

Abstract
The mechanisms involved in radiation-induced cellular injury and death remain incompletely understood. In addition to the direct formation of highly reactive hydroxyl radicals (HO·) by radiolysis of water, oxidative stress events in the cytoplasm due to formation of H2O2 may also be important. Since the major pool of low-mass redox-active intracellular iron seems to reside within lysosomes, arising from the continuous intralysosomal autophagocytotic degradation of ferruginous materials, formation of H2O2 inside and outside these organelles may cause lysosomal labilization with release to the cytosol of lytic enzymes and low-mass iron. If of limited magnitude, such release may induce ‘reparative autophagocytosis’, causing additional accumulation of redox-active iron within the lysosomal compartment. We have used radio-resistant histiocytic lymphoma (J774) cells to assess the importance of intralysosomal iron and lysosomal rupture in radiation-induced cellular injury. We found that a 40 Gy radiation dose increased the ‘loose’ iron content of the (still viable) cells approx. 5-fold when assayed 24 h later. Cytochemical staining revealed that most redox-active iron was within the lysosomes. The increase of intralysosomal iron was associated with ‘reparative autophagocytosis’, and sensitized cells to lysosomal rupture and consequent apoptotic/necrotic death following a second, much lower dose of radiation (20 Gy) 24 h after the first one. A high-molecular-mass derivative of desferrioxamine, which specifically localizes intralysosomally following endocytic uptake, added to the culture medium before either the first or the second dose of radiation, stabilized lysosomes and largely prevented cell death. These observations may provide a biological rationale for fractionated radiation.
Keywords: apoptosis, ionizing radiation, iron chelator, lysosome, macrophage, oxidative stress
Abbreviations: AO, Acridine Orange; DMEM, Dulbecco's modified Eagle's medium; FBS, fetal bovine serum; HBSS, Hanks balanced salt solution; HMW-DFO, high-molecular-mass desferrioxamine mesylate; IR, ionizing radiation; pHPA, p-hydroxyphenylacetic acid; PI, propidium iodide; SSM, sulphide silver method; TCA, trichloroacetic acid; TEM, transmission electron microscopy
INTRODUCTION
Cells contain small amounts of ‘loose’ redox-active iron, which can promote the formation of the highly reactive hydroxyl radical (HO·), or similarly reactive iron-centred (ferryl and perferryl) radicals. These highly reactive radicals can attack almost all cellular constituents and create chain reactions that lead to cell death [1–4]. Although neither hydrogen peroxide (H2O2) nor superoxide (O2·−) is particularly reactive, in the presence of redox-active iron even low doses of these reactive oxygen species are cytotoxic. It has been recognized that lysosomes are responsible for the autophagic degradation of many iron-containing metalloproteins and iron-rich particulates, such as ferritin, various metalloproteins and mitochondria [5–7]. Elsewhere, we have argued that the resultant accumulation of redox-active iron within lysosomes predisposes them to oxidant-induced damage and rupture, followed by either apoptosis or necrosis, depending on the magnitude of lysosomal damage [8]. If so, the lysosomal content of redox-active iron may well be of great importance to the overall sensitivity of cells to oxidant challenge [8–14] and, as argued herein, IR (ionizing radiation).
In attempts to explain the cytotoxic effects of radiation damage, HO· has been repeatedly invoked as an important intermediate, because HO· forms by radiolysis of water. It is generally assumed that HO·-mediated DNA damage, particularly in S-phase cells, which are less able to repair DNA damage, is responsible for cell death caused by IR [15]. However, site-specific HO· formation due to iron-catalysed homolytic cleavage of H2O2, another product of radiolytic cleavage of water, may be significant, since it is well established that iron loading of target cells can greatly increase radiation-induced cell damage and death [16–19].
Exposure of cells to a dose of IR that is not immediately lethal engenders a wave of reparative autophagic activity, probably initiated by limited lysosomal rupture that is not sufficient to induce apoptosis or necrosis [8], during which damaged proteins and whole organelles are autophagocytosed and digested [20–23]. One very likely and potentially hazardous consequence of such digestion would be lysosomal accumulation of ‘loose’ redox-active iron when iron-containing metalloproteins are degraded. Consequently, the lysosomes of previously irradiated cells might accumulate excess iron, which could sensitize the lysosomes to attack by additional radiation events.
As a test of this general concept, we exposed radio-resistant lysosome-rich murine histiocytic lymphoma (J774) cells to a large (but, within the time-frame of the present study, non-lethal) dose of IR (40 Gy). This led to a substantial increase in intralysosomal redox-active iron after 24 h. When these cells (now containing iron-enriched lysosomes through the natural process of autophagic clearance of damaged organelles and iron-containing metalloproteins) were exposed to a second dose of radiation (20 Gy) 24 h after the first one, extensive, mainly apoptotic, cell death occurred secondary to lysosomal rupture. The importance of intralysosomal iron in this radiation sensitization was indicated by experiments employing HMW-DFO (high-molecular-mass desferrioxamine mesylate), a derivative of the iron-chelator desferrioxamine that exclusively localizes and remains within lysosomes following fluid-phase endocytosis [24,25]. Cells pre-treated with HMW-DFO before the second radiation event were powerfully protected against lethal effects, and even more so if cells were exposed to HMW-DFO before the first radiation event. Overall, our results indicate that intralysosomal redox-active iron interacts with IR to cause cell death, and provide a rational basis for the use of fractionated radiation.
