

Abstract
Multinuclear non-heme iron dependent oxidative enzymes (MNIOs), formerly known as domain of unknown function 692 (DUF692), are involved in the post-translational modification of peptides during the biosynthesis of peptide-based natural products. These enzymes catalyze highly unusual and diverse chemical modifications. Several class-defining features of this large family (>14,000 members) are beginning to emerge. Structurally, the enzymes are characterized by a TIM-barrel fold and a set of conserved residues for a di- or tri-iron binding site. They use molecular oxygen to modify peptide substrates in a four-electron oxidation, often taking place at a cysteine residue. This review summarizes the current understanding of MNIOs. Four modifications are discussed in detail: oxazolone-thioamide formation, β-carbon excision, hydantoin-macrocycle formation, and 5-thiooxazole formation. Briefly discussed are two reactions that do not take place on Cys residues.
Keywords: Metalloenzyme, RiPPs, natural product biosynthesis, metallophore, dinuclear/trinuclear iron enzyme, specialized metabolism, secondary metabolism
Graphical Abstract
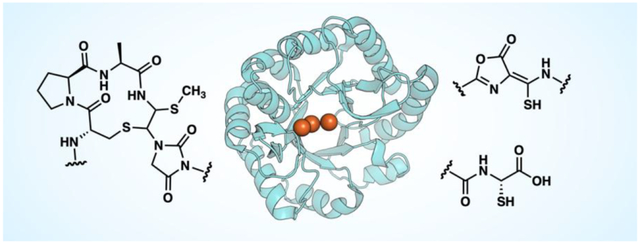
Introduction
Several families of non-heme diiron enzymes are involved in the biosynthesis of natural products, such as the ferritin-like oxidases and oxygenases (FDOs), the heme oxygenase-like oxidases and oxygenases (HDOs), and the mixed-valent diiron oxygenases (MVDOs) [1–4]. Recently, members from a different of iron-dependent enzymes, initially annotated as domain of unknown function 692 (DUF692), have been identified in the biosynthesis of ribosomally synthesized and post-translationally modified peptides (RiPPs). These natural products display a diverse array of bioactivities [5–7]. RiPPs are made following a common biosynthetic logic in which a precursor peptide is modified by one or more post-translational modifying enzymes, and then proteolytically processed to yield the mature natural product. Several classes of post-translational modifying enzymes have been characterized in RiPP biosynthesis [8], with the DUF692 family enzymes implicated relatively recently. The first member to be characterized was MbnB, involved in the biosynthesis of the copper-chelating peptide methanobactin [9]. Following recent characterization of more members, DUF692 enzymes were renamed multinuclear non-heme iron dependent oxidative enzymes (MNIOs) [10]. While most characterized MNIOs have been shown to be oxidases, some MNIOs may function as monooxygenases or dioxygenases (Figure 1A).
Figure 1.
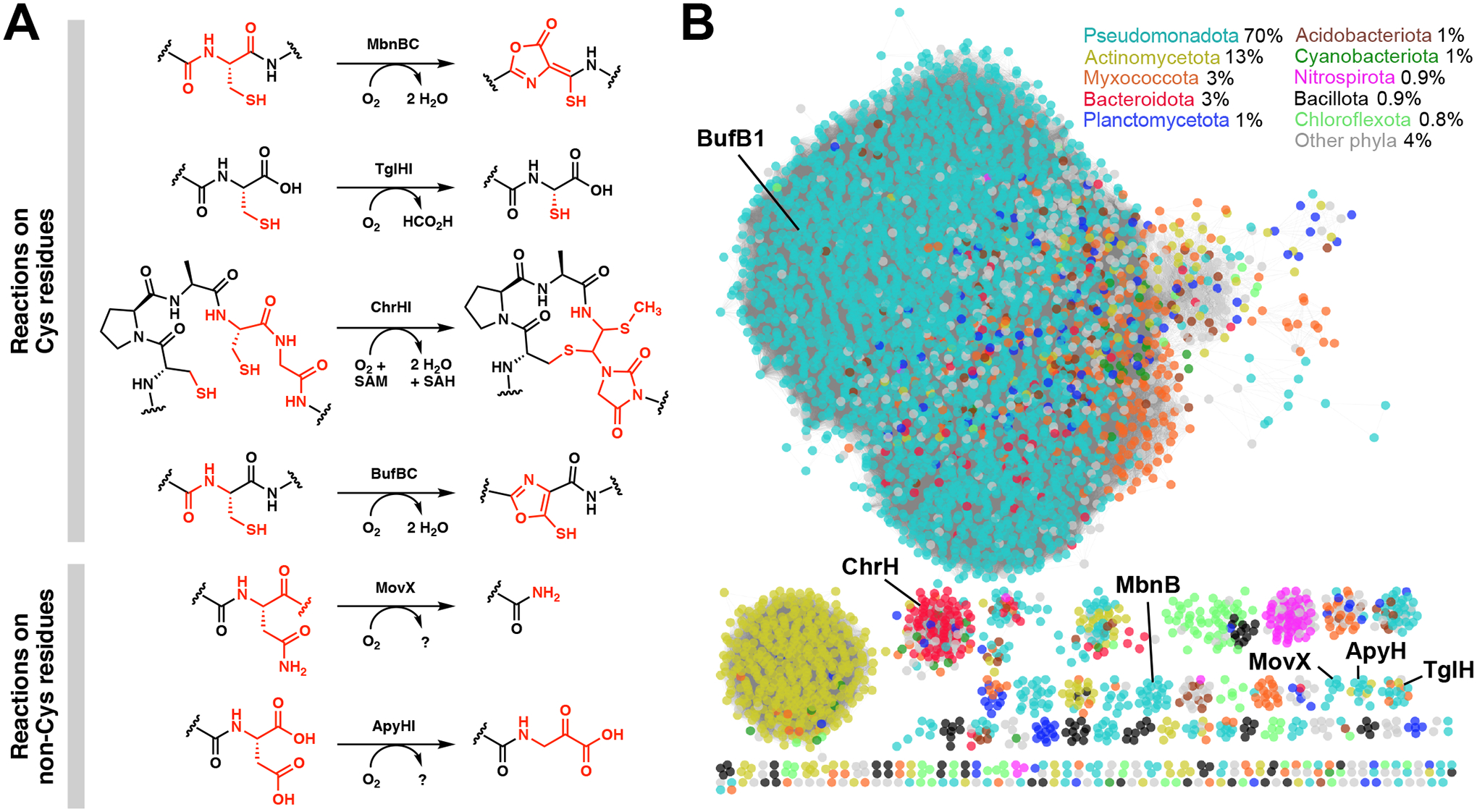
Diverse post-translational modifications performed by members of the MNIO family. (A) Reactions catalyzed by MbnBC, TglHI, ChrHI, BufBC, MovX, and ApyHI. (B) Sequence similarity network (SSN) of MNIOs (DUF692), colored by the top ten phyla in which MNIOs are most frequently found. Percentages represent the distribution of MNIO-containing species across phyla. The SSN was generated by using the Enzyme Function Initiative-Enzyme Similarity Tool with over 14 000 members [15,16].
