

Abstract
Several recent studies have suggested that linkage disequilibrium (LD) in the human genome has a fundamentally “blocklike” structure. However, thus far there has been little formal assessment of how well the haplotype block model captures the underlying structure of LD. Here we propose quantitative criteria for assessing how blocklike LD is and apply these criteria to both real and simulated data. Analyses of several large data sets indicate that real data show a partial fit to the haplotype block model; some regions conform quite well, whereas others do not. Some improvement could be obtained by genotyping higher marker densities but not by increasing the number of samples. Nonetheless, although the real data are only moderately blocklike, our simulations indicate that, under a model of uniform recombination, the structure of LD would actually fit the block model much less well. Simulations of a model in which much of the recombination occurs in narrow hotspots provide a much better fit to the observed patterns of LD, suggesting that there is extensive fine-scale variation in recombination rates across the human genome.
Introduction
There is currently great interest in the prospect of genomewide association studies to identify the genetic factors underlying complex diseases. To design these studies appropriately, it is important to have a detailed description of linkage disequilibrium (LD) in the human genome (Kruglyak 1999). Information about the distribution and extent of LD is critical for (1) estimating how many markers will be needed to achieve acceptable power in genomewide studies, (2) selecting markers appropriately so that they reflect local patterns of LD, and (3) designing statistical methods of analysis that make optimal use of the data.
The extent of LD is highly variable across the genome (e.g., Clark et al. 1998; Dunning et al. 2000; Taillon-Miller et al. 2000; Reich et al. 2001, 2002; Wall and Pritchard 2003), and the determinants of LD are not yet fully understood (Pritchard and Przeworski 2001). However, several recent empirical studies suggest that the seemingly complex patterns of LD can be represented as a series of “haplotype blocks”—that is, consecutive sites that are in complete (or nearly complete) LD with each other (Daly et al. 2001; Patil et al. 2001; Dawson et al. 2002; Gabriel et al. 2002; Phillips et al. 2003). Adjacent blocks are separated by sites that show evidence of historical recombination. This model has important implications for association mapping, because it implies that, by identifying haplotype blocks, it is possible to predict the likely configurations of alleles at unobserved sites. The International HapMap Project aims to produce a genomewide haplotype map that can be used to streamline association mapping.
It has generally been assumed that the presence of haplotype blocks provides evidence for fine-scale variation in recombination rates, with blocks corresponding to regions of reduced recombination and interblock regions corresponding to recombination hotspots (Daly et al. 2001; Gabriel et al. 2002). Consistent with this view, a few studies have found direct experimental evidence for recombination hotspots (Lien et al. 2000; Jeffreys et al. 2001; May et al. 2002; Schneider et al. 2002), and there are a small number of examples in which boundaries of LD correspond roughly to experimentally determined hotspots (Chakravarti et al. 1984; Jeffreys et al. 2001; May et al. 2002; Li and Stephens, in press). However, these studies provide examples of recombination hotspots in just a few genetic regions; it is not yet clear whether fine-scale rate variation is a general feature of the human genome, nor whether the reported phenomenon of haplotype blocks implies that it is (Wang et al. 2002; Phillips et al. 2003).
Thus far, a range of operational definitions has been used to identify haplotype blocks in genotype data (Daly et al. 2001; Patil et al. 2001; Dawson et al. 2002; Gabriel et al. 2002; Wang et al. 2002; Zhang et al. 2002; Phillips et al. 2003). However, there has been no attempt to outline formal criteria that haplotype blocks should meet if they are to be considered a good description of the underlying structure of LD. Here we propose three such criteria and apply them to existing data. As presented, our criteria relate specifically to the Gabriel et al. (2002) block definition, which breaks blocks when D′, a measure of LD between pairs of sites (Lewontin 1964), is substantially <1. This block definition was chosen because it is directly related to the goal of detecting historical recombination, which is central to the block concept, and because it can be applied directly to diploid data. The block definition of Gabriel et al. (2002) also seems to perform reasonably well at controlling the random noise inherent in D′. However, our three criteria can be modified to treat other haplotype block definitions, as well.
We report the results of applying these criteria to three large human data sets: SNP data from Gabriel et al. (2002) and resequencing data from the Seattle SNP study (described, in part, by Carlson et al., in press) and from the Environmental Genome Project (EGP) SNP study. We also report the results of applying these criteria to simulated data obtained under models with uniform recombination rates and under simple hotspot models. Comparison of the actual and simulated data can tell us whether observed patterns of LD provide evidence for widespread fine-scale variation in recombination rates.
Our article aims to address three issues. (1) To what extent does the haplotype block model capture the underlying structure of LD? (2) Are recombination hotspots necessary to explain the observed patterns of LD in human data? (3) What is the impact of experimental design on the inferred haplotype block structure?
Material and Methods
Data Sets
Data from Gabriel et al. (2002)
The data were downloaded from the Whitehead Institute Web site. Regions 49a and 12b were excluded because the authors reported systematic departures from Hardy-Weinberg equilibrium and/or widespread genotyping failure, region 12b was excluded by the authors because of high map instability, and regions 5a and 6a were unavailable. This left 50 regions spread over a total of 12.2 Mb of sequence. The data consisted of four populations: Utah CEPH families, unrelated African Americans, unrelated East Asians, and parent-offspring trios from Nigeria. Trios 502, 583, and 903 from the Nigerian sample were excluded because of apparent non-Mendelian inheritance patterns (S. Gabriel, personal communication). Our analyses considered only unrelated individuals, leaving sample sizes of 48, 50, 42, and 58 individuals, respectively, for the four populations. In these populations, there were a total of 1,932, 1,950, 1,736, and 1,821 SNPs, respectively, with minor allele frequency (MAF) ⩾0.1, and an average spacing between neighboring SNPs of 6.2, 6.1, 6.7, and 6.5 kb, respectively, in the different populations.