MATERIALS AND METHODS
Chemicals
DMEM (Dulbecco's modified Eagle's medium), FBS (fetal bovine serum), penicillin and streptomycin were from Gibco BCL (Paisley, Renfrewshire, Scotland, U.K). HMW-DFO was kindly given from the company Biomedical Frontiers (Minneapolis, MN, U.S.A.). HMW-DFO has a molecular mass of ≈75 kDa, and the stock solution is 40 mM in DFO. PI (propidium iodide), Trypan Blue, TCA (trichloroacetic acid), pHPA (p-hydroxyphenylacetic acid) and Ferene-S [3-(2-pyridyl)-5,6-bis-[2-(5-furylsulphonic acid)]-1,2,4-triazine] were from Sigma Chemical Co. (St. Louis, MO, U.S.A.). AO (Acridine Orange) base was from Gurr (Poole, Dorset, U.K.) and the Amplex Red Hydrogen Peroxide/Peroxidase Assay Kit was from Molecular Probes (Eugene, OR, U.S.A.). Giemsa and sodium cacodylate (dimethylarsinic acid sodium salt) were from Merck (Darmstadt, Germany). Osmium tetroxide was from Johnson Matthey (Royston, Herts., U.K.), whereas glutaraldehyde and Epon-812 were purchased from Fluka AG (Buchs, Switzerland).
Cell culture
Lysosome-rich murine histiocytic lymphoma (J774) cells, well known to be resistant to IR [26], were cultured in DMEM, supplemented with 10% FBS, 100 units/ml penicillin and 100 μg/ml streptomycin at 37 °C in humidified air with 5% CO2. Cells were sub-cultivated twice a week.
IR and treatment with HMW-DFO
Before radiation exposure, cells were grown to confluence in 75 cm2 flasks, sub-cultivated in 35 mm Petri dishes [4×105 cells/2 ml of medium (≡50000 cells/cm2)] and kept for 6 h under standard culture conditions (37 °C; 5% CO2). Non-attached cells were removed by a change of medium immediately before radiation.
IR was performed at the Department of Radiation Oncology, University Hospital of Linköping, Sweden using a 4 MV linear accelerator (Varian Clinac 600C). Cells in complete culture medium were exposed in closed dishes to 10–80 Gy at 22 °C. Immediately following radiation, cells were returned to standard culture conditions for a further 24 h and then irradiated, or not, a second time. On the basis of initial trials, a first dose of 40 Gy and a second one of 20 Gy, both radiation events delivered as 2 Gy/min, were selected for the double irradiation experiments. After each dose of radiation, culture medium was replaced by new medium, and cells were followed under standard conditions for up to 64 h.
Optimal concentrations and exposure times for HMW-DFO were established in preliminary experiments. A stock solution of HMW-DFO in PBS, pH 7.4, was diluted to 2 mM HMW-DFO (expressed as DFO equivalents) in complete culture medium. Cells were exposed to this dose of HMW-DFO for 3 h once, either before the first 40 Gy or the second 20 Gy radiation events.
H2O2 assays
Petri dishes (35 mm-diameter) containing HBSS (Hanks balanced salt solution; 2 ml/dish) but no cells were irradiated with 2 Gy/min or 5 Gy/min to a total dose of 40 Gy, and H2O2 formation was measured using: (i) an Amplex Red Hydrogen Peroxide/Peroxidase Assay Kit according to the manufacturer's instructions, and (ii) the horseradish-peroxidase-mediated H2O2-dependent pHPA oxidant technique, as described previously [13]. HBSS was used instead of complete culture medium in order to avoid interference with any catalase possibly present in the serum-containing medium.
Cell viability assay
Cells were stained (5 min; 22 °C) with 0.2% (w/v) Trypan Blue in PBS and counted (103 cells/dish) in pre-selected areas using an inverted microscope (×250 magnification). Non-stained cells were considered viable, or in the early stages of apoptosis. Less than 3% of the control cells stained blue, and these were considered as being post-apoptotic necrotic (a normal value for J774 cells). After the first and second radiation exposure, non-stained cells/initial number of cells within pre-selected areas were determined. Since detached cells always stained blue (necrotic), this method gave accurate numbers for viable or early apoptotic (non-necrotic) cells.
Apoptosis assays
Using the hypotonic PI nuclear staining technique described by Nicoletti et al. [27] and Giemsa staining, the proportions of cells with fragmented apoptotic DNA and apoptotic morphology were evaluated respectively. Cells (pre-exposed to HMW-DFO or not), irradiated with 40 Gy, kept under standard culture conditions for a further 24 h and then exposed to HMW-DFO (or not; no cells were exposed to HMW-DFO more than once) were again irradiated with 20 Gy and returned to standard culture conditions until analysed (see above). Appropriate controls were also checked.
Briefly, cells for DNA analysis were suspended in a cell-rupturing PI solution [50 μg/ml in 0.1% sodium citrate with 0.2% (v/v) Triton X-100]. After incubation in the dark overnight at 4 °C, nuclei were analysed by flow cytofluorimetry. Red (FL3 channel) fluorescence was recorded in a logarithmic scale. CellQuest software was used for acquisition and analyses.
Cells fixed in 4% formaldehyde and stained in Giemsa, showing signs of apoptosis (cellular shrinkage and nuclear condensation), were counted and documented using a Nikon Microphot SA (Nikon, Tokyo, Japan) microscope equipped with a Hamamatsu C4742-95 (Bridgewater, NJ, U.S.A.) digital camera and Adobe Photoshop software.
Cytochemical assay of lysosomal reactive iron
For evaluation of intralysosomal iron we used ‘autometallography’ or the SSM (sulphide silver method), as described previously [6]. Briefly, cells seeded on coverslips were irradiated with 40 Gy (or not), medium was renewed, and cells were then returned to standard conditions. Cells were rinsed in PBS (22 °C) 24 h later, fixed in 2% glutaraldehyde in 0.1 M sodium cacodylate buffer with 0.1 M sucrose, pH 7.2, for 2 h at 22 °C, rinsed 5 times in glass-distilled water at 22 °C, and sulphidated for 15 min at 22 °C, pH ≈9, in a 70% (v/v) ethanol/1% (w/v) ammonium sulphide solution. Following careful rinsing in glass-distilled water for 10 min at 22 °C, cells were ‘developed’ using a physical colloid-protected developer containing silver lactate. The reaction was performed in the dark at 26 °C for 20, 30 and 40 min for all specimens (dependent on lysosomal amounts of low-molecular-mass iron, shorter or longer development is required for precipitation of silver). Following dehydration in a graded series of ethanol, cells were mounted in reducing Canada balsam (to avoid oxidation of atomic silver to white silver oxide), and examined by transmitted light in a Nikon Microphot SA microscope (no counterstaining). Results (precipitation of silver granules) were documented using a Hamamatsu C4742-95 digital camera and Adobe Photoshop software.