MNIOs are around 30–45 kDa in size and typically form a heterodimer with a partner protein, although the identity of the partner protein is not conserved. The structures of several MNIOs have been elucidated, revealing a triose-phosphate isomerase (TIM)-barrel fold with a di- or tri-iron binding site located within the barrel [11–13]. MNIOs use molecular oxygen, appear to function without the need of external reductant, and in most cases catalyze a four-electron oxidation of substrate [9,10,14].
The growing family of MNIOs contains over 14 000 members across both Gram-negative and Gram-positive bacteria. A sequence similarity network (SSN) [15,16] shows that the majority of MNIOs are found in Pseudomonadota (Proteobacteria), followed by Actinomycetota (Actinobacteria), Myxococcota, Bacteroidota, and other phyla (Figure 1B). Several subfamilies remain to be characterized, the majority of which are predicted to be involved in RiPP biosynthesis (for a co-occurrence SSN of MNIO-RiPPs, see refs [17,18]).
In this review, we summarize the unusual rearrangements catalyzed by currently characterized MNIOs (Figure 1A) and discuss patterns across MNIO structures, substrates, and mechanistic proposals.
Cysteine-modifying MNIOs
MbnB
The first characterized MNIO was MbnB, an enzyme involved in the maturation of the RiPP methanobactin, a copper-chelator (chalkophore) that is produced by methanotrophic bacteria under copper-limiting conditions [19,20]. The peptide is secreted into the environment in its apo-form, then taken back up by the cell in its copper-bound form [21]. Structurally, most methanobactins contain a pair of oxazolone-thioamide moieties (Figure 1A) that bind copper [19,22].
Biosynthetic gene clusters (BGCs) responsible for methanobactin biosynthesis encode MbnB and MbnC as well as the gene for the precursor peptide MbnA [23]. In vitro assays demonstrated that MbnB, together with its partner protein MbnC, installs the thioamide and oxazolone moieties onto MbnA [9]. An MbnC deletion mutant in Methylosinus trichosporium OB3b showed that while both MbnBC are required for the formation of the C-terminal oxazolone-thioamide, the N-terminal oxazolone-thioamide could be formed without MbnC [24]. Furthermore, in contrast to MsMbnB, which is only stable in a complex with MbnC, homologs from Vibrio fluvalis (MovB and MovC) could be purified independently [9,18].
The active form of MbnB is believed to be a mixed-valent Fe(II)/Fe(III) diiron species instead of a triiron species, supported by spectroscopic data detailed below [12,25]. The mechanism is proposed to feature coordination of the cysteine thiolate of MbnA to Fe(III) (Figure 2A), which was observed crystallographically in an MbnABC complex [11] and supported by electron nuclear double resonance spectroscopy [25]. Then, in a mechanism reminiscent of isopenicillin N synthase (IPNS) [26,27], binding of O2 to the ferrous iron has been proposed to generate an Fe(III)-superoxo intermediate, which abstracts a hydrogen atom from the β-carbon of cysteine. The radical is then oxidized to a thioaldehyde that is attacked by the adjacent amide-nitrogen, forming a β-lactam and an Fe(IV)-oxo (ferryl) intermediate. This potent oxidizing species performs a second H-atom abstraction from the β-lactam to form a thio analog of a cyclic imide, which was proposed to be opened by an enzyme nucleophile [11,12]. Substitution and tautomerization form the final oxazolone-thioamide moiety [12].
Figure 2.
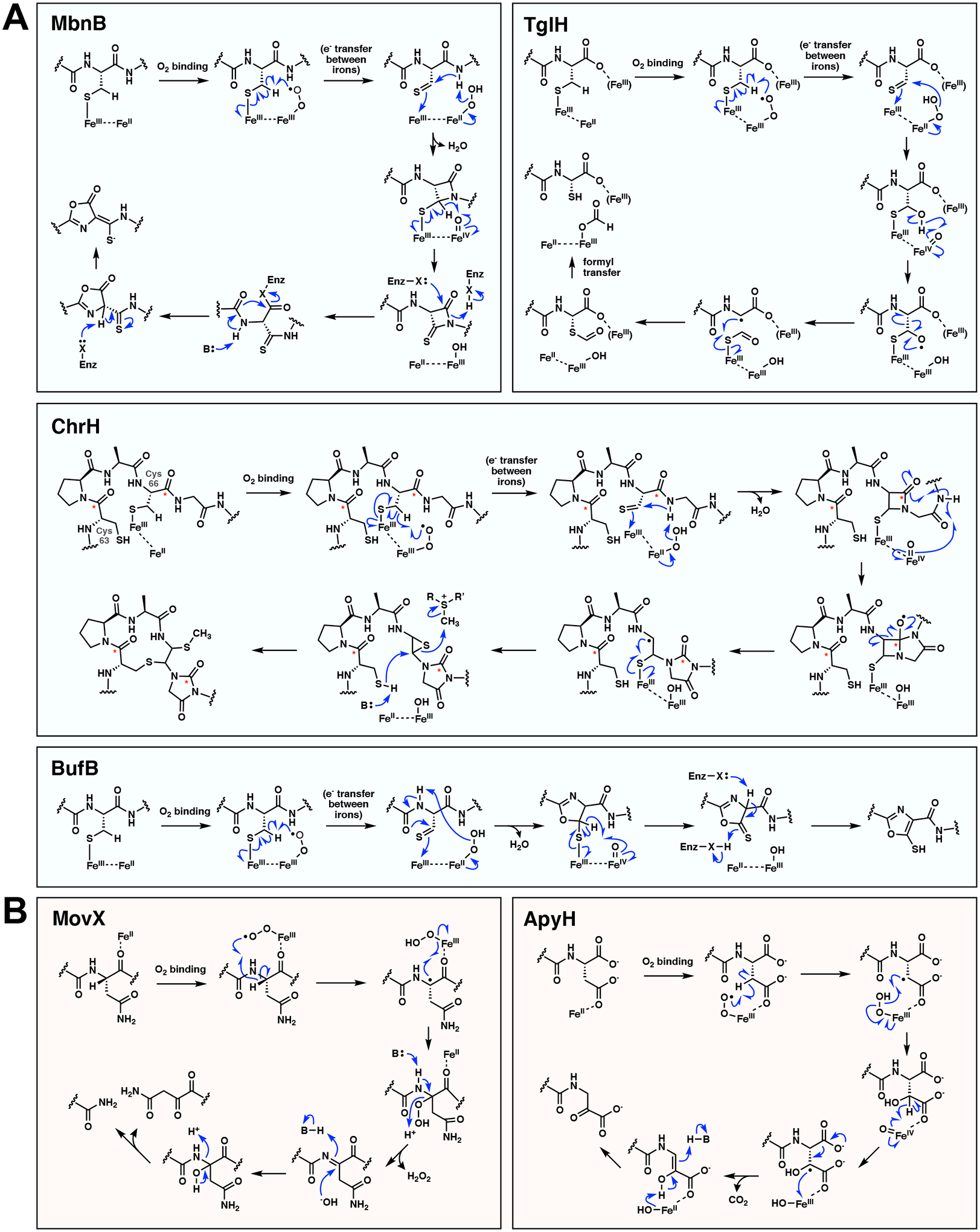
Proposed reaction mechanisms of MNIOs. (A) Proposed mechanisms of the cysteine modifying MNIOs MbnB, TglH, ChrH, and BufB. The mechanisms depicted here are consistent with the current evidence and are drawn to show possible similarities across the family. For example, all four mechanisms involving cysteine are drawn with a thiolate-coordinating ferric iron being different from the ferrous iron that activates O2. Other mechanisms can be drawn in which the same Fe coordinates Cys and activates O2 [14]. The fate of the carbonyl carbons during the ChrH reaction is indicated with an asterisk; X = enzyme residue. (B) Proposed mechanisms of the asparagine/aspartate modifying MNIOs MovX and ApyH. B = enzyme base. We note that for all reactions shown, other mechanisms are possible, with several steps having both homolytic or heterolytic possibilities.