Seattle SNP data
Polymorphism data (both SNPs and indels) were downloaded from the University of Washington Fred Hutchinson Cancer Research Center (UW-FHCRC) Variation Discovery Resource Web site on October 5, 2002. A total of 85 loci were available on that date. Some loci were completely resequenced, whereas others have small gaps. The data were obtained by DNA resequencing of 24 unrelated African Americans and 23 unrelated European Americans from the Coriell Cell Repository (see Carlson et al. [in press] for more details). In the African American (European American) sample, there were a total of 2,502 (2,023) segregating polymorphisms with MAF ⩾0.1, producing an average marker spacing of 616 bp (820 bp) spread over 1.5 Mb (1.4 Mb) of sequence.
EGP SNP data
The EGP SNP Web site was accessed on October 5, 2002, and data (both SNP and indel) from 90 loci were downloaded at that time. As above, some loci have small gaps in the regions that were resequenced. Overall, they contained 1,886 segregating mutations with MAF ⩾0.1, with an average marker spacing of 946 bp spread over 1.7 Mb of sequence. The data were obtained by resequencing 90 unrelated individuals of mixed ethnicity from the DNA Polymorphism Discovery Resource.
Definitions
Coverage criterion
We followed the haplotype block definition of Gabriel et al. (2002) with minor modifications, as follows. We considered only biallelic variants with MAF ⩾0.1. Given a pair of such variants, there is uncertainty in the “true” value of 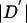 , because of the finite sample size and the lack of phase information in double heterozygotes. To deal with this, we followed the method of Gabriel et al. (2002) for constructing approximate confidence intervals on
, because of the finite sample size and the lack of phase information in double heterozygotes. To deal with this, we followed the method of Gabriel et al. (2002) for constructing approximate confidence intervals on 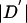 (see Ayres and Balding [2001] for a Bayesian alternative).
(see Ayres and Balding [2001] for a Bayesian alternative).
For each site, we assumed that the true allele frequencies equal the sample allele frequencies. Then, for each pair of sites, we calculated the likelihood of the diploid genotype data as a function of 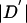 , in increments of 0.01. For k=0,1,…,100, call this likelihood
, in increments of 0.01. For k=0,1,…,100, call this likelihood 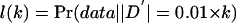 . Define CL as the largest value of (0.01×k) such that
. Define CL as the largest value of (0.01×k) such that 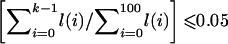 , and define CU as the smallest value of (0.01×k) such that
, and define CU as the smallest value of (0.01×k) such that 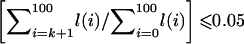 . Each pair of SNPs was then classified into one of three categories. If CL⩾0.7 and CU⩾0.98, then the pair was considered to be in “strong LD.” If CU<0.9, then the pair was considered to have “historical evidence of recombination.” All other pairs were categorized as “other.” These latter pairs include both pairs with intermediate levels of LD and pairs that are uninformative about the true value of
. Each pair of SNPs was then classified into one of three categories. If CL⩾0.7 and CU⩾0.98, then the pair was considered to be in “strong LD.” If CU<0.9, then the pair was considered to have “historical evidence of recombination.” All other pairs were categorized as “other.” These latter pairs include both pairs with intermediate levels of LD and pairs that are uninformative about the true value of 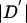 . A group of two or more consecutive markers was considered to be a haplotype block if (1) the endpoint markers were in “strong LD” and (2) the number of pairs of markers in “strong LD” was at least 19 times the number of pairs of markers with “historical evidence of recombination” (Gabriel et al. 2002). Markers were not permitted to be members of more than one block.
. A group of two or more consecutive markers was considered to be a haplotype block if (1) the endpoint markers were in “strong LD” and (2) the number of pairs of markers in “strong LD” was at least 19 times the number of pairs of markers with “historical evidence of recombination” (Gabriel et al. 2002). Markers were not permitted to be members of more than one block.
This definition does not guarantee a unique solution for parsing the data into disjoint blocks (as, for example, when the “overlapping blocks” criterion described below is violated). The algorithm that we implemented finds the leftmost marker contained in a block and takes the largest block containing this marker. The algorithm continues by repeating the process, using only the markers to the right of the previously defined block, until no more blocks can be found. We define the length of a haplotype block as the physical distance from the leftmost marker to the rightmost marker. Then, the coverage proportion is defined as the sum of the haplotype block lengths divided by the total length of sequence. We tried two other simple algorithms for assigning markers to haplotype blocks; both yielded similar results.
Hole criterion
Consider triplets of markers, labeled “A,” “B,” and “C” (in that physical order), where A and C are in “strong LD.” We define a “hole” as occurring when either A and B or B and C (or both) show “historical evidence of recombination.” Conversely, the data are considered consistent if both pairs are in “strong LD.” (All other situations are treated as uninformative and are not considered.) As a function of the distance between markers A and C, we tabulated the proportion of comparisons that have a hole, summing across all trios of sites within each region. Note that population genetics theory predicts that low-frequency variants can potentially be in strong LD over long distances, even across relative hotspots of recombination. Therefore, the hole criterion and the overlapping blocks criterion below are primarily of interest for SNPs with reasonably high MAF (hence, the cutoff of 0.1 used here).