Chemical analysis of cellular ‘loose’ and total iron
Cells were irradiated with 40 Gy (or not), medium was renewed, and cells were returned to standard conditions. After 24 h, the cells were rinsed in PBS and a colorimetric assay for labile cellular iron was employed [28,29]. Following rinsing in PBS, the cells (2×106 cells/analysis) were precipitated in 0.5 ml of ice-cold 10% ultra-pure TCA, kept on ice for 60 min and mechanically scraped from the plate. The precipitates were centrifuged at 12000 g for 5 min, and 0.4 ml of the supernatant was added to 0.1 ml of sodium ascorbate (0.25 M). Following a further 5 min on ice, 0.4 ml of ammonium acetate (40%) and 0.1 ml of the ironspecific chromogenic reagent Ferene-S was added, and the Ferene-S–Fe2+ complex absorbance was measured at 594 nm [molar absorption coefficient (ϵ) of 35500 M−1·cm−1] using an Ultrospec 3000 UV/visible spectrophotometer (Cambridge, England).
Total iron was measured in cells lysed in Triton X-100 using a Z-8270 Polarized Zeeman atomic absorption spectrophotometer (Hitachi, Japan) equipped with an iron lamp (243.3 nm). Both the PBS and the water used for all solutions were treated with Chelex-100 to minimize adventitious iron.
Assays of lysosomal integrity
AO is a metachromatic fluorophore and a lysosomotropic base (pKa 10.3). It is retained in its charged form (AOH+) by proton trapping inside the acidic vacuolar compartment, preferentially in secondary lysosomes (pH 4–5). Cells (chelator-protected or not), which had been irradiated with 40 Gy and were then cultured for a further 24 h, were again irradiated with 20 Gy, and returned to standard culture conditions before analyses were evaluated with the AO-uptake technique [11,14,24]. Cells with a reduced number of intact AO-accumulating lysosomes (here termed ‘pale’ cells) were detected up to 10 h following the second radiation event. In other experiments, the AO relocation method [11,14,24] was applied. In this case, cells were stained with AO immediately before the second radiation event, and early lysosomal rupture, resulting in increased green cytoplasmic and nuclear fluorescence, was analysed for up to 3 h after start of radiation. Green (FL1 channel) and red (FL3 channel) fluorescence was recorded in a logarithmic scale using a BD LSR Flow Cytometer (Becton-Dickinson, Mountain View, CA, U.S.A.) equipped with a 488 nm exciting argon laser. CellQuest software was used for data acquisition and analyses.
Evaluation of ‘reparative autophagocytosis’
Cells, irradiated with 40 Gy, were kept under standard culture conditions for a further 24 h and then evaluated for ‘reparative autophagocytosis’. Changes in the volume of the acidic vacuolar compartment, reflecting ‘reparative autophagocytosis’, were estimated and compared with appropriate controls using flow cytofluorimetry of AO-stained cells, as described above. AO-induced red fluorescence intensity was considered proportional to the volume of the lysosomal compartment.
In other experiments employing TEM (transmission electron microscopy), similarly treated cells were fixed in situ in a cacodylate-buffered (0.1 M, pH 7.2) 2% glutaraldehyde solution with 0.1 M sucrose at 4 °C, post-fixed in 1% osmium tetroxide (in 0.15 M cacodylate buffer) at room temperature, stained en bloc with 2% uranyl acetate in 50% ethanol, dehydrated in a graded series of ethanol, and embedded in Epon-812. Ultrathin sections were cut with a diamond knife parallel to the growth substrate, stained with lead citrate, and examined in a JEOL 1200-EX electron microscope (Tokyo, Japan) operated at 80 kV. Micrographs were taken randomly from different sections of two cultures for each experimental condition. Electron microscopy images were photographed.
Statistical analysis
Results are presented as means±S.D. Statistical comparisons were made using ANOVA. *P<0.05; **P<0.01; ***P<0.001.
RESULTS
Ionizing radiation sensitizes cells to a second round of radiation
Within 64 h following a dose of 40 Gy (2 Gy/min), almost no cell death had yet occurred, whether the cells were protected by HMW-DFO or not (Figure 1). Proliferation, however, was diminished in a dose-dependent fashion, and was fully inhibited for at least 44 h by 40 Gy. At a higher dose of 80 Gy, approx. 20–30% of unprotected cells died within 36 h following radiation (results not shown).
Figure 1. Cell viability following one or two radiation events (40+20 Gy; 2 Gy/min) to cells protected or not by exposure to the iron chelator HMW-DFO.

Viability was assessed using Trypan Blue (TB) exclusion. Cells were initially irradiated with 40 Gy. Following renewal of the medium, cells were kept under standard culture conditions for a further 24 h and then, in some cases, irradiated a second time with 20 Gy. Following renewal of the medium, cells were then followed for a further 40 h under standard culture conditions. Pre-exposure to HMW-DFO (2 mM for 3 h) was before the first or the second radiation event. Values shown represent means±1 S.D., (n=4). Pairwise comparisons were made using ANOVA. All groups were significantly different from the 40 Gy+20 Gy group from 48 h after the first radiation (24 h after the second radiation).
Unprotected cultures, irradiated twice (40 Gy+20 Gy; 24 h between the events), contained approx. 50% viable (or early apoptotic) cells (i.e. not stained by Trypan Blue) 24 h after the second radiation event (Figure 1). This loss of viability appeared to involve intralysosomal redox-active iron inasmuch as cells initially irradiated with 40 Gy and then exposed to HMW-DFO before the second radiation event showed significantly increased viability compared with unprotected cells when analysed 24 h later (Figure 1). Cells already protected before the first event of radiation (HMW-DFO+40 Gy+20 Gy) did not demonstrate any cell death within 36 h following the second radiation event. Since HMW-DFO is not degradable, it would have been present intralysosomally during both radiation events in this case. Furthermore, cells protected with HMW-DFO before a 40 Gy dose, or unprotected cells irradiated with the same radiation dosage (HMW-DFO+40 Gy or 40 Gy respectively), showed only insignificant cell death, whereas proliferation returned partially after the end of the inhibition period. Owing to induced iron starvation, HMW-DFO by itself caused inhibition of proliferation and eventually apoptotic cell death, although this occurred beyond the time period indicated in Figure 1.