TglH
The second MNIO to be characterized, TglH, was discovered as part of a RiPP pathway that produces 3-thiaglutamate (or a derivative) in the plant pathogen Pseudomonas syringae [14]. TglH, together with its partner protein TglI, performs an unusual excision of the β-carbon of a C-terminal cysteine, releasing the carbon as formate and reattaching the thiol group to the ⍺-carbon (Figure 1A) [14]. These findings suggest that TglH could be an oxygenase if molecular oxygen is incorporated into formate. Use of a substrate analog perdeuterated at the ⍺- and β-carbon of cysteine resulted in only one deuterium in the peptide product, suggesting that the ⍺ hydrogen is likely not removed in the reaction. These findings support a mechanism with similarity to myo-inositol oxygenase (MIOX), 2-hydroxyethylphosphonate dioxygenase (HEPD), and IPNS catalyzed reactions [26,28–30]. The proposed mechanism again involves H-atom abstraction from the β-carbon of cysteine by an Fe(III)-superoxo (Figure 2A). Hydration of the thioaldehyde generates a thioacetal and Fe(IV)-oxo. Proton-coupled electron transfer (PCET) of the hydroxyl group to the ferryl generates an oxygen radical that undergoes β-scission resulting in a resonance stabilized radical at the ⍺-carbon. Recombination of the sulfur of the thioformyl group with the ⍺-carbon radical generates the S-C⍺ bond. Finally, hydrolysis of the thioester by an iron-bound hydroxide results in the production of formate and the peptide product [14].
A follow up study showed that an analog of the precursor peptide TglA in which cysteine was replaced with selenocysteine inhibited TglHI [31]. How selenocysteine acts as an inhibitor, and whether this strategy could be applied to generate other MNIO inhibitors remains to be explored. In another study, a homolog of TglHI, TmoHI from Tistrella mobilis, was found to perform the same reaction on a C-terminal cysteine of a scaffold peptide [32]. In this case the later biosynthetic steps diverged, resulting in the production of 3-thiahomoleucine (instead of 3-thiaglutamate) attached to the scaffold peptide TmoA [32].
ChrH
ChrH from Chryseobacterium sp. JM1 is part of another MNIO subfamily that performs vastly different chemistry compared to MbnB and TglH (Figure 1A). ChrH performs a remarkably complex rearrangement of a peptide ChrA containing two conserved Cys residues to install a peptide macrocycle, a hydantoin (imidazolidine-2,4-dione) heterocycle, and a thiomethyl group (Figure 1A) [10]. 13C-labelling showed that the carbonyl carbon of the 2nd Cys in the sequence migrates to the hydantoin moiety in the product (Figure 2A). Furthermore, S-adenosylmethionine (SAM) is required for the reaction in vitro [10]. ChrH copurifies with ChrI, annotated as a member of the anthrone oxygenase-like protein family (formerly DUF1772) [33]. Whereas the original report suggested that ChrI was required for ChrH activity, our recent unpublished work shows that ChrI is not needed. Furthermore, while the structure of the ChrH-modified peptide has been determined, the final product of the chr BGC has yet to be elucidated.
Several possible mechanisms have been proposed for the complex rearrangement by ChrH [6]. Here, we propose a modified mechanism that is more similar to those proposed for MbnB and TglH. The first step again invokes hydrogen atom abstraction from the β-carbon of Cys66 by an Fe(III)-superoxo intermediate (Figure 2A). Formation of a thioaldehyde, subsequent β-lactam formation, and formation of a ferryl could occur as in the MbnB mechanism [12]. Then, N-H bond cleavage at a Gly amide nitrogen by the Fe(IV)-oxo could result in a rearrangement to generate a bicyclic system and oxygen radical. Next, β-scission akin to the C-C bond cleavage step in TglHI would form the hydantoin heterocycle and a radical on the former ⍺-carbon of the initial cysteine residue. Radical recombination with an iron-bound thiolate could then generate an epi-sulfide. Finally, the thiol of the second cysteine (Cys63) attacks the epi-sulfide, which undergoes ring opening and S-methylation by SAM to generate the final product.
BufB
BufB is part of the largest subfamily of MNIOs and is involved in the biosynthesis of bufferins (Figure 1), which are copper-chelating metallophores [34]. Unlike methanobactins, which are involved in copper import, bufferins are thought to be involved in copper export during copper-induced stress. BufB1 from Caulobacter vibrioides, together with its partner protein BufC1 (a DUF2063 family protein), converts cysteine residues to 5-thiooxazoles on the precursor peptide BufA1 (a DUF2282 family protein) (Figure 1A) [34]. BufA1 contains four cysteine residues, the middle two of which are modified to 5-thiooxazoles that enable copper binding.
While little is known about the mechanism of BufB, the reaction is proposed to similarly start from a mixed-valent state that involves oxygen activation and H-atom abstraction from the β-carbon of Cys (Figure 2A). Cyclization followed by ring oxidation and tautomerization is proposed to form the 5-thiooxazole [34]. Interestingly, this motif is precedented in RiPP biosynthesis, but is formed by a radical SAM enzyme [35].