Overlapping blocks criterion
Suppose that pair A and B and pair B and C are each in “strong LD.” The data are considered consistent if A and C are also in “strong LD.” If instead A and C show “historical evidence of recombination,” then we have overlapping haplotype blocks. (We do not consider triplets where A and C are uninformative.) We calculated the proportion of comparisons that result in overlapping haplotype blocks, as a function of the distance between markers A and C. The estimated rates of holes and overlapping blocks obtained from the data tended to be more noisy than the coverage results, because there is statistical dependence among many of the trios from a given region, especially when the outermost markers are widely separated.
Simulations
We used the coalescent with recombination (Hudson 1983) to simulate data for comparison with the Nigerian data from Gabriel et al. (2002). Like the data, the simulations had a sample size of n = 58 unphased diploid individuals. We assumed a constant population size and no population structure. Given the protocol of the Gabriel et al. (2002) study, it is not immediately clear how to model the ascertainment bias of the SNPs that were used. We used a model in which only polymorphisms that segregated in the first eight chromosomes were included. This particular model was chosen over several others because it provides a reasonably good fit between the observed and simulated distributions of MAFs (see fig. 1A). In contrast, models with more-stringent ascertainment schemes (which might be more plausible a priori), with or without recent population growth, led to substantially worse fits between observed and simulated MAF distributions but to similar haplotype block patterns, as measured by our three criteria (results not shown). We used the same frequency cutoff (MAF ⩾0.1) as in the analyses of the actual data and considered population sizes of N=10,000, 15,000, and 20,000 individuals. For most of our simulations, we chose the population mutation parameter θ (which is equal to 4Nμ, where μ is the mutation rate per site per generation) in such a way that the expected number of ascertained SNPs in the simulations (after applying the frequency cutoff) equals the actual number observed across the 50 regions. This corresponds to θ=7.836×10-5/bp and yields a distribution of marker spacing that is close to the empirical distribution (see fig. 1B).
Figure 1.

Comparison of the actual (African sample from Gabriel et al. 2002) and simulated distributions of MAFs (A) and distances between neighboring markers with MAF ⩾0.1 (B). The distribution of allele frequencies was obtained by tabulating the first 80 informative chromosomes at each segregating site (i.e., the first 80 chromosomes that did not have missing data) and was compared with simulations of 80 chromosomes subject to the SNP ascertainment scheme described in the text. The simulated distribution of intermarker distances is an average over 100 replicates.
Each replicate simulation was matched to the real data by simulating 50 independent regions whose physical lengths and average recombination rates matched the corresponding values for each of the 50 regions sampled by Gabriel et al. (2002). For each region, an estimate of the average recombination rate was obtained from the results of Kong et al. (2002). These recombination rate estimates are based on observed meioses in pedigree data and represent average rates between markers that flank the sampled regions.
We considered three different models of local variation in recombination rate. The uniform recombination model assumes that r (the recombination rate per base pair per generation) is constant within regions but varies between regions, as described above. To examine the effects of fine-scale variation in recombination rates (in addition to regional variation in recombination rates), we implemented a model with recombination hotspots. We assumed a two-rate model for recombination in which hotspots of length 1 kb are separated by coldspots whose lengths are drawn from an exponential distribution. This roughly corresponds to hotspots being randomly distributed throughout the sequence. Our medium hotspot model has 50% of all recombination events happening in hotspots and an average of one hotspot every 30 kb. The strong hotspot model has 75% of all recombination events happening in hotspots, with an average of one hotspot every 50 kb. Within each region, all three models were scaled to produce the same average value of r as estimated for the corresponding region in the original data.
Simulation of the coalescent with variation in recombination rates was performed by simulating the coalescent with uniform recombination rates and varying mutation rates, with the appropriate scaling. The physical distances were then rescaled to produce the appropriate final model (cf. Li and Stephens, in press).
To examine the effects of marker density and sample size on haplotype block patterns, we ran further simulations of the medium hotspot model, with the same ascertainment scheme and N=104. This model was chosen because it provides a reasonably good fit to the data (see the “Results” section). These simulations used sample sizes of n/2, 2n, and 4n individuals (with all other parameters the same) or set θ=k/2, 2k, 4k, and 8k, where k=7.836×10-5/bp (this multiplies the average marker density by .5, 2, 4, and 8, respectively, while keeping all the other parameters the same).
We ran ⩾100 replicates for all of the coverage-criterion simulations and ⩾20 replicates for the other two criteria. Programs for the analyses were written in C and are available from the authors on request.
Results
Haplotype Block Criteria
To measure how well real and simulated data fit the haplotype block model, we developed three criteria (fig. 2). Informal descriptions and brief justification are given below. See the “Material and Methods” section for quantitative definitions.
Figure 2.

Schematic diagrams for the three haplotype block criteria. Sites in strong LD with each other are indicated by the same color. The coverage criterion calculates the proportion of sequence that is contained in haplotype blocks, the “hole” criterion measures how often blocks are disrupted by internal SNPs not in LD with their neighbors, and the “overlapping blocks” criterion measures how ambiguous haplotype block boundaries are. In the bottom diagram, the black/green site is in strong LD with both black and green sites. See the “Material and Methods” and “Results” sections for more details.