IR induces ‘reparative autophagocytosis’ and increases intracellular ‘loose’ iron, mainly intralysosomally
As revealed by light microscopy, morphological changes following radiation, i.e. spreading and vacuolization, started to be apparent in unprotected cells 8 h after the 40 Gy radiation event, and were obvious at 24 h, whereas the morphology remained almost normal in cells exposed to HMW-DFO before radiation. Because HMW-DFO ends up intralysosomally, this observation supports our earlier suggestion that radiation damage may be promoted by intralysosomal iron [16,17].
TEM revealed prominent ‘reparative autophagocytosis’ following radiation of unprotected cells with 40 Gy (Figure 2). This was accompanied by an expansion of the lysosomal compartment as reflected by an approx. 50% increase in AO uptake by unprotected irradiated cells as compared with control and HMW-DFO-protected cells (Figure 3). The efficient, although not complete, prevention of radiation-induced ‘reparative autophagocytosis’ by the intralysosomal presence of HMW-DFO strongly supports the notion that this phenomenon is mainly secondary to intralysosomal iron-catalysed peroxidative damage to lysosomal membranes with ensuing leakage of harmful lysosomal contents into the cytosol, in turn leading to ‘reparative autophagocytosis’.
Figure 2. Radiation-induced ‘reparative autophagocytosis’ evaluated using TEM.

Cells irradiated with 40 Gy (2 Gy/min), and then kept under standard culture conditions for a further 24 h, were prepared for TEM. One micrograph, as a representative of randomly taken micrographs from two experiments, shows cells with signs of ‘reparative autophagocytosis’ following radiation. Autophagic material is shown by the arrows.
Figure 3. Changes in the volume of the acidic vacuolar compartment, as an indication of radiation-induced ‘reparative autophagocytosis’, estimated by flow cytofluorimetry.

AO-induced red fluorescence is proportional to the volume of the lysosomal compartment. Radiation (40 Gy; 2 Gy/min) of unprotected cells was accompanied by an approx. 50% expansion of the lysosomal compartment, whereas HMW-DFO-protected irradiated cells showed significantly less expansion of the AO-stained lysosomal compartment.
In order to investigate possible changes in reactive intralysosomal iron following radiation-induced ‘reparative autophagocytosis’, we employed the SSM. This is an extremely sensitive semiquantitative technique, which can be used to cytochemically demonstrate the presence of loosely bound, reactive iron and several other heavy transition metals, such as copper [6]. The SSM is based on a catalytic reaction, which causes metal-sulphide-dependent precipitation of metallic silver in a time-dependent fashion. Normal cells showed distinct, lysosome-sized granular silver precipitations (Figure 4A), which were much more pronounced in irradiated non-protected cells (Figure 4B), indicating considerably increased amounts of intralysosomal redox-active iron.
Figure 4. Cytochemical demonstration of iron by the SSM (autometallography).

(A) Non-irradiated, control cells. (B) Unprotected cells irradiated with 40 Gy (2 Gy/min) and then kept under standard culture conditions for a further 24 h. Note lysosomal-sized silver precipitates which are larger and more numerous following radiation (developing time of 30 min for both groups).
As would be expected from the cytochemical analyses described above, irradiated cells showed a massive increase (approx. 500%) in intracellular ‘loose’ iron following a 40 Gy radiation event compared with control non-irradiated cells (Table 1). In contrast, total cellular iron was moderately decreased (approx. 30%) in unprotected cells following a 40 Gy radiation. In cultures pre-treated with HMW-DFO, total iron levels were unaffected by the 40 Gy radiation, although ‘loose’ iron could not be determined, since HMW-DFO interferes with the assay.
Table 1. Control cells and cells pre-treated with the iron-chelator HMW-DFO (2 mM; 3 h) were irradiated (or not) with 40 Gy.
Following renewal of medium, cells were kept under standard culture conditions for a further 24 h, rinsed and analysed for ‘loose’ and total cellular iron, as described in the Materials and methods section (n=3 experiments). ‘Loose’ iron could not be determined on protected cells, since the iron chelator interferes with the assay. Significant differences compared with non-irradiated control cells are indicated (*P<0.05, ***P<0.001).
| Non-irradiated cells | Irradiated cells (40 Gy) | |||
|---|---|---|---|---|
| Assay (nmol of Fe/106 cells) | Control | HMW-DFO | Control | HMW-DFO |
| ‘Loose’ cellular Fe | 0.52±0.20 | Not assayed | 2.65±0.13*** | Not assayed |
| Total cellular Fe | 17.99±1.32 | 16.83±0.51 | 12.28±0.30* | 16.52±0.17 |
HMW-DFO stabilizes lysosomal membranes against radiation and prevents apoptosis
A significant loss of lysosomal integrity was observed (using the AO relocation technique) in non-protected cells 3 h after the start of the second (20 Gy) radiation event [given 24 h after the initial (40 Gy) irradiation] (Figure 5A), whereas HMW-DFO endocytosed by the cells before the second radiation event significantly stabilized lysosomes. The preservation of lysosomes by chelation of lysosomal iron was confirmed further using the AO uptake method, by which the proportion of ‘pale’ cells, i.e. cells with a reduced number of intact lysosomes, was evaluated at 10 h after the second radiation (Figure 5B).
Figure 5. Lysosomal integrity following various courses of radiation in the presence and absence of HMW-DFO.