Asparagine/Aspartate-modifying MNIOs
MovX
MovX (previously termed MbnX) is part of a methanobactin-like gene cluster in Vibrio fluvalis [23]. MovX was shown to act on an asparagine residue, performing a N-C⍺ bond cleavage to generate a C-terminal amide (Figure 1A) [18]. This discovery showed that MNIOs can modify residues other than cysteine. Furthermore, MovX can catalyze this transformation without the need for a partner protein, likely because it encodes a C-terminal extension that is predicted to recruit substrate [18].
One proposed mechanism for this transformation involves oxygen activation to generate a Fe(III)-superoxo that abstracts the ⍺-hydrogen atom from asparagine (Figure 2B) [18]. Peroxide rebound is proposed to be followed by H2O2 elimination and generation of an imine. Hydrolysis of the imine generates the C-terminally amidated peptide and a diketone dipeptide. Although the dipeptide coproduct eluded detection, several lines of evidence are consistent with this mechanism. The reaction required O2, hydrogen peroxide was detected, and use of 15N- labelled asparagine (at the side chain amide) resulted in loss of the label in the C-terminally amidated peptide product [18]. This mechanism deviates from the other characterized MNIOs in that the enzyme would perform a two-electron oxidation of substrate.
ApyH
ApyH from Burkholderia thailandensis is another example of an MNIO that modifies a non-Cys residue. ApyH, together with its partner protein ApyI, converts a C-terminal aspartate on the precursor peptide ApyA to an ⍺-keto acid moiety during aminopyruvatide biosynthesis (Figure 1A) [36]. In addition to the conserved C-terminal aspartate, ApyA homologs contain a highly conserved cysteine and glycine in the middle of the sequence. While no modification was detected at these residues, various mutations of the cysteine abolished activity, suggesting that the cysteine may be involved in substrate binding or substrate-assisted catalysis [36].
The ApyHI mechanism is thought to occur by oxygen activation and H-atom abstraction from the β-carbon of aspartate (Figure 2B) [36]. Like the Cys-modifying enzymes, a Fe(III)-hydroperoxo is proposed to initiate hydroxylation of the β-carbon with generation of an Fe(IV)-oxo. The ferryl may then abstract the remaining hydrogen atom from the β-carbon. Decarboxylation involving an electron transfer to Fe(III) then produces the enol form of the ketoacid. Electron transfer coupled to decarboxylation to form an alkene has been previously proposed in other iron-dependent enzymes such as iron(II)/2-oxoglutarate-dependent and radical SAM enzymes [37–39]. Finally, tautomerization of the enol would form the C-terminal ⍺-ketoacid [36].
Structural basis of catalysis and substrate binding
Several MNIO structures have been solved in the iron-free, diiron, or triiron forms, or in a complex with their partner protein and substrate (Table 1; Figure 3A) [11–13]. The active site structure generally shows that iron 1 (Fe1) and Fe2 are six-coordinate, with Fe3 (when it is present) having five or six-coordinate geometry (Figure 3B). Fe1 is coordinated by the side chains of two His and one Glu, which is bidentate and bridges Fe1 and Fe2. Fe2 is coordinated by two Glu, one His, one Asp residue, and Fe3 is coordinated by His, Asp, and Asn. Combined structural and mutagenesis data have also shown that the active site residues Asp240 in MbnB and Asn73 in TglH are essential, likely playing a role as a base or for substrate positioning, respectively [11,13].
Table 1.
Summary of the characterized MNIOs to date.
| MNIO | Organism | Modification | Modified residue | Representative structure(s) (PDB ID) | Pathway |
|---|---|---|---|---|---|
| U.C.a | Haemophilus somnus (Histophilus somni) strain 129pt | Unknown | Unknown | 3BWW (diiron) | Unknown |
| MbnB | Methylosinus trichosporium OB3b, Rugamonas rubra ATCC-43154, Vibrio caribbeanicus ATCC BAA-2122 | Oxazolone-thioamide installation | Cysteine | 7DZ9, 7FC0 (triiron MbnABC complex) | Methanobactin biosynthesis |
| TglH | Pseudomonas syringae pv. maculicola ES4326 | β-carbon excision | Cysteine | 8HCI (apo TglHI) 8HI7 (diiron TglHI) 8HI8 (triiron TglHI) |
3-Thiaglutamate biosynthesis |
| TmoH | Tistrella mobilis KA081020-065 | β-carbon excision | Cysteine | N.A.a | 3-Thiahomoleucine biosynthesis |
| ChrH | Chryseobacterium sp. JM1 | Hydantoin-macrocycle formation | Cysteine | N.A. | Unknown |
| ApyH | Burkholderia thailandensis E264 | Aminopyruvic acid formation | Aspartate | N.A. | Aminopyruvatide biosynthesis |
| MovX (MbnX) | Vibrio fluvalis 33809 | C-terminal amidation via N-C⍺ bond cleavage | Asparagine | N.A. | Unknown |
| BufB | Caulobacter vibrioides NA1000 | 5-thiooxazole installation | Cysteine | N.A. | Bufferin biosynthesis |
U.C., uncharacterized; N.A., not available
Figure 3.