-
1.
Coverage: Most or all of the physical length of the sequence should be contained in identified haplotype blocks. Clearly, the utility of a haplotype map would be limited if a substantial part of the total sequence is not actually contained in identified haplotype blocks. The proportion of coverage by haplotype blocks depends both on the underlying structure of LD and on the experimental details, including marker density and sample size.
-
2.
Absence of “holes”: If a pair of SNPs is in strong LD, then both SNPs should be in strong LD with SNPs that lie in between. A key prediction of the haplotype block model (and the strategy of the HapMap Project) is that it is possible to make predictions about LD with unobserved SNPs from surrounding SNPs that are assigned to the same block. This is central to the concept of haplotype blocks, as well as to the goal of association mapping with a limited marker set arranged in haplotype blocks.
-
3.
Absence of “overlapping blocks”: There should be few SNPs that can be assigned to more than one block—that is, if an SNP is in strong LD with SNPs to the left and right, then those SNPs should also be in strong LD with each other. This criterion measures the extent to which blocks are discrete and unambiguous. If there are many overlapping blocks, then this indicates that any particular assignment of sites to mutually exclusive blocks is arbitrary and not a natural description of the data.
Fit of the Data to the Haplotype Block Criteria
We first examined the coverage properties for the four large sets of SNP data reported by Gabriel et al. (2002). We found that the haplotype block coverage varies tremendously across different genomic regions. Some are almost completely covered by haplotype blocks, whereas others have essentially no pairs of SNPs in strong LD with each other (Wall and Pritchard 2003). Pairwise D′ plots for all regions (and all three studies), as well as haplotype block comparisons across populations, are available online at the Pritchard Lab Web site (click on the “data Archive” link). These plots incorporate a frequency cutoff of 0.1 for the minor allele, since the vast majority of pairs with at least one low-frequency variant are uninformative (results not shown).
Overall, the observed coverage proportions are strikingly low (fig. 3A). In each population, more than half of the total region studied is not contained in identified blocks: in Europeans, 54% is not in blocks; in African Americans, 76%; in East Asians, 59%; and in sub-Saharan Africans, 73%. The proportion of sequence contained in long blocks (>30 kb) varies considerably across populations, ranging from 25%, in the European American sample, to only 5%, in the African American and sub-Saharan African samples. The distributions of haplotype block sizes for the East Asian and African American samples are very similar to those for the European American and sub-Saharan African samples, respectively, and therefore are not shown here.
Figure 3.

Analyses of three data sets using the three haplotype block criteria. A, The proportion of sequence contained in haplotype blocks of various sizes or not contained in blocks. The five samples presented are the European American Gabriel et al. (2002) sample, the African Gabriel et al. (2002) sample, the European American Seattle SNP sample, the African American Seattle SNP sample, and the EGP SNP sample. We define the size of a haplotype block to be the distance between the outermost markers, and the length of a region as the distance between the most distant pair of markers. B and C, Probability of violating the “hole” (B) and “overlapping blocks” (C) criteria as a function of the distance between the outermost pair in a set of three markers. See the “Material and Methods” section for details.
Given the marker spacing in the Gabriel et al. (2002) data sets (one SNP per 6.1–6.7 kb), it is possible that the resolution is too low to detect small haplotype blocks. Therefore, we conducted a similar analysis of two publicly available large-scale resequencing studies from D. Nickerson’s laboratory (see the “Material and Methods” section). Resequencing studies ascertain essentially all the common SNPs, so they have the highest possible resolution for a given number of sampled individuals. Again, we found that a substantial part of the sequence is not contained in identified blocks, although the coverage is higher than for the Gabriel et al. (2002) data (fig. 3A). Indeed, as shown below, our simulations indicate that increasing either the sample size or the marker density generally leads to greater coverage for the block definition used here.
The EGP SNP study examined a mixed sample of different ethnicities. Resampling experiments that we have performed with the Gabriel et al. (2002) data suggest that the haplotype block patterns in mixed samples are likely to be biased towards groups with smaller blocks. For example, with a sample of half European Americans and half African Americans (a total of 48 individuals from Gabriel et al. 2002), 70% of the sequence is not contained in haplotype blocks, compared with 54% and 76%, respectively, for the European Americans and African Americans exclusively (results not shown). Thus, the EGP SNP study is more comparable to the sub-Saharan African sample (Gabriel et al. 2002) in figure 3A. Note that, unlike the Gabriel et al. (2002) data, in which the effect is minimal, the proportion of sequence contained in long blocks is underestimated in the two resequencing studies, because of the limited sizes of the regions sequenced.
We next looked at the frequency with which trios of SNPs violate the “hole” and “overlapping blocks” criteria (fig. 3B and 3C). For all data sets, the results indicate that a substantial fraction of trios violate the hole criterion, especially when the outermost pair of markers is widely spaced (see fig. 3B). However, even two nearby SNPs are frequently unreliable indicators of the patterns of LD between them. For example, for SNPs in strong LD that are separated by <10 kb, ∼10%–20% of the intervening markers show “historical evidence of recombination” (see the “Material and Methods” section) with one of the two SNPs.
In contrast (fig. 3C), we found that the frequency of “overlapping blocks” was low at all distances (although this rate also increases with distance). Thus, unlike the previous two criteria, the data analyzed fit the overlapping blocks criterion reasonably well, indicating that the ambiguity of block boundaries is relatively modest.