Assays were performed using flow cytofluorimetry. (A) Cells were loaded with AO immediately prior to the second radiation event (AO-relocation test), and green fluorescence (representing the release of previously intralysosomal AO into the cell cytoplasm) is shown, expressed as the percentage of control mean values. Progressive lysosomal rupture after the second (40+20 Gy; 2 Gy/min) radiation exposure is not seen in cells protected with HMW-DFO. (B) Cells were loaded with AO 10 h following radiation (AO-uptake method), and the percentage of ‘pale’ cells (i.e. cells with a reduced number of intact lysosomes) was analysed. Note the marked loss of intact lysosomes in cells irradiated twice (40+20 Gy; 2 Gy/min) in the absence of HMW-DFO, and improved lysosomal integrity in chelator-treated cells. Values are means±1 S.D. (n≥3). Pairwise comparisons were made using ANOVA. Significant differences are indicated (*P<0.05; **P<0.01).
No signs of apoptosis were detected 24 h following the first radiation event (40 Gy), regardless of whether cells were protected or not. In contrast, apoptosis appraised 14 h after the second radiation event as the proportion of nuclei with hypo-diploid DNA (according to the method of Nicoletti et al. [27]), in combination with an evaluation of apoptotic morphology in Giemsa-stained cells, demonstrated a marked increase in apoptotic cells (Figures 6A and 6B). Importantly, HMW-DFO significantly reduced this short-term apoptosis.
Figure 6. Apoptosis following varying courses of radiation in the presence and absence of HMW-DFO.

Control and chelator-protected cells were radiated as indicated, and the frequency of apoptosis was assayed 14 h following the second (40+20 Gy; 2 Gy/min) radiation event. (A) Flow-cytofluorimetric analysis of hypo-diploid apoptotic DNA following PI staining. (B) Fraction of cells with apoptotic morphology (assessed in Giemsa-stained preparations). Values are means±1 S.D. (n≥3). Pairwise comparisons were made using ANOVA. Significant differences are indicated (*P<0.05; **P<0.01).
Formation of H2O2 during radiation
It is generally considered that radiation-induced cell damages are events secondary to the primary formation of the highly reactive HO· radical following radiolytic cleavage of water. In order to test the hypothesis that significant amounts of H2O2 are also formed during radiation, giving rise to intralysosomal Fenton-type chemistry and lysosomal rupture, we assayed formation of H2O2 in HBSS (no cells present), thus reflecting radiation-induced generation all over the cells. Regardless of the dosage rate (2 or 5 Gy/min), radiation up to a total dosage of 40 Gy generated H2O2 to a final concentration of approx. 15 μM (Figure 7).
Figure 7. Formation of radiation-induced extracellular H2O2.

Petri dishes (35 mm-diameter) containing 2 ml of HBSS/dish (no cells) were irradiated up to a total dose of 40 Gy using a dosage rate of either 2 or 5 Gy/min. Formation of H2O2 was assayed using the Amplex Red Hydrogen/Peroxidase Assay Kit and the horseradish-peroxidase-mediated H2O2-dependent pHPA oxidant technique. Both methods gave identical results. A single representative experiment is shown.
DISCUSSION
It is generally considered that the cytotoxic and cytocidal effects of IR arise by radiolysis of water with random intracellular production of HO· and attendant DNA damage, particularly to actively dividing cells. In this case, the rationale for the use of fractionated radiation would be based on the assumption that the second dose would be more likely to affect a new population of cells entering S-phase, a point at which actively dividing DNA may be more at risk of lethal double-strand breaks [15]. However, additional mechanisms, and cytoplasmic ionization events in particular, may also be important, which in turn may secondarily influence nuclear DNA [30].
IR not only generates HO· by radiolysis of water (reaction 1), but also generates O2·− and H2O2 (reactions 2 and 3):
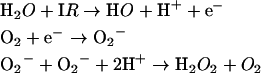 |
1 |
The objective of the present study was to test the hypothesis that IR, by inducing intracellular oxidative stress, causes intralysosomal Fenton-type peroxidation with ensuing lysosomal rupture, and relocation of lysosomal enzymes and low-mass redox-active iron. Such relocation, in turn, may induce apoptosis and site-specific iron-mediated DNA damage [8,12,14,25,31,32]. In order to obtain clear results after one or two IR sessions on cells in culture, we used a histiocytotic lysosome-rich type of cell and high IR doses.
The O2·− and H2O2 species produced during radiation would be specifically harmful in the event that it occurs in relation to targets that contain reactive transition metals, such as iron. Since lysosomes appear to contain the major proportion of intracellular redox-active iron [13,14,24,25,31], the diffusion of H2O2 into, or the direct formation of H2O2 within, that compartment would give rise to iron-mediated oxidative reactions that may rupture lysosomes. Since lysosomes do not contain H2O2-splitting enzymes, even low interior concentrations of O2·− and H2O2 are expected to generate abundant HO·, if the lysosomes are rich in redox-active iron. Our findings of H2O2 formation in an irradiated salt solution would suggest that substantial amounts of this oxidant are generated within the lysosomal compartment during a radiation dose of 40 Gy (Figure 7). Indeed, concentrations of cytosolic H2O2 from 0.7 to 3 μM, corresponding to approx. 2–20 μM extracellular H2O2 under steady-state conditions, and even higher concentrations following the addition of a bolus dose of H2O2, are sufficient to induce lysosomal rupture and ensuing apoptosis in Jurkat T-cells [33,34]. IR will induce H2O2 formation anywhere in the cell, and since intralysosomal degradation might be slight, oxidative stress within this compartment would be expected to be prominent, even considering a rapid diffusion of H2O2 into the surrounding cytosol. In the case that the lysosomal compartment contains substantial amounts of redox-active iron, e.g. after pronounced ‘reparative autophagocytosis’ with degradation of iron-containing metalloproteins, oxidative damage and ensuing permeabilization of lysosomal surrounding membranes should be substantial.