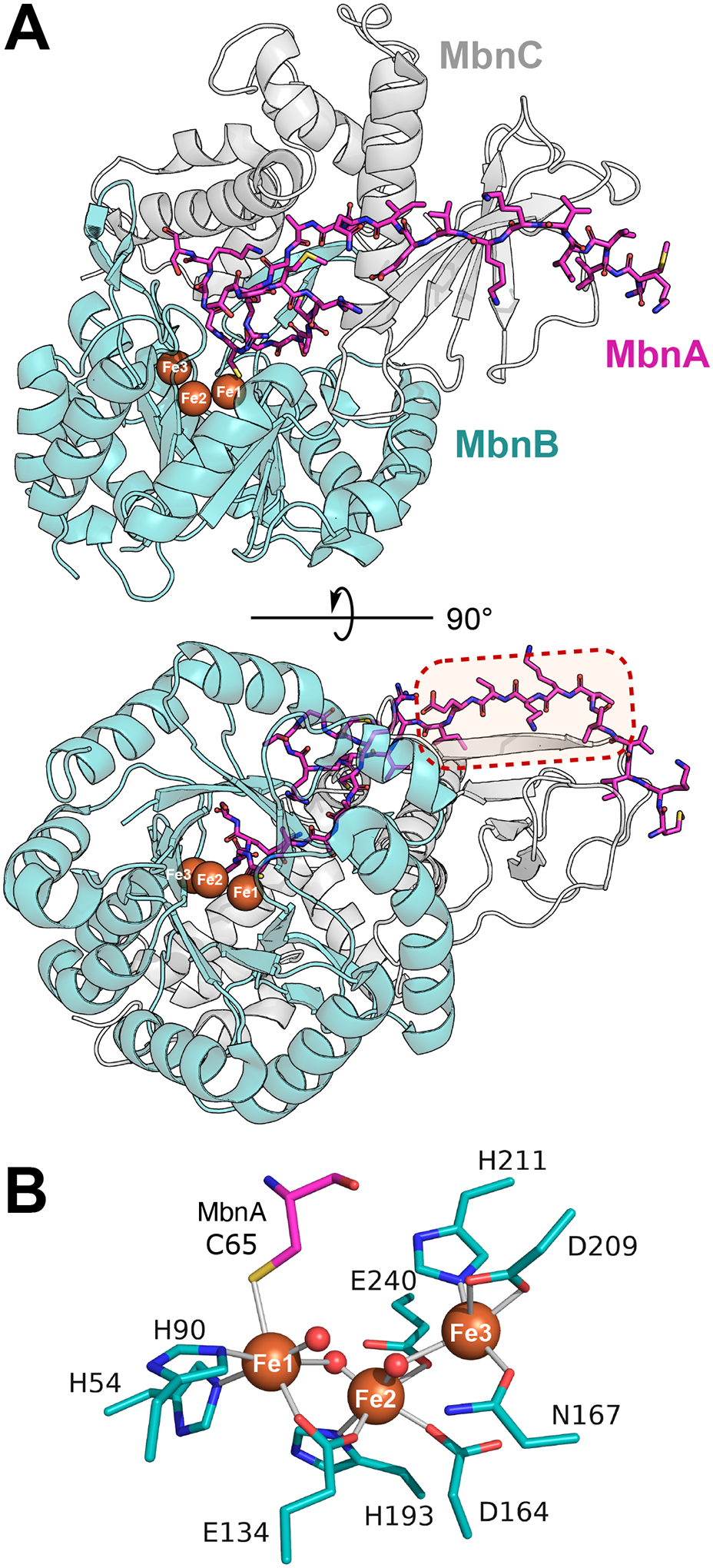
Structural insights into MNIO catalysis and substrate binding. (A) Structure of the MbnABC complex from Rugamonas rubra (PDB:7FC0), showing the common TIM-barrel fold shared across MNIOs and the three bound irons (orange spheres). The dotted box highlights the contact between MbnA and the MbnC partner protein that is required for substrate binding [11]. (B) Active site structure of RrMbnB showing coordinating side chains (teal sticks) and water molecules (red spheres). The cysteine residue of the substrate MbnA coordinating with Fe1 is shown in pink. A water molecule occupies this coordination site in the absence of substrate.
RiPP BGCs frequently encode a RiPP recognition element (RRE) to recruit substrates [40]. RREs contain three ⍺-helices and three β-sheets that bind an N-terminal leader sequence of the precursor peptide. The partner proteins TglI, ApyI, and BufC1 contain RREs that recruit their respective substrates. Furthermore, MovX contains a C-terminal extension with a winged helix-turn-helix fold that is predicted to act as an RRE. However, both MbnB and ChrH lack an obvious RRE encoded in their clusters [9,10,14]. The structure of the MbnABC complex shows that while a canonical RRE is not present, the leader peptide of MbnA binds to the partner protein MbnC in an analogous fashion to RRE recognition (Figure 3A) [11]. Notably, contacts between MbnH and TglH and their substrates also seem to be important. This observation may help to explain why ChrH can function without a partner protein to recruit substrate. The mechanism of substrate binding of ChrA by ChrH is still unclear due to lack of a co-crystal structure, but is likely to occur via a different mechanism than TglAHI and MbnABC.
MNIO substrates
In general, MNIOs appear to have relaxed substrate specificity with respect to peptide length and sequence. TglH can modify a truncated substrate consisting of the last 19 amino acids of the precursor peptide TglA, and mutations at several positions within this 19-mer were well tolerated [31]. BufBC could modify the core peptide lacking the N-terminal signal sequence, albeit with lower efficiency [34]. Similarly, N-terminal truncation of MbnA to a 22-mer still resulted in oxazolone-thioamide formation by MbnBC [9]. MbnBC from Ms. trichosporium was also shown to modify MbnA precursor peptides from four out of the five phylogenetic classes of methanobactin-like operons, despite substantially different sequences [9].
While initial studies of MNIOs described modifications of cysteine residues [41], characterization of MovX and ApyH showed that MNIOs can also act on asparagine and aspartate residues. These observations suggest that the breadth of MNIO reactivity is larger than currently realized, and that other amino acid residues could potentially be substrates. At present, no examples have been reported of MNIOs that use a non-peptide substrate.
Mechanistic commonalities and differences
Despite the seemingly highly diverse reactions in Figure 1A, some parallels can be drawn from the transformations that involve Cys residues. MbnB, TglH, ChrH, and BufB all carry out a four-electron oxidation of the substrate and initial H-atom abstraction from the β-carbon of cysteine [9,10,14]. In addition, all three enzymes are proposed to use an Fe(III) ion as a Lewis acid to activate the initially formed thioaldehyde. Other similarities between TglH and ChrH are that they both catalyze transfer of the sulfur of Cys from the β-carbon to the α-carbon. As shown in Figure 2A, Fe(III)-superoxo and Fe(IV)-oxo species have been proposed as the oxidizing species in the catalytic cycle, but both have yet to be detected spectroscopically. Prior studies on oxygen activation in a variety of non-heme iron enzymes have established several spectroscopic features that may inform future studies on O2 activation in MNIOs [1,4]. Currently also unresolved is whether oxygen binding is substrate triggered as observed for other non-heme diiron enzymes [42–44]. At present, any similarities of MovX and ApyH with other MNIOs are less obvious [18,36].
The mechanisms of the cysteine modifying MNIOs all invoke one ferrous ion in the resting state of the enzyme that activates oxygen to form both Fe(III)-superoxo and Fe(IV)-oxo intermediates (Figure 2A). Such use of a single iron center to generate consecutive oxidizing species has precedent in a subset of non-heme iron-dependent enzymes, including IPNS and HEPD [1,45]. The other irons in the MNIO active site are proposed to not be used for oxygen activation but for electron transfer, Lewis acid activation of sulfur, and potentially substrate binding and orientation. EPR and Mössbauer studies on MbnB coupled to activity assays have revealed that a mixed valent Fe(II)/Fe(III) is the active species [12]. The Fe(II)/Fe(III) irons in MbnB are antiferromagnetically coupled, giving a S=1/2 rhombic signal with a gave-value of 1.9. The sample also showed the presence of a rhombic S=5/2 Fe(III) signal at geff=4.3 [9,12]. Although no external reductant is needed in the proposed MNIO mechanisms, the addition of ascorbate to MbnB has been shown to increase enzyme activity in vitro [12,18]. This observation may be attributed to the reduction of a ferric state to the active mixed-valent species, and is consistent with the Fe(III) EPR signal decreasing as ascorbate was titrated into MbnBC.