Comparison of the Data to Simulations
The haplotype block paradigm implicitly assumes that recombination rates are low within blocks and higher in the regions between blocks (Daly et al. 2001; Jeffreys et al. 2001; Gabriel et al. 2002; Cardon and Abecasis 2003). To assess how patterns of LD are affected by variation in recombination rates, we ran simulations under the assumption of either a uniform recombination rate or localized “hotspots” of recombination. The simulation parameters were chosen to allow comparison with the sub-Saharan African data from the Gabriel et al. (2002) study, since the patterns of variation in this population seem to fit a relatively simple demographic model (see the “Material and Methods” section) better than do patterns in other populations (Frisse et al. 2001; Reich et al. 2001; Pluzhnikov et al. 2002). In a sense, this choice turns out to be conservative, because the signal reported below would be even stronger if we were comparing simulations to the non-African samples.
We ran simulations with three different population sizes: N=10,000, 15,000, and 20,000. Since the expected amount of LD is inversely correlated with the population recombination rate ρ (which is equal to 4Nr) (e.g., Ohta and Kimura 1969; Long and Langley 1999; Pritchard and Przeworski 2001), we expect there to be more LD (and thus longer haplotype blocks and greater coverage) with smaller values of N. Estimates of N from population genetic data tend to be ∼10,000–15,000 (e.g., Takahata 1993; Frisse et al. 2001; Yu et al. 2001).
Figure 4 shows comparisons between the simulated data and the actual data for the three block criteria. We find that although only a minority of the genome is contained in haplotype blocks in the actual data (27%), even less (10%–20%) is contained in blocks in the simulations with uniform recombination rates. Similarly, under the assumption of uniform recombination rates, we also find that the “holes” and “overlapping blocks” criteria are violated much more often than in the real data. Therefore, although the actual data do not fit the haplotype block model particularly well (i.e., the data do not perform well under two of three criteria), the fit under a model of uniform recombination rates is substantially worse.
Figure 4.

Comparison of simulation results (under different recombination rate variation models) to the African population from Gabriel et al. (2002), using the three haplotype block criteria. A–C, Proportion of sequence contained in haplotype blocks of various sizes or not contained in blocks. D–F, Probability of violating the “hole” criterion as a function of the distance between the outermost markers. G–I, Probability of violating the “overlapping blocks” criterion as a function of the distance between the outermost markers. The simulations assume different population sizes: N=10,000 (A, D, and G); N=15,000 (B, E, and H); and N=20,000 (C, F, and I). See the “Material and Methods” section for more details.

Under a hotspot model, recombination is concentrated into recombination hotspots, whereas most of the genome experiences lower recombination rates than the average. This makes long regions of strong LD much more likely. Consequently, coverage levels increase under the hotspot models, whereas the frequencies of holes and overlapping blocks both decrease (fig. 4). When N=10,000, the medium hotspot model seems to fit the data well, at least for the coverage and hole criteria. As N increases, more rate variation is needed for the simulated data to match the actual data. For N=15,000, the observed results lie between the predictions of the medium and strong hotspot models, whereas, for N=20,000, the strong hotspot model agrees most closely with the actual data (for the coverage and hole criteria). Curiously, the simulated frequencies of overlapping blocks seem to be greater than or equal to the actual frequencies for all parameter values. Although this might have biological significance, it may also have arisen by chance, because of the considerable stochasticity associated with estimates of overlapping block frequencies from limited data (see above). In any case, our simulations provide strong indirect evidence that there is widespread fine-scale variation in recombination rates.
Impact of Experimental Parameters on Inferred Blocks
Finally, we used simulations to explore the impact of study design on the characteristics of haplotype blocks. One important question is whether the relatively poor performance of the block model under two of our criteria is due simply to insufficient sample sizes or marker densities.
To explore this issue, we performed further simulations of the medium hotspot model with N=104 which, as described above, produces results very similar to the Nigerian data of Gabriel et al. (2002). We examined how changes in sample size (fig. 5) or marker density (fig. 6) affect patterns of LD, as measured by our three criteria. Increasing the sample size leads to only a slight increase in coverage (fig. 5A) but also to a slight increase in the frequency of holes (fig. 5B) and a large increase in the frequency of overlapping blocks (fig. 5C). For example, when n=232 individuals (a fourfold increase), the proportion of sequence contained in haplotype blocks increases from 26% to 32%, while the frequencies of holes and overlapping blocks (for different distances) increase by ∼20% and ∼100%, respectively.
Figure 5.

The effect of varying sample size on the “coverage” (A), “hole” (B), and “overlapping blocks” (C) criteria. The simulations assume a population size of N=10,000 and the medium hotspot model (see the “Material and Methods” section for details). n refers to the number of individuals sampled. The n=58 simulations are roughly comparable to the sub-Saharan African data from Gabriel et al. (2002).
Figure 6.

The effect of varying marker density on the “coverage” criterion. The simulations assume a population size of N=10,000 and the medium hotspot model (see the “Material and Methods” section for details). The simulated marker density is proportional to the mutation rate parameter θ. The simulations with θ=k are roughly comparable to the sub-Saharan African data from Gabriel et al. (2002), where k=7.836×10-5/bp. The simulations with θ=8k produce a marker density only slightly lower than would be obtained by complete resequencing in an ascertainment sample of eight chromosomes (Frisse et al. 2001).
