, particularly for lower ranks when there were many zero or negative scores. Marshfield (“Mfd”) locations are shown for the marker at the distal boundary of each bin. Bin boundaries were selected at ∼30-cM spacing on the Généthon map; mean bin width is 29.1 cM on the Marshfield map. Peds = number of pedigrees; Aff = number of genotyped affected cases. See table 1 for the references associated with each study.