Diiron or triiron enzyme?
An unresolved question is whether MNIOs are di- or triiron enzymes, or whether examples exist of both within the family. For BufB1, enzyme that modified the precursor peptide in vitro co-purified with two irons [34]. Similarly, in MbnB, higher activity was correlated with higher abundance of the diiron form, and has led to its functional classification as an MVDO [12,25]. However, MbnB and TglH can bind up to three irons based on reported structures, and Mössbauer and mass spectrometry data [11–13]. A co-crystal structure of the MbnABC complex indicated ligation of the substrate cysteine thiolate to Fe1, identifying it as crucial to the reaction (Figure 3B) [11]. Both Fe1 and Fe2 sites showed full occupancy whereas the Fe3 site often shows partial occupancy [11,12]. Thus, the importance and role of Fe3 remain unresolved.
In both MbnB and TglH, mutating some of the Fe3 ligands to alanine or serine decreases activity but does not completely abolish it, suggesting Fe3 may not be critical [12,13]. However, mutation of His ligands to Fe1 or Fe2 in TglH to Ala also decreased but did not completely abolish activity [13]. Thus, Fe3 could still play a role in facilitating substrate positioning, or orientating side chains of active site residues in TglH. Detailed spectroscopic studies on TglH, ChrH, MovX, ApyH, and BufB will be needed to investigate whether the active species differs from MbnB. If MNIOs are diiron enzymes, it begs the question of why the ligand set for binding three irons is conserved across most MNIOs.
Conclusions and Outlook
Recent reports have demonstrated the remarkable breadth of MNIO-catalyzed reactions, all housed within a common TIM-barrel fold. MNIOs have been shown to modify cysteine, asparagine, and aspartate residues in RiPP biosynthesis. Several questions remain in our understanding of this enzyme family. Firstly, what other chemical transformations are performed by MNIOs? Despite their prevalence across different phyla, several subfamilies of MNIOs remain functionally uncharacterized (Figure 1B) [17]. The diverse and unprecedented reactivity of the enzymes characterized thus far suggests that exciting new post-translational modifications are to be discovered. Secondly, what is the mechanism behind these transformations, and what determines the type of modification performed? Probing the mechanisms may not only reveal fundamental insights into an unusual di-/tri-iron catalytic center, but will be key for the potential development of MNIOs as biocatalysts or the design of mechanism-based inhibitors. Finally, which biological processes are mediated by the products of MNIO-containing BGCs? Methanobactins play a key role in copper acquisition in methanotrophic bacteria, while bufferins have been shown to provide protection against copper toxicity [9,34]. Recent studies also suggest that an MNIO-modified RiPP is a virulence factor in the human respiratory pathogen, Haeomophilus influenzae [46,47]. On the other hand, the physiological roles for 3-thia-⍺-amino acids, aminopyruvatides, and the chr product remain unknown.
Reports on the few MNIOs characterized thus far have demonstrated their emerging role in performing unusual and challenging peptide modifications. Future genome mining efforts, functional characterizations, and protein engineering studies will elucidate the full catalytic breadth of this enzyme family.
Highlights.
Remarkable chemistry catalyzed by multinuclear non-heme iron dependent oxidative enzymes (MNIOs)
MNIOs convert Cys to an oxazolone-thioamide in methanobactin biosynthesis
MNIOs excise the β-carbon of Cys in 3-thiaglutamate biosynthesis
MNIOs convert Cys residues into macrocyclic thioaminals and hydantoins
MNIOs convert Cys residues to 5-thiooxazoles in bufferin biosynthesis
MNIOs cleave the peptide backbone at Asn to generate a C-terminal amide
MNIOs convert C-terminal Asp into aminopyruvic acid
Acknowledgments
This work was supported by a grant from the National Institutes of Health (R37 GM058822 to W.A.v.d.D.) and an NSERC PGS-D to J.Y.C.
Abbreviations
- BGC
biosynthetic gene cluster
- DUF692
domain of unknown function 692
- EPR
electron paramagnetic resonance
- HEPD
2-hydroxyethylphosphonate dioxygenase
- IPNS
isopenicillin N synthase
- MIOX
myo-inositol oxygenase
- MNIO
multinuclear non-heme iron dependent oxidative enzyme
- PCET
proton-coupled electron transfer
- RiPP
ribosomally synthesized and post-translationally modified peptides
- RRE
RiPP recognition element
- SAM
S-adenosylmethionine
- SSN
sequence similarity network
- TIM
triose-phosphate isomerase
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability
No data was used for the research described in the article.
References
Papers of particular interest, published within the period of review, have been highlighted as:
* of special interest
* * of outstanding interest
- 1.Rajakovich LJ, Zhang B, McBride MJ, Boal AK, Krebs C, Bollinger JM Jr.: Emerging Structural and Functional Diversity in Proteins with Dioxygen-Reactive Dinuclear Transition Metal Cofactors. In: Comprehensive Natural Products III: Chemistry and Biology. Liu H-w, Begley TP (Eds), Elsevier, Amsterdam: (2020):215–250. [Google Scholar]
- 2.Ushimaru R, Abe I: Unusual dioxygen-dependent reactions catalyzed by nonheme iron enzymes in natural product biosynthesis. ACS Catal (2023) 13(2):1045–1076. [Google Scholar]
- 3.Feig AL, Lippard SJ: Reactions of non-heme iron(II) centers with dioxygen in biology and chemistry. Chem Rev (1994) 94(3):759–805. [Google Scholar]
- 4.Jasniewski AJ, Que L: Dioxygen activation by nonheme diiron enzymes: diverse dioxygen adducts, high-valent intermediates, and related model complexes. Chem Rev (2018) 118(5):2554–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Y, Rebuffat S: The manifold roles of microbial ribosomal peptide-based natural products in physiology and ecology. J Biol Chem (2020) 295(1):34–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cao L, Do T, Link AJ: Mechanisms of action of ribosomally synthesized and posttranslationally modified peptides (RiPPs). J Ind Microbiol Biotechnol (2021) 48(3–4):kuab005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ongpipattanakul C, Desormeaux EK, DiCaprio A, van der Donk WA, Mitchell DA, Nair SK: Mechanism of action of ribosomally synthesized and post-translationally modified peptides. Chem Rev (2022) 122(18):14722–14814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Montalbán-López M, Scott TA, Ramesh S, Rahman IR, van Heel AJ, Viel JH, Bandarian V, Dittmann E, Genilloud O, Goto Y, Grande Burgos MJ et al. : New developments in RiPP discovery, enzymology and engineering. Nat Prod Rep (2021) 38(1):130–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kenney GE, Dassama LMK, Pandelia ME, Gizzi AS, Martinie RJ, Gao P, DeHart CJ, Schachner LF, Skinner OS, Ro SY, Zhu X et al. : The biosynthesis of methanobactin. Science (2018) 359(6382):1411–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]; [**] This paper describes the characterization of the first MNIO (MbnB) that installs oxazolone-thioamide moieties during the biosynthesis of methanobactin. The substrate specificity of MbnB was explored, and preliminary EPR and Mössbauer characterization are reported.