Increasing the marker density has a different effect: the levels of coverage increase sharply (fig. 6), whereas the frequencies of holes and overlapping blocks are roughly unchanged (results not shown). With a fourfold increase in the number of markers, the coverage level increases from 26% to 57%; an eightfold increase in marker density (close to the theoretical maximum that could be achieved by a full resequencing study) yields a coverage level of 71%. If one had the choice of increasing either the sample size or the marker density, increasing the latter is clearly more desirable.
Discussion
The first large-scale haplotype block studies showed that some characteristics of LD data are blocklike, but they did not assess the extent to which patterns of LD can be fully described by the haplotype block model (Patil et al. 2001; Dawson et al. 2002; Gabriel et al. 2002). Thus, it has been unclear to what degree the notion of haplotype blocks is an approximate heuristic for interpreting complex patterns of LD, as opposed to a model that genuinely captures the true structure of LD across the genome. To address this issue, we proposed three quantitative criteria for measuring how well the structure of LD in human data fits a strict model of haplotype blocks. Our results were somewhat mixed, since we found that, for existing studies, coverage proportions were quite low, and also that the frequency of “holes” is nonnegligible. In contrast, the rate of overlapping blocks (and, hence, the ambiguity involved in assigning sites to blocks) was fairly low. Overall, however, despite these departures from a strict haplotype block model, the real data conform to haplotype blocks much better than would be predicted by simulating data with a uniform recombination rate. This was true for all populations.
Although a few recombination hotspots have been characterized experimentally (Lien et al. 2000; Jeffreys et al. 2001; May et al. 2002; Schneider et al. 2002), there is no direct experimental information yet about how widespread fine-scale variation in recombination rates is. Moreover, even if there were widespread variation in local recombination rates, it does not follow that the structure of LD would be strongly blocklike. Our simulations indicate that rate heterogeneity must be extremely pronounced to produce data that really fit the haplotype block model. For example, even our model in which half of all recombination events happen in just 3% of the sequence can provide a poor fit to the haplotype block model when all three of our criteria are used (see fig. 4). Thus, we find strong support for rate heterogeneity while still finding substantial departures from the haplotype block model.
In a sense, our results can be viewed as showing that there is an excess of medium- and long-range LD compared with what would be expected with uniform recombination rates (see, e.g., the frequencies of medium and large blocks in fig. 4A–4C); one natural explanation for this excess of LD is widespread heterogeneity in recombination rates (Reich et al. 2002). Although other studies have found an excess of long-range LD (Dunning et al. 2000; Abecasis et al. 2001; Reich et al. 2001, 2002; Dawson et al. 2002; Innan et al. 2003), they have done so with European or ethnically mixed samples. It has been postulated that European populations have experienced a recent bottleneck (i.e., a temporary sharp reduction in effective population size) (Tishkoff et al. 1996; Reich et al. 2001), and bottlenecks are known to increase levels of LD (Reich et al. 2001; Wall et al. 2002). Therefore, it is not known whether the results of previous studies are due to the effects of population history, or whether they reflect the underlying recombinational landscape.
We have improved upon previous studies by considering a Nigerian population that is more likely to be at equilibrium and by explicitly examining how recombination rate heterogeneity affects patterns of LD. Studies of a Hausa population (sampled in neighboring Cameroon) found no departures from a simple equilibrium model (Frisse et al. 2001; Pluzhnikov et al. 2002). Also, our model of a constant-sized population with N=10,000 is actually conservative for our purposes; more-realistic models that incorporate either a larger effective population size or recent population growth lead to substantially less LD (results not shown). Although there remains the possibility that there is some unknown population structure or admixture in the Gabriel et al. (2002) Nigerian sample, both of which are known to increase levels of LD (Wall 2000; Pritchard and Przeworski 2001), this seems unlikely given the relatively low levels of LD found at short scales in the nearby Hausa (Frisse et al. 2001; L. Frisse and A. Di Rienzo, personal communication).
With a demographic explanation unlikely, the global excess of LD probably results from fine-scale variation in recombination rates. The parameters in our hotspot model are comparable to what has been estimated from sperm typing studies (Jeffreys et al. 2001); clearly, however, more empirical data must be gathered before we can be confident that our models are a reasonable approximation. We point out that the apparent conflict in patterns of LD at short scales (Frisse et al. 2001) versus medium and long scales (present study; see also Pritchard and Przeworski 2001) may be explained in part by high rates of intragenic gene conversion without exchange of flanking markers (Ardlie et al. 2001; Frisse et al. 2001; Pritchard and Przeworski 2001; Przeworski and Wall 2001). Since mean tract lengths are thought to be small (e.g., <1 kb), gene conversion increases effective recombination rates at short scales while having a negligible effect on LD at longer distances (Andolfatto and Nordborg 1998). The higher-than-expected frequencies of holes at short distances in the actual data (figs. 3 and 4) are also consistent with this hypothesis.
In summary, our results show that the structure of LD is “blocklike” in some regions but not in others. This suggests that the usefulness of the haplotype block concept for future association studies will be uneven and will depend on the patterns of LD in the specific genomic regions that are considered. One likely determinant of how blocklike patterns of LD are is how much variation there is in local recombination rates. Although our analyses suggest that a substantial degree of rate variation is necessary to explain the overall patterns of LD in the data, further empirical studies will be required to determine the extent to which the variation in blockiness from region to region reflects differences in the underlying recombinational landscapes.