We have argued elsewhere that the destabilization of lysosomes (especially by oxidants), and the consequent release of lysosomal hydrolases and redox-active iron, may be an important intermediate step in ultimate apoptotic or necrotic cell death [8–14,25,31,35]. We therefore sought to determine whether lysosomes and, especially, intralysosomal redox-active iron might play a role in cell damage secondary to IR. Indeed, our previous studies [9,16,17] and another one published recently [36] support the concept that lysosomal damage may occur during radiation-induced damage. In the present study, we exposed radio-resistant histiocytic lymphoma (J774) cells to two separate challenges of radiation in the presence or absence of HMW-DFO localized intralysosomally. HMW-DFO is a high-molecular-mass neutral polymer that is undegradable by lysosomal enzymes, and thus remains permanently inside the lysosomal compartment after endocytic uptake [25]. Unfortunately, this high-affinity iron chelator acts as a ‘sink’ for cellular iron, most of which circulates through the lysosomal compartment because of autophagocytotic turnover of iron-containing biomolecules [14,24], eventually causing apoptotic cell death from iron starvation [25]. Therefore long-term effects on cellular proliferation are impossible to assess in cells treated with this agent.
The initial high-dose radiation event (40 Gy; 2 Gy/min) caused significant ‘reparative autophagocytosis’, stopped cell proliferation for approx. 44 h, but did not cause significant cell death within 64 h, regardless of whether cells were exposed to HMW-DFO or not. The same effects on cell viability and proliferation were found following 20 Gy of radiation delivered as 5 Gy/min. These findings are almost identical with those of Gallin et al. [26]. Considering that a lower dose rate requires longer radiation time for the same formation of H2O2, and that a continuous degradation of H2O2 takes place intracellularly, the dose rate would influence the H2O2-mediated toxicity of a given dose and explain the almost equal cytotoxicity of a 20 Gy radiation event, administered as 5 Gy/min, and a 40 Gy radiation event delivered as 2 Gy/min.
Although an initial 40 Gy (2 Gy/min) radiation event did not cause significant cell death, but considerable ‘reparative autophagocytosis’, a second lower dose (20 Gy; 2 Gy/min) administered 24 h after the first one induced significant loss of lysosomal membrane integrity and ensuing apoptotic cell death. This greatly enhanced susceptibility to the second dose of radiation was accompanied by a marked increase in the amounts of redox-active ‘loose’ iron detected within lysosomes cytochemically and in cell extracts by direct analysis. This increased reactive iron appears to be important in the sensitization of target cells to the second dose of radiation, because the exposure to HMW-DFO, targeted to the lysosomal apparatus, provided significant (although not complete) protection against the second radiation exposure. Overall, these results support the idea that iron-catalysed lysosomal rupture may be an important consequence of radiation-induced oxidative stress.
It might be argued that the remarkable cytoprotective effect of HMW-DFO against the second round of radiation could arise from a secondary effect on the cellular pool of labile iron, leading, for example, to inhibition of ribonucleotide reductase and, consequently, forcing the cells into the G0/G1 phase of the cell cycle [3]. This possibility, however, was excluded by the finding that cells pre-treated with HMW-DFO demonstrated the same cell cycle distribution as control cells prior to the second event of radiation (results not shown).
The remarkable increase in intralysosomal labile iron seems to result from ‘reparative autophagocytosis’ secondary to radiation-induced lysosomal damage, with release of lytic enzymes and low-mass iron and consequent cellular damage. Such ‘reparative autophagocytosis’ enhances degradation of various iron-containing structures, such as ferritin, mitochondria and iron-containing metalloproteins [16,17,20,21]. The occurrence of ‘reparative autophagocytosis’ was evident both from electron microscopy and from a significant expansion of the volume of the acidic vacuolar compartment. The observed moderate decrease in total cellular iron that paralleled the striking increase in labile iron could result from release of low-mass iron from the cells. This might reflect a temporary lack of intracellular iron-storing capacity, arising from the increased amounts of intracellular ‘loose’ iron. Because HMW-DFO given prior to the initial 40 Gy (2 Gy/min) radiation event efficiently prevented lysosomal rupture promoted by intralysosomal iron-catalysed oxidative reactions following either diffusion of H2O2 into the lysosomes or formation within the lysosomes of O2·− and H2O2, intralysosomal oxidation and destabilization, rather than random intracellular formation of HO· by radiolytic cleavage of water, seems to be the major event inducing ‘reparative autophagocytosis’. It should be noted that even small amounts of H2O2 formed intralysosomally should induce substantial Fenton-type chemistry. Although a single radiation event with 40 Gy (2 Gy/min) induced repair in our cell culture system, it did not kill the cells within the period of time studied. However, a second lower dose of radiation administered when ‘reparative autophagocytosis’ had resulted in increased intralysosomal low mass iron caused marked lysosomal labilization and cell death.
The results of the present study support the concept that IR-mediated cell damage is, in vitro and perhaps in vivo, a consequence of intralysosomal iron-catalysed oxidative processes leading to lysosomal rupture with release of hydrolytic enzymes and redox-active iron. Released lysosomal redox-active iron may partly relocate to nuclear and mitochondrial DNA, causing site-specific HO· production in the presence of oxidative stress [25,37]. Such site-specific HO· induction would be much more powerful with respect to DNA damage than random formation of HO· due to radiolysis of water.
In the use of fractionated IR, it is typically assumed that each successive dose results in equivalent cell killing. However, there are isolated reports that this may not always be so. For example, Scott et al. [38] have recently reported that two of three prostate cancer lines studied had significantly lower-than-predicted survival rates upon exposure to secondary doses of IR. Although these authors focused on the possible importance of changes in the cell cycle, the reasons for this effect are not completely clear. Here, by exposing radio-resistant murine histiocytic lymphoma (J774) cells to a very high dose of IR (40 Gy), we show that irradiated cells are sensitized to killing by a much lower dose (20 Gy) administered 24 h later. Our results have led us to the tentative conclusion that this sensitization to a secondary dose of IR probably arises from an increase in the amounts of lysosomal redox-active iron. These results may provide a rationale for the use of fractionated radiation, and also raise the possibility of using appropriately targeted iron chelators to minimize radiation damage to normal tissues.
