

Abstract
Dynamic programming algorithms guarantee to find the optimal alignment between two sequences. For more than a few sequences, exact algorithms become computationally impractical, and progressive algorithms iterating pairwise alignments are widely used. These heuristic methods have a serious drawback because pairwise algorithms do not differentiate insertions from deletions and end up penalizing single insertion events multiple times. Such an unrealistically high penalty for insertions typically results in overmatching of sequences and an underestimation of the number of insertion events. We describe a modification of the traditional alignment algorithm that can distinguish insertion from deletion and avoid repeated penalization of insertions and illustrate this method with a pair hidden Markov model that uses an evolutionary scoring function. In comparison with a traditional progressive alignment method, our algorithm infers a greater number of insertion events and creates gaps that are phylogenetically consistent but spatially less concentrated. Our results suggest that some insertion/deletion “hot spots” may actually be artifacts of traditional alignment algorithms.
Keywords: insertion/deletion, progressive algorithm, sequence alignment
Sequence alignment is a central tool in molecular biology. High sequence similarity between a pair of molecules usually implies significant structural and functional similarities, such that information on a known molecule can often be assigned to an unknown molecule that shows high sequence conservation in a pairwise alignment. Also, related molecules represent semi-independent realizations of an evolutionary process and possess information regarding the structural constraints enabling the element to maintain its function. The reconstruction of the evolutionary history of a set of molecules requires an assessment of homology among their characters, i.e., a multiple alignment.
Pairwise and Multiple Alignment
The comparison of two biological sequences closely resembles the edit transcript problem in computer science (1), although biologists traditionally focus more on the product than the process and call the result an alignment. The first dynamic programming algorithm for pairwise alignment of biological sequences was described by Needleman and Wunsch (2), and modifications reducing its time complexity from O(L3) to O(L2) (where L is the sequence length) soon followed (see ref. 3 for a review). In real life, insertion/deletion (indel) events affect sequence regions of very different lengths, and the early methods' gap costs (proportional to gap length) were unsatisfactory: the gap cost is either so high that long gaps never appear or so low that the alignment gets fragmented by numerous small-length gaps. An elegant O(3L2)-complexity solution was proposed by Gotoh (4) by the separation of the gap opening and gap extension costs (leading to so-called affine gap scores). Importantly, by using Hirschberg's divide-and-conquer recursion (5, 6), these algorithms can all be implemented in memory that grows linearly with L at the cost of only doubling their computation time.
Although multiple sequence alignment can be considered a generalization of the pairwise alignment problem, it is much harder both conceptually and computationally. A set of homologous biological sequences has been produced through an evolutionary process, and when considering more than three sequences the multiple alignment should pay regard to the phylogenetic tree connecting the sequences. The tree alignment problem can be solved in exponential time using dynamic programming (7), but this solution is inadequate for most biological problems, and practical applications rely on the use of heuristic algorithms (8–10). Many of these methods are variants of iterated pairwise alignment (11), also called progressive multiple alignment, and perform pairwise alignments according to a guide tree (9, 12, 13).
Progressive Alignment and Insertions
In a pairwise alignment, an insertion is not distinguished from a deletion, but in a progressive multiple alignment the two events are drastically different: deletions are naturally penalized only at the place where they occur, whereas insertions have to be handled in every alignment performed between their original occurrence and the root of the guide tree (Fig. 1). The penalty for each insertion event should, however, be incurred only once.
Fig. 1.