Acknowledgments
We thank M. Przeworski and two anonymous reviewers for comments on a previous version of this manuscript, as well as D. Altshuler, M. Daly, S. Gabriel, D. Nickerson, and S. Schaffner for help accessing and interpreting their data. This work was supported by National Institutes of Health grant HG 2772 (to J.K.P.).
Electronic-Database Information
The URLs for data presented herein are as follows:
- EGP SNPs, http://egp.gs.washington.edu/
- Pritchard Lab, http://pritch.bsd.uchicago.edu/ (click on the “Data Archive” link)
- UW-FHCRC Variation Discovery Resource, http://pga.gs.washington.edu/ (for the Seattle SNPs)
- Whitehead Institute Center for Genome Research, http://www-genome.wi.mit.edu/mpg/hapmap/hapstruc.html
References
- Abecasis GR, Noguchi E, Heinzmann A, Traherne JA, Bhattacharya S, Leaves NI, Anderson GG, Zhang Y, Lench NJ, Carey A, Cardon LR, Moffat MF, Cookson WO (2001) Extent and distribution of linkage disequilibrium in three genomic regions. Am J Hum Genet 68:191–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andolfatto P, Nordborg M (1998) The effect of gene conversion on intralocus associations. Genetics 148:1397–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardlie K, Liu-Cordero SN, Eberle MA, Daly M, Barrett J, Winchester E, Lander ES, Kruglyak L (2001) Lower-than-expected linkage disequilibrium between tightly linked markers in humans suggests a role for gene conversion. Am J Hum Genet 69:582–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayres KL, Balding DJ (2001) Measuring gametic disequilibrium from multilocus data. Genetics 157:413–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardon LR, Abecasis GR (2003) Using haplotype blocks to map human complex trait loci. Trends Genet 19:135–140 [DOI] [PubMed] [Google Scholar]
- Carlson CS, Eberle MA, Rieder M, Smith JD, Kruglyak L, Nickerson DA (2003) Additional SNPs and linkage-disequilibrium analysis in whole-genome association studies in humans. Nat Genet 33:518–521 [DOI] [PubMed] [Google Scholar]
- Chakravarti A, Buetow KW, Antonarakis SE, Waber PG, Boehm CD, Kazazian HH (1984) Nonuniform recombination within the human β-globin gene cluster. Am J Hum Genet 36:1239–1258 [PMC free article] [PubMed] [Google Scholar]
- Clark AG, Weiss KM, Nickerson DA, Taylor SL, Buchanan A, Stengard J, Salomaa V, Vartianinen E, Perloa M, Boerwinkle E, Sing CF (1998) Haplotype structure and population genetic inferences from nucleotide-sequence variation in human lipoprotein lipase. Am J Hum Genet 63:595–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly MJ, Rioux JD, Schaffner SF, Hudson TJ, Lander ES (2001) High-resolution haplotype structure in the human genome. Nat Genet 29:229–232 [DOI] [PubMed] [Google Scholar]
- Dawson E, Abecasis GR, Bumpstead S, Chen Y, Hunt S, Beare DM, Pabilal J, et al (2002) A first-generation linkage disequilibrium map of human chromosome 22. Nature 418:544–548 [DOI] [PubMed] [Google Scholar]
- Dunning, AM, Durocher F, Healey CS, Teare MD, McBride SE, Carlomagno F, Xu CF, Dawson E, Rhodes S, Ueda S, Lai E, Luben RN, Van Rensburg EJ, Mannermaa A, Kataja V, Rennart G, Dunham I, Purvis I, Easton D, Ponder BAJ (2000) The extent of linkage disequilibrium in four populations with distinct demographic histories. Am J Hum Genet 67:1544–1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisse L, Hudson RR, Bartoszewicz A, Wall JD, Donfack J, Di Rienzo A (2001) Gene conversion and different population histories may explain the contrast between polymorphism and linkage disequilibrium levels. Am J Hum Genet 69:831–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly MJ, Altshuler D (2002) The structure of haplotype blocks in the human genome. Science 296:2225–2229 [DOI] [PubMed] [Google Scholar]
- Hudson RR (1983) Properties of a neutral-allele model with intergenic recombination. Theor Popul Biol 23:183–201 [DOI] [PubMed] [Google Scholar]
- Innan H, Padhukasahasram B, Nordborg M (2003) The pattern of polymorphism on human chromosome 21 Genome Res 13:1158–1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffreys AJ, Kauppi L, Neumann R (2001) Intensely punctuate meiotic recombination in the class II region of the major histocompatibility complex. Nat Genet 29:217–222 [DOI] [PubMed] [Google Scholar]
- Kong, A, Gudbjartsson DF, Sainz J, Jonsdottir GM, Gudjonsson SA, Richardsson B, Sigurdardottir S, Barnard J, Hallbeck B, Masson G, Shlien A, Palsson ST, Frigge ML, Thorgeirsson TE, Gulcher JR, Stefansson K (2002) A high-resolution recombination map of the human genome. Nat Genet 31:241–247 [DOI] [PubMed] [Google Scholar]
- Kruglyak L (1999) Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nat Genet 22:139–144 [DOI] [PubMed] [Google Scholar]