- 10.Ayikpoe RS, Zhu L, Chen JY, Ting CP, van der Donk WA: Macrocyclization and backbone rearrangement during RiPP biosynthesis by a SAM-dependent domain-of-unknown-function 692. ACS Cent Sci (2023) 9(5):1008–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]; [**] This investigation shows that ChrH, an MNIO enriched in the phylum Bacteroidota, modifies two cysteines in the precursor ChrA to form a macrocycle, a hydantoin heterocycle, and a thiomethyl group.
- 11.Dou C, Long Z, Li S, Zhou D, Jin Y, Zhang L, Zhang X, Zheng Y, Li L, Zhu X, Liu Z et al. : Crystal structure and catalytic mechanism of the MbnBC holoenzyme required for methanobactin biosynthesis. Cell Res (2022) 32(3):302–314. [DOI] [PMC free article] [PubMed] [Google Scholar]; [**] This study presents the only crystal structure of an MNIO with its substrate bound (the MbnABC complex from two different species). The structure shows the thiolate of MbnA bound to Fe1 of MbnB, as well as the molecular basis for substrate recognition by MbnBC.
- 12.Park YJ, Jodts RJ, Slater JW, Reyes RM, Winton VJ, Montaser RA, Thomas PM, Dowdle WB, Ruiz A, Kelleher NL, Bollinger JM Jr. et al. : A mixed-valent Fe(II)Fe(III) species converts cysteine to an oxazolone/thioamide pair in methanobactin biosynthesis. Proc Natl Acad Sci U S A (2022) 119(13):e2123566119. [DOI] [PMC free article] [PubMed] [Google Scholar]; [*] In this work, the first detailed spectroscopic characterization of MbnB is presented, showing that the active form is likely a Fe(II)/Fe(III) mixed-valent diiron species. Furthermore, the structure of MbnBC was solved with two and three irons.
- 13.Zheng Y, Xu X, Fu X, Zhou X, Dou C, Yu Y, Yan W, Yang J, Xiao M, van der Donk WA, Zhu X et al. : Structures of the holoenzyme TglHI required for 3-thiaglutamate biosynthesis. Structure (2023) 31(10):1220–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]; [*] The paper reports the structure of TglHI, solved in its apo-, di-, and triiron forms. Considerable conformational shifts were observed upon iron binding. Mutation of the iron ligands to alanine revealed that certain mutants could still perform β-carbon excision with reduced activity.
- 14.Ting CP, Funk MA, Halaby SL, Zhang Z, Gonen T, van der Donk WA: Use of a scaffold peptide in the biosynthesis of amino acid-derived natural products. Science (2019) 365(6450):280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]; [**] This report demonstrates that TglH (the second characterized MNIO) performs β-methylene excision on a C-terminal cysteine in 3-thiaglutamate biosynthesis. Isotopic labelling was used to show that formate is produced and MicroED was used to elucidate the structure of the final product.
- 15.Zallot R, Oberg N, Gerlt JA: The EFI web resource for genomic enzymology tools: Leveraging protein, genome, and metagenome databases to discover novel enzymes and metabolic pathways. Biochemistry (2019) 58(41):4169–4182. [DOI] [PMC free article] [PubMed] [Google Scholar]; [*] This report describes a very useful resource used for generating sequence similarity networks (SSNs), exploring gene neighbourhoods, and obtaining taxonomic information about a protein family and has been used extensively in MNIO research.
- 16.Oberg N, Zallot R, Gerlt JA: EFI-EST, EFI-GNT, and EFI-CGFP: Enzyme Function Initiative (EFI) web resource for genomic enzymology tools. J Mol Biol (2023) 435(14):168018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clark KA, Seyedsayamdost MR: Bioinformatic atlas of radical SAM enzyme-modified RiPP natural products reveals an isoleucine-tryptophan crosslink. J Am Chem Soc (2022) 144(39):17876–17888. [DOI] [PubMed] [Google Scholar]
- 18.Chioti VT, Clark KA, Ganley JG, Han EJ, Seyedsayamdost MR: N–Cα bond cleavage catalyzed by a multinuclear iron oxygenase from a divergent methanobactin-like RiPP gene cluster. J Am Chem Soc (2024) 146(11):7313–7323. [DOI] [PMC free article] [PubMed] [Google Scholar]; [*] This study presents a new transformation for a MNIO family member, the cleavage of the Cα-N bond of an Asn in the peptide substrate.