Alignment of sequences with insertions. (A)(Top) Example of four sequences undergoing insertion events (boxed). (Middle) Alignments at nodes x and y, with flags (•) for preexisting gaps (compared with ○, indicating nongapped positions), are shown on top and left of the matrix, respectively. Their alignment (x, y, and m for insertions in upper and lower subtrees, and a match, respectively) is depicted in gray, with steps penalized multiple times in light gray. The gaps skipped over without repeated penalty by our method are shown by an arrow. (Bottom) In practice, the dynamic programming is performed with a stack of matrices among which some moves, indicated by light gray arrows and squares, are without penalty. The matrices in front, middle, and back correspond to standard moves, moves skipping over gaps in sequence y (vertical), and moves skipping over gaps in sequence x (horizontal), respectively. A flagged two-base insertion in the subtree to the right of x is skipped over, and the alignment continues with an insertion in edge 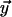 leading from z to y. The example corresponds to a simple linear gap cost algorithm with two storage matrices, and only one vector of flags is shown because x and y do not have grandchild sequences. (B) Here, we illustrate a special case where independent insertions have occurred at the same site in both subtrees. (C) Four sequences are aligned by applying our gap-flagging approach. In the sequences for nodes a, b, and c, the height of each character represents its relative probability, and flags show gaps in the current sequence (bottom) and overlapping gaps in its existing child sequence (top). In a, a one-character gap is opened; in b, the gap is skipped over and extended by one character at each side, with the double flag at the fourth site denoting the overlapping insertion and deletion; and in c, only the one-character insertion in the grandchild is skipped over. If c were to be aligned to its sister sequence, only the one site in the middle could be skipped over, because the sites adjacent to it (denoted by gray circles) were inferred as deletions and are now hidden. The inferred alignment is shown at the bottom (gray box). See text for further notation details.
leading from z to y. The example corresponds to a simple linear gap cost algorithm with two storage matrices, and only one vector of flags is shown because x and y do not have grandchild sequences. (B) Here, we illustrate a special case where independent insertions have occurred at the same site in both subtrees. (C) Four sequences are aligned by applying our gap-flagging approach. In the sequences for nodes a, b, and c, the height of each character represents its relative probability, and flags show gaps in the current sequence (bottom) and overlapping gaps in its existing child sequence (top). In a, a one-character gap is opened; in b, the gap is skipped over and extended by one character at each side, with the double flag at the fourth site denoting the overlapping insertion and deletion; and in c, only the one-character insertion in the grandchild is skipped over. If c were to be aligned to its sister sequence, only the one site in the middle could be skipped over, because the sites adjacent to it (denoted by gray circles) were inferred as deletions and are now hidden. The inferred alignment is shown at the bottom (gray box). See text for further notation details.
Traditional progressive algorithms inherently consider all gaps as deletions and use heuristics to correct for the cost of repeated handling of insertions; for example, clustalw (9) encourages alignment gaps to overlap by lowering the gap penalty at sites where gaps already exist and increasing it at sites that do not have gaps but are within a short distance of an existing gap. Such heuristic rules do reduce the cost of insertion events and discourage nonhomologous characters from matching inserted sites but also discard the phylogenetic information that indel events contain and try to explain all spatially close length differences with the same few gapped sites. In addition, methods that do not distinguish insertions from deletions necessarily infer ancestral sequences that do not represent the actual sequence history but, unrealistically, get longer the deeper into the tree the progressive algorithm proceeds.
Progressive Alignment Algorithm for Sequences with Insertions
Whereas a single pairwise alignment does not indicate whether length differences between two sequences are caused by insertion or deletion, information from an evolutionarily related outgroup sequence can differentiate the events; in progressive alignment the comparison between the first pair and the related outgroup is, inherently, performed in their subsequent alignment. We describe an algorithm that allows for any substring of a sequence marked as a gap in previous alignments to be skipped, thereby incurring no additional penalty. This procedure is appropriate for an insertion; our algorithm also considers whether a better alignment may be obtained by matching the characters normally, corresponding to a gap created by a deletion. Our method has some resemblance to the parsimony alignment of sequence graphs (14), although we prefer to see it as an extension of the standard alignment algorithm by the use of additional matrices. A similar idea of gap penalty adjustment was proposed by Hogeweg and Hesper (11) but, to our knowledge, has never been formally described. If insertions are seen as dummy edges in sequence graphs (15) (or matrix pointers; see below), the complexity of our method is not significantly higher than that of a standard method. However, the problem is computationally more attractive if insertions are skipped via “storage” matrices that for Gotoh's method require additional O(4L2) space; using Hirschberg's algorithm, the extra space is reduced to O(8L). Given that the guide tree is known, the computation scales linearly with the number of sequences to be aligned.
Hidden Markov Model (HMM) for Probabilistic Alignment
We illustrate the modification in the dynamic programming algorithm by implementing it with an evolutionary scoring function similar to that described in ref. 16. This approach allows for a realistic modeling of the substitution process and, by considering the information contained in partial multiple alignments of descendant sequences that are already solved, makes the alignment algorithm more robust against locally ambiguous solutions or uncertainty of character states in ancestral sequences. A normalization of the match and gap probabilities ensures that matching two random sites is not favored over two independent gaps and significantly reduces the chances of erroneous alignment of unrelated sequences. We incorporate the scoring function into a probabilistic alignment algorithm and can thus describe the whole model as a pair HMM (15).