- Lewontin RC (1964) The interaction of selection and linkage. I. General considerations: heterotic models. Genetics 49:49–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Stephens M (2003) A new multilocus model for linkage disequilibrium, with application to exploring variations in recombination rate. Genetics (in press) [Google Scholar]
- Lien S, Szyda J, Schechinger B, Rappold G, Arnheim N (2000) Evidence for heterogeneity in recombination in the human pseudoautosomal region: high resolution analysis by sperm typing and radiation-hybrid mapping. Am J Hum Genet 66:557–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long AD, Langley CH (1999) The power of association studies to detect the contribution of candidate genetic loci to variation in complex traits. Genome Res 9:720–731 [PMC free article] [PubMed] [Google Scholar]
- May, CA, Shone AC, Kalaydjieva L, Sajantila A, Jeffreys AJ (2002) Crossover clustering and rapid decay of linkage disequilibrium in the Xp/Yp pseudoautosomal gene SHOX. Nat Genet 31:272–275 [DOI] [PubMed] [Google Scholar]
- Ohta T, Kimura M (1969) Linkage disequilibrium at steady state determined by random genetic drift and recurrent mutation. Genetics 63:229–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil N, Berno AJ, Hinds DA, Barrett WA, Doshi JM, Hacker CR, Kautzer CR, Lee DH, Marjoribanks C, McDonough DP, Nguyen BTN, Norris MC, Sheehan JB, Shen N, Stern D, Stokowski RP, Thomas DJ, Trulson MO, Vyas KR, Frazer KA, Fodor SPA, Cox DR (2001) Blocks of limited haplotype diversity revealed by high-resolution scanning of human chromosome 21. Science 294:1719–1723 [DOI] [PubMed] [Google Scholar]
- Phillips MS, Lawrence R, Sachidanandam R, Morris AP, Balding DJ, Donaldson MA, Studebaker JF, et al (2003) Chromosome-wide distribution of haplotype blocks and the role of recombination hotspots. Nat Genet 33:382–387 [DOI] [PubMed] [Google Scholar]
- Pluzhnikov A, Di Rienzo A, Hudson RR (2002) Inferences about human demography based on multilocus analyses of noncoding sequences. Genetics 161:1209–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Przeworski M (2001) Linkage disequilibrium in humans: models and data. Am J Hum Genet 69:1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przeworski M, Wall JD (2001) Why is there so little intragenic linkage disequilibrium in humans? Genet Res 77:143–151 [DOI] [PubMed] [Google Scholar]
- Reich DE, Cargill M, Bolk S, Ireland J, Sabeti PC, Ritcher DJ, Lavery T, Kouyoumjian R, Farhadian SF, Ward R, Lander ES (2001) Linkage disequilibrium in the human genome. Nature 411:199–204 [DOI] [PubMed] [Google Scholar]
- Reich DE, Schaffner SF, Daly MJ, McVean G, Mullikin JC, Higgins JM, Richter DJ, Lander ES, Altshuler D (2002) Human genome sequence variation and the influence of gene history, mutation and recombination. Nat Genet 32:135–142 [DOI] [PubMed] [Google Scholar]
- Schneider JA, Peto TE, Boone RA, Boyce AJ, Clegg JB (2002) Direct measurement of the male recombination fraction in the human beta-globin hot spot. Hum Mol Genet 11:207–215 [DOI] [PubMed] [Google Scholar]
- Taillon-Miller P, Bauer-Sardina I, Saccone NL, Putzel J, Laitinen T, Cao A, Kere J, Pilia G, Rice JP, Kwok PY (2000) Juxtaposed regions of extensive and minimal linkage disequilibrium in human Xq25 and Xq28. Nat Genet 25:324–328 [DOI] [PubMed] [Google Scholar]
- Takahata N (1993) Allelic genealogy and human evolution. Mol Biol Evol 10:2–22 [DOI] [PubMed] [Google Scholar]
- Tishkoff SA, Dietzsch E, Speed W, Pakstis AJ, Kidd JR, Cheung K, Bonné-Tamir B, Benerecetti ASS, Moral P, Krings M, Pääbo S, Watson E, Risch N, Jenkins T, Kidd KK (1996) Global patterns of linkage disequilibrium at the CD4 locus and modern human origins. Science 271:1380–1387 [DOI] [PubMed] [Google Scholar]
- Wall JD (2000) Detecting ancient admixture in humans using sequence polymorphism data. Genetics 154:1271–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall JD, Andolfatto P, Przeworski M (2002) Testing models of selection and demography in Drosophila simulans. Genetics 162:203–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall JD, Pritchard JK (2003) Haplotype blocks and linkage disequilibrium in the human genome. Nat Rev Genet 4:587–597 [DOI] [PubMed] [Google Scholar]
- Wang N, Akey JM, Zhang K, Chakraborty R, Jin L (2002) Distribution of recombination crossovers and the origin of haplotype blocks: the interplay of population history, recombination, and mutation. Am J Hum Genet 71:1227–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu N, Zhao Z, Fu YX, Sambuughin N, Ramsay M, Jenkins T, Leskinen E, Patthy L, Jorde LB, Kuromori T, Li WH (2001) Global patterns of human DNA sequence variation in a 10-kb region on chromosome 1. Mol Biol Evol 18:214–222 [DOI] [PubMed] [Google Scholar]
- Zhang K, Deng M, Chen T, Waterman MS, Sun F (2002) A dynamic programming algorithm for haplotype block partitioning. Proc Natl Acad Sci USA 99:7335–7339 [DOI] [PMC free article] [PubMed] [Google Scholar]