Acknowledgments
We thank Ms Lotta Jonsson for help with cell culture radiation, and Professor Helge Dalen for the TEM studies. These studies were supported by the Linköping University Hospital Research Funds and funding from the Swedish Society of Physicians (Svenska Läkarsällskapet) (to H.L.P.), Swedish Cancer Foundation grant no. 4296 (to U.T.B.), and the National Institutes of Health (grant DK58882), funding from the Kentucky Lung Cancer Research Program, and the Commonwealth of Kentucky Research Challenge Trust Fund (to J.W.E.). H.L.P. and J.W.E. were recipients of a Postdoctoral Research Fellowship and a Visiting Professorship, respectively, from the Linköping University Hospital.
References
- 1.Meneghini R. Iron homeostasis, oxidative stress, and DNA damage. Free Radical Biol. Med. 1997;23:783–792. doi: 10.1016/s0891-5849(97)00016-6. [DOI] [PubMed] [Google Scholar]
- 2.Lipinski P., Drapier J. C., Oliveira L., Retmanska H., Sochanowicz B., Kruszewski M. Intracellular iron status as a hallmark of mammalian cell susceptibility to oxidative stress: a study of L5178Y mouse lymphoma cell lines differentially sensitive to H2O2. Blood. 2000;95:2960–2966. [PubMed] [Google Scholar]
- 3.Kakhlon O., Cabantchik Z. I. The labile iron pool: characterization, measurement, and participation in cellular processes. Free Radical Biol. Med. 2002;33:1037–1046. doi: 10.1016/s0891-5849(02)01006-7. [DOI] [PubMed] [Google Scholar]
- 4.Richardson D. R., Ponka P. The molecular mechanisms of the metabolism and transport of iron in normal and neoplastic cells. Biochim. Biophys. Acta. 1997;1331:1–40. doi: 10.1016/s0304-4157(96)00014-7. [DOI] [PubMed] [Google Scholar]
- 5.Brun A., Brunk U. Histochemical indications for lysosomal localization of heavy metals in normal rat brain and liver. J. Histochem. Cytochem. 1970;18:820–827. doi: 10.1177/18.11.820. [DOI] [PubMed] [Google Scholar]
- 6.Zdolsek J. M., Roberg K., Brunk U. T. Visualization of iron in cultured macrophages: a cytochemical light and electron microscopic study using autometallography. Free Radical Biol. Med. 1993;15:1–11. doi: 10.1016/0891-5849(93)90120-j. [DOI] [PubMed] [Google Scholar]
- 7.Petrat F., de Groot H., Rauen U. Subcellular distribution of chelatable iron: a laser scanning microscopic study in isolated hepatocytes and liver endothelial cells. Biochem. J. 2001;356:61–69. doi: 10.1042/0264-6021:3560061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brunk U. T., Neuzil J., Eaton J. W. Lysosomal involvement in apoptosis. Redox Rep. 2001;6:91–97. doi: 10.1179/135100001101536094. [DOI] [PubMed] [Google Scholar]
- 9.Abok K., Hirth T., Ericsson J. L., Brunk U. Effect of iron on the stability of macrophage lysosomes. Virchows Arch. B. Cell Pathol. Incl. Mol. Pathol. 1983;43:85–101. doi: 10.1007/BF02932947. [DOI] [PubMed] [Google Scholar]
- 10.Sakaida I., Kyle M. E., Farber J. L. Autophagic degradation of protein generates a pool of ferric iron required for the killing of cultured hepatocytes by an oxidative stress. Mol. Pharmacol. 1990;37:435–442. [PubMed] [Google Scholar]
- 11.Zdolsek J., Zhang H., Roberg K., Brunk U. H2O2-mediated damage to lysosomal membranes of J-774 cells. Free Radical Res. Commun. 1993;18:71–85. doi: 10.3109/10715769309147344. [DOI] [PubMed] [Google Scholar]
- 12.Öllinger K., Brunk U. T. Cellular injury induced by oxidative stress is mediated through lysosomal damage. Free Radical Biol. Med. 1995;19:565–574. doi: 10.1016/0891-5849(95)00062-3. [DOI] [PubMed] [Google Scholar]
- 13.Persson H. L., Nilsson K. J., Brunk U. T. Novel cellular defenses against iron and oxidation: ferritin and autophagocytosis preserve lysosomal stability in airway epithelium. Redox Rep. 2001;6:57–63. doi: 10.1179/135100001101536049. [DOI] [PubMed] [Google Scholar]
- 14.Yu Z., Persson H. L., Eaton J. W., Brunk U. T. Intralysosomal iron: a major determinant of oxidant-induced cell death. Free Radical Biol. Med. 2003;34:1243–1252. doi: 10.1016/s0891-5849(03)00109-6. [DOI] [PubMed] [Google Scholar]
- 15.Scherer E., Streffer C., Trott K.-R. New York: Springer-Verlag; 1991. Radiopathology of Organs and Tissues; pp. 1–404. [Google Scholar]
- 16.Abok K., Blomquist E., Ericsson J., Brunk U. Macrophage radiosensitivity in culture as a function of exposure to ionic iron. Virchows Arch. B. 1983;42:119–129. doi: 10.1007/BF02890375. [DOI] [PubMed] [Google Scholar]
- 17.Abok K., Rundquist I., Forsberg B., Brunk U. Dimethylsulfoxide increases the survival and lysosomal stability of mouse peritoneal macrophages exposed to low-LET ionizing radiation and/or ionic iron in culture. Virchows Arch. B. Cell Pathol. Incl. Mol. Pathol. 1984;46:307–320. doi: 10.1007/BF02890319. [DOI] [PubMed] [Google Scholar]
- 18.Nelson J. M., Stevens R. G. Ferritin-iron increases killing of Chinese hamster ovary cells by X-irradiation. Cell Prolif. 1992;25:579–585. doi: 10.1111/j.1365-2184.1992.tb01461.x. [DOI] [PubMed] [Google Scholar]
