Jian-Liang Li
Jian-Liang Li
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
1,3,8,
Michael R Hayden
Michael R Hayden
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
9,
Elisabeth W Almqvist
Elisabeth W Almqvist
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
9,
Ryan R Brinkman
Ryan R Brinkman
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
9,
Alexandra Durr
Alexandra Durr
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
10,
Catherine Dodé
Catherine Dodé
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
11,
Patrick J Morrison
Patrick J Morrison
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
12,13,
Oksana Suchowersky
Oksana Suchowersky
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
14,
Christopher A Ross
Christopher A Ross
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
15,16,17,
Russell L Margolis
Russell L Margolis
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
15,17,
Adam Rosenblatt
Adam Rosenblatt
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
15,
Estrella Gómez-Tortosa
Estrella Gómez-Tortosa
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
18,
David Mayo Cabrero
David Mayo Cabrero
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
18,
Andrea Novelletto
Andrea Novelletto
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
19,
Marina Frontali
Marina Frontali
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
20,
Martha Nance
Martha Nance
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
21,
Ronald J A Trent
Ronald J A Trent
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
22,
Elizabeth McCusker
Elizabeth McCusker
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
23,
Randi Jones
Randi Jones
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
24,
Jane S Paulsen
Jane S Paulsen
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
25,
Madeline Harrison
Madeline Harrison
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
26,
Andrea Zanko
Andrea Zanko
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
27,
Ruth K Abramson
Ruth K Abramson
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
28,
Ana L Russ
Ana L Russ
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
1,
Beth Knowlton
Beth Knowlton
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
1,
Luc Djoussé
Luc Djoussé
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
2,
Jayalakshmi S Mysore
Jayalakshmi S Mysore
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
5,
Suzanne Tariot
Suzanne Tariot
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
5,
Michael F Gusella
Michael F Gusella
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
5,
Vanessa C Wheeler
Vanessa C Wheeler
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
5,
Larry D Atwood
Larry D Atwood
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
1,
L Adrienne Cupples
L Adrienne Cupples
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
4,
Marie Saint-Hilaire
Marie Saint-Hilaire
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
1,
Jang-Ho J Cha
Jang-Ho J Cha
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
6,
Steven M Hersch
Steven M Hersch
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
6,
Walter J Koroshetz
Walter J Koroshetz
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
6,
James F Gusella
James F Gusella
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
5,7,
Marcy E MacDonald
Marcy E MacDonald
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
5,
Richard H Myers
Richard H Myers
1Department of Neurology and 2Section of Preventive Medicine and Epidemiology, Evans Department of Medicine, Boston University School of Medicine, 3Bioinformatics Program and 4Department of Biostatistics, School of Public Health, Boston University, 5Molecular Neurogenetics Unit, and 6Department of Neurology, Massachusetts General Hospital, and 7Department of Genetics, Harvard Medical School, Boston; 8Department of Genomics, Wyeth Research, Cambridge, MA; 9Centre for Molecular Medicine & Therapeutics and Department of Medical Genetics, University of British Columbia, Vancouver; 10INSERM U289, Hôpital de la Salpêtrière, and 11Service de Biochimie et de Génétique Moléculaire, Hôpital Cochin, Paris; 12Department of Medical Genetics, Belfast City Hospital, Belfast; 13School of Biomedical Science, University of Ulster, Coleraine, United Kingdom; 14Departments of Clinical Neurosciences and Medical Genetics, University of Calgary, Calgary; Departments of 15Psychiatry and 16Neuroscience and 17Program in Cellular and Molecular Medicine, John Hopkins University, Baltimore; 18Servicio de Neurología y Genética, Fundación Jiménez Díaz, Madrid; 19Department of Cell Biology, University of Calabria, Rende, Italy; 20Institute of Neurobiology and Molecular Medicine, Consiglio Nazionale delle Ricerche, Rome; 21Department of Neurology, Hennepin County Medical Center, Minneapolis; 22Department of Medicine, University of Sydney, and 23Neurology Department, Westmead Hospital, Sydney; 24Neurology Department, Emory University, Atlanta; 25Department of Psychiatry, University of Iowa, Iowa City; 26Health Sciences Center, University of Virginia, Charlottesville; 27Division of Medical Genetics, University of California San Francisco, San Francisco; and 28William S. Hall Psychiatric Institute, Columbia, SC
1,3


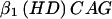 +
+ 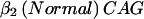 +
+ 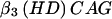 ×
× 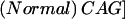 (Djoussé et al. 2003).
(Djoussé et al. 2003).
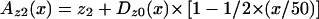 , where x is the distance in cM from the HD locus and may vary from 0 cM—where all of Dz0(x) is allocated to z2—to 50 cM—where none of the Dz0(x) is allocated to both z1 and z2. Az1(x) and Az2(x) are the adjusted estimates of z1 and z2, respectively, at the x position.
, where x is the distance in cM from the HD locus and may vary from 0 cM—where all of Dz0(x) is allocated to z2—to 50 cM—where none of the Dz0(x) is allocated to both z1 and z2. Az1(x) and Az2(x) are the adjusted estimates of z1 and z2, respectively, at the x position.