- 19.Kim HJ, Graham DW, DiSpirito AA, Alterman MA, Galeva N, Larive CK, Asunskis D, Sherwood PM: Methanobactin, a copper-acquisition compound from methane-oxidizing bacteria. Science (2004) 305(5690):1612–1615. [DOI] [PubMed] [Google Scholar]
- 20.Kenney GE, Rosenzweig AC: Chemistry and biology of the copper chelator methanobactin. ACS Chem Biol (2011) 7(2):260–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dassama LMK, Kenney GE, Ro SY, Zielazinski EL, Rosenzweig AC: Methanobactin transport machinery. Proc Natl Acad Sci USA (2016) 113(46):13027–13032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.El Ghazouani A, Baslé A, Gray J, Graham DW, Firbank SJ, Dennison C: Variations in methanobactin structure influences copper utilization by methane-oxidizing bacteria. Proc Natl Acad Sci USA (2012) 109(22):8400–8404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kenney GE, Rosenzweig AC: Genome mining for methanobactins. BMC Biol (2013) 11(1):17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dershwitz P, Gu W, Roche J, Kang-Yun CS, Semrau JD, Bobik TA, Fulton B, Zischka H, DiSpirito AA: MbnC is not required for the formation of the N-terminal oxazolone in the methanobactin from Methylosinus trichosporium OB3b. Appl Environ Microbiol (2022) 88(2):e01841–01821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jodts RJ, Ho MB, Reyes RM, Park YJ, Doan PE, Rosenzweig AC, Hoffman BM: Initial steps in methanobactin biosynthesis: substrate binding by the mixed-valent diiron enzyme MbnBC. Biochemistry (2024):doi: 10.1021/acs.biochem.1024c00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tamanaha E, Zhang B, Guo Y, Chang WC, Barr EW, Xing G, St Clair J, Ye S, Neese F, Bollinger JM Jr., Krebs C: Spectroscopic evidence for the two C-H-cleaving intermediates of Aspergillus nidulans isopenicillin N synthase. J Am Chem Soc (2016) 138(28):8862–8874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roach PL, Clifton IJ, Hensgens CM, Shibata N, Schofield CJ, Hajdu J, Baldwin JE: Structure of isopenicillin N synthase complexed with substrate and the mechanism of penicillin formation. Nature (1997) 387(6635):827–830. [DOI] [PubMed] [Google Scholar]
- 28.Peck SC, Wang C, Dassama LM, Zhang B, Guo Y, Rajakovich LJ, Bollinger JM Jr., Krebs C, van der Donk WA: O-H activation by an unexpected ferryl intermediate during catalysis by 2-hydroxyethylphosphonate dioxygenase. J Am Chem Soc (2017) 139(5):2045–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xing G, Diao Y, Hoffart LM, Barr EW, Prabhu KS, Arner RJ, Reddy CC, Krebs C, Bollinger JM Jr.: Evidence for C-H cleavage by an iron-superoxide complex in the glycol cleavage reaction catalyzed by myo-inositol oxygenase. Proc Natl Acad Sci USA (2006) 103(16):6130–6135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bollinger JM Jr., Diao Y, Matthews ML, Xing G, Krebs C: myo-Inositol oxygenase: a radical new pathway for O2 and C-H activation at a nonheme diiron cluster. Dalton Trans (2009) 6):905–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McLaughlin MI, Yu Y, van der Donk WA: Substrate recognition by the peptidyl-(S)-2-mercaptoglycine synthase TglHI during 3-thiaglutamate biosynthesis. ACS Chem Biol (2022) 17(4):930–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu Y, van der Donk WA: Biosynthesis of 3-thia-α-amino acids on a carrier peptide. Proc Natl Acad Sci U S A (2022) 119(29):e2205285119. [DOI] [PMC free article] [PubMed] [Google Scholar]; [*] This report demonstrates that TmoHI (a homolog of TglHI) also performs a β-methylene excision on a C-terminal cysteine during the biosynthesis of 3-thiahomoleucine.
- 33.Griffiths S, Mesarich CH, Saccomanno B, Vaisberg A, De Wit PJGM, Cox R, Collemare J: Elucidation of cladofulvin biosynthesis reveals a cytochrome P450 monooxygenase required for anthraquinone dimerization. Proc Natl Acad Sci USA (2016) 113(25):6851–6856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leprevost L, Jünger S, Lippens G, Guillaume C, Sicoli G, Oliveira L, Rivera-Millot A, Billon G, Henry C, Antoine R, Dubiley S et al. : A widespread family of ribosomal peptide metallophores involved in bacterial adaptation to copper stress. bioRxiv (2024): 10.1101/2024.1103.1118.585515. [DOI] [PMC free article] [PubMed] [Google Scholar]; [**] This study reports the discovery of a new family of metallophores, the bufferins that are suggested to help detoxify copper. A member of the largest family of MNIOs is shown to convert Cys to a thioxazole.
- 35.Lewis JK, Jochimsen AS, Lefave SJ, Young AP, Kincannon WM, Roberts AG, Kieber-Emmons MT, Bandarian V: New role for radical SAM enzymes in the biosynthesis of thio(seleno)oxazole RiPP natural products. Biochemistry (2021) 60(45):3347–3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nguyen DT, Zhu L, Gray DL, Woods TJ, Padhi C, Flatt KM, Mitchell DA, van der Donk WA: Biosynthesis of macrocyclic peptides with C-terminal β-amino-α-keto acid groups by three different metalloenzymes. ACS Cent Sci (2024): 10.1021/acscentsci.1024c00088. [DOI] [PMC free article] [PubMed] [Google Scholar]; [*] In this study, the authors discovered a new reaction for an MNIO, the oxidative decarboxylation of a C-terminal Asp residue resulting in an aminopyruvate structure.
- 37.Yu C-P, Tang Y, Cha L, Milikisiyants S, Smirnova TI, Smirnov AI, Guo Y, Chang W-c: Elucidating the reaction pathway of decarboxylation-assisted olefination catalyzed by a mononuclear non-heme iron enzyme. J Am Chem Soc (2018) 140(45):15190–15193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Copeland RA, Davis KM, Shoda TKC, Blaesi EJ, Boal AK, Krebs C, Bollinger JM Jr.: An iron(IV)–oxo intermediate initiating L-arginine oxidation but not ethylene production by the 2-oxoglutarate-dependent oxygenase, ethylene-forming enzyme. J Am Chem Soc (2021) 143(5):2293–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bruender NA, Bandarian V: The radical S-adenosyl-L-methionine enzyme MftC catalyzes an oxidative decarboxylation of the C-terminus of the MftA peptide. Biochemistry (2016) 55(20):2813–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burkhart BJ, Hudson GA, Dunbar KL, Mitchell DA: A prevalent peptide-binding domain guides ribosomal natural product biosynthesis. Nat Chem Biol (2015) 11(8):564–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao Y, Zhu Y, Awakawa T, Abe I: Unusual cysteine modifications in natural product biosynthesis. RSC Chem Biol (2024) 5(4):293–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fox BG, Lyle KS, Rogge CE: Reactions of the diiron enzyme stearoyl-acyl carrier protein desaturase. Acc Chem Res (2004) 37(7):421–429. [DOI] [PubMed] [Google Scholar]
- 43.Zhang B, Rajakovich LJ, Van Cura D, Blaesi EJ, Mitchell AJ, Tysoe CR, Zhu X, Streit BR, Rui Z, Zhang W, Boal AK et al. : Substrate-triggered formation of a peroxo-Fe2(III/III) intermediate during fatty acid decarboxylation by UndA. J Am Chem Soc (2019) 141(37):14510–14514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Manley OM, Tang H, Xue S, Guo Y, Chang W-C, Makris TM: BesC initiates C–C cleavage through a substrate-triggered and reactive diferric-peroxo intermediate. J Am Chem Soc (2021) 143(50):21416–21424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peck SC, van der Donk WA: Go it alone: four-electron oxidations by mononuclear non-heme iron enzymes. J Biol Inorg Chem (2017) 22(2–3):381–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ahearn CP, Kirkham C, Chaves LD, Kong Y, Pettigrew MM, Murphy TF: Discovery and contribution of nontypeable Haemophilus influenzae NTHI1441 to human respiratory epithelial cell invasion. Infect Immun (2019) 87(11):e00462–00419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nasreen M, Fletcher A, Hosmer J, Zhong Q, Essilfie A-T, McEwan AG, Kappler U: The alternative sigma factor RpoE2 is involved in the stress response to hypochlorite and in vivo survival of Haemophilus influenzae. Front Microbiol (2021) 12(Feb 12):637213. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data was used for the research described in the article.