- 19.Vile G. F., Tyrrell R. M. UVA radiation-induced oxidative damage to lipids and proteins in vitro and in human skin fibroblasts is dependent on iron and singlet oxygen. Free Radical Biol. Med. 1995;18:721–730. doi: 10.1016/0891-5849(94)00192-m. [DOI] [PubMed] [Google Scholar]
- 20.Hamberg H., Brunk U., Ericsson J. L., Jung B. Cytoplasmic effects of X-irradiation on cultured cells 2. Alterations in lysosomes, plasma membrane, Golgi apparatus, and related structures. Acta Pathol. Microbiol. Scand. 1977;85:625–639. [PubMed] [Google Scholar]
- 21.Hamberg H. Cellular autophagocytosis induced by X-irradiation and vinblastine. On the origin of the segregating membranes. Acta Pathol. Microbiol. Immunol. Scand. 1983;91:317–327. doi: 10.1111/j.1699-0463.1983.tb02762.x. [DOI] [PubMed] [Google Scholar]
- 22.Piao Y. J., Ogawa K., Ono K., Abe M. The relationship between heterophagy and autophagy in the splenic macrophage of rats after gamma-ray irradiation. Acta Histochem. Cytochem. 1983;16:353–367. [Google Scholar]
- 23.Telbisz A., Kovacs A. L., Somosy Z. Influence of X-ray on the autophagic-lysosomal system in rat pancreatic acini. Micron. 2002;33:143–151. doi: 10.1016/s0968-4328(01)00005-1. [DOI] [PubMed] [Google Scholar]
- 24.Persson H. L., Yu Z., Tirosh O., Eaton J. W., Brunk U. T. Prevention of oxidant-induced cell death by lysosomotropic iron chelators. Free Radical Biol. Med. 2003;34:1295–1305. doi: 10.1016/s0891-5849(03)00106-0. [DOI] [PubMed] [Google Scholar]
- 25.Kurz T., Leake A., von Zglinicki T., Brunk U. T. Relocalized redox-active lysosomal iron is an important mediator of oxidative-stress-induced DNA damage. Biochem. J. 2004;378:1039–1045. doi: 10.1042/BJ20031029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gallin E. K., Green S. W., Sheehy P. A. Enhanced activity of the macrophage-like cell line J774.1 following exposure to gamma radiation. J. Leukocyte Biol. 1985;38:369–381. doi: 10.1002/jlb.38.3.369. [DOI] [PubMed] [Google Scholar]
- 27.Nicoletti I., Migliorati G., Pagliacci M. C., Grignani F., Riccardi C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods. 1991;139:271–279. doi: 10.1016/0022-1759(91)90198-o. [DOI] [PubMed] [Google Scholar]
- 28.Artiss J. D., Vinogradov S., Zak B. Spectrophotometric study of several sensitive reagents for serum iron. Clin. Biochem. 1981;14:311–315. doi: 10.1016/s0009-9120(81)91065-1. [DOI] [PubMed] [Google Scholar]
- 29.Qian M. W., Eaton J. W. Tobacco-borne siderophoric activity. Arch. Biochem. Biophys. 1989;275:280–288. doi: 10.1016/0003-9861(89)90374-3. [DOI] [PubMed] [Google Scholar]
- 30.Mikkelsen R. B., Wardman P. Biological chemistry of reactive oxygen and nitrogen and radiation-induced signal transduction mechanisms. Oncogene. 2003;22:5734–5754. doi: 10.1038/sj.onc.1206663. [DOI] [PubMed] [Google Scholar]
- 31.Doulias P. T., Christoforidis S., Brunk U. T., Galaris D. Endosomal and lysosomal effects of desferrioxamine: protection of HeLa cells from hydrogen peroxide-induced DNA damage and induction of cell-cycle arrest. Free Radical Biol. Med. 2003;35:719–728. doi: 10.1016/s0891-5849(03)00396-4. [DOI] [PubMed] [Google Scholar]
- 32.Nakagami Y., Ito M., Hara T., Inoue T., Matsubara S. Nuclear translocation of DNase II and acid phosphatase during radiation-induced apoptosis in HL60 cells. Acta Oncol. 2003;42:227–236. doi: 10.1080/02841860310010745. [DOI] [PubMed] [Google Scholar]
- 33.Antunes F., Cadenas E. Cellular titration of apoptosis with steady state concentrations of H2O2: submicromolar levels of H2O2 induce apoptosis through Fenton chemistry independent of the cellular thiol state. Free Radical Biol. Med. 2001;30:1008–1018. doi: 10.1016/s0891-5849(01)00493-2. [DOI] [PubMed] [Google Scholar]
- 34.Antunes F., Cadenas E., Brunk U. T. Apoptosis induced by exposure to a low steady-state concentration of H2O2 is a consequence of lysosomal rupture. Biochem. J. 2001;356:549–555. doi: 10.1042/0264-6021:3560549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu Z., Eaton J. W., Persson H. L. The radioprotective agent, amifostine, suppresses the reactivity of intralysosomal iron. Redox Rep. 2003;8:347–355. doi: 10.1179/135100003225003384. [DOI] [PubMed] [Google Scholar]
- 36.Ogawa Y., Kobayashi T., Nishioka A., Kariya S., Ohnishi T., Hamasato S., Seguchi H., Yoshida S. Reactive oxygen species-producing site in radiation-induced apoptosis of human peripheral T cells: involvement of lysosomal membrane destabilization. Int. J. Mol. Med. 2004;13:69–73. [PubMed] [Google Scholar]
- 37.Tenopoulou M., Doulias P. T., Barbouti A., Brunk U. T., Galaris D. The role of compartmentalized redox-active iron on hydrogen peroxide-induced DNA damage and apoptosis. Biochem. J. 2004;387:703–710. doi: 10.1042/BJ20041650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scott S. L., Gumerlock P. H., Beckett L., Li Y., Goldberg Z. Survival and cell cycle kinetics of human prostate cancer cell lines after single- and multifraction exposures to ionizing radiation. Int. J. Radiat. Oncol. Biol. Phys. 2004;59:219–227. doi: 10.1016/j.ijrobp.2004.01.027. [DOI] [PubMed] [Google Scholar]