We believe that both the distinction between insertions and deletions and the realistic match/gap scoring help our method to avoid overmatching of the sequences aligned and thus produce improved alignments. Our approach provides an efficient means of combining the clarity of a true pairwise alignment method with the robustness of traditional methods for progressively aligning alignments or profiles (9, 13) and permits us to employ evolutionary modeling techniques, already established in phylogenetics (17), in the sequence alignment. Furthermore, our ability to identify insertion events permits us to locate affected sites in order to infer ancestral sequences and get a more realistic picture of the evolutionary history.
Methods
Progressive algorithms build the multiple alignment by the iteration of pairwise alignment in the internal nodes of a predefined guide tree. The alignment progresses from the terminal nodes toward the root of tree, such that the alignment at each internal node is performed before the node itself is aligned with its sister node. We describe our method of correcting for insertion sites by using a probabilistic alignment algorithm as an example, although with minor modifications the method is applicable to any type of dynamic programming algorithm used in a progressive manner.
Notation. The alignment is performed according to a binary guide tree that for N extant sequences consists of 2N - 1 nodes and 2N - 2 edges connecting them. We describe the tree with its root node drawn to the extreme left; a node is terminal if it is not connected to nodes on its right side; otherwise it is internal. An internal node is the parent of two child nodes that are sisters to each other. Terminal nodes are associated with extant sequences, and internal nodes are associated with conditional probabilities given their descendants (see below). We denote sequence and node with x, and the edge leading to the node with 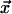 (see Fig. 1 for examples).
(see Fig. 1 for examples).
Alignment HMM and Evolutionary Scoring Function. We implement an affine gap pairwise alignment method by using a HMM loosely based on refs. 15 and 16, consisting of three nonsilent states, X, Y, and M, where either a character is matched against a gap, a gap is matched against a character, or two characters are matched, respectively. It is convenient to draw our model with an additional silent wait state W that links the character-emitting states X, Y, and M (see Scheme 1).
Scheme 1.

Alignment HMM.
Transitions to character-emitting states are then simply defined by the gap opening probability δ, and transitions back to the wait state by the gap and match extension probabilities ε and γ. Of these probabilities, ε and γ are fixed, and δ is defined by an indel rate r and the evolutionary distance 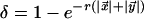 where
where 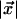 and
and 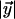 denote the lengths of the edges
denote the lengths of the edges 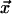 and
and 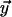 between the parent node and the sister nodes to be aligned.
between the parent node and the sister nodes to be aligned.
We consider a pairwise alignment of sequences x and y consisting of characters x1... xn and y1... ym. Sequence sites are described with vectors of probabilities, pa(xi), that site i in sequence x has character a. At a terminal node, the observed character is given a probability of 1 and others are set to 0. At internal nodes, the pa(xi) are defined from the pairwise alignment of the child nodes as the conditional probability of all possible parent characters given the child sites and all their descendants related by a phylogenetic tree. This approach resembles the standard maximum likelihood computation in phylogenetics (15, 18).
Character emission is defined by an evolutionary substitution model [such as that of Jukes and Cantor (19) or Hasegawa, Kishino, and Yano (20)] and the evolutionary distance between the parent and the child sequences. In state M, the conditional probability for a parent character a at ancestral position zk, given the child character distributions, is defined by
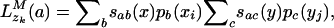 |
[1] |
where sab(x) is the substitution probability between characters a and b given 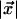 (the evolutionary distance between sequence x and its immediate ancestor) and the evolutionary substitution model [and similarly for sac(y)]. Because zk cannot be known, the match probability is computed over all possible character assignments a at the parent site. Finally, we define dxi,yj, a normalized evolutionary score for matching sites xi and yj, by dividing the probability of the observed character emission by the probability of the process emitting the same output randomly,
(the evolutionary distance between sequence x and its immediate ancestor) and the evolutionary substitution model [and similarly for sac(y)]. Because zk cannot be known, the match probability is computed over all possible character assignments a at the parent site. Finally, we define dxi,yj, a normalized evolutionary score for matching sites xi and yj, by dividing the probability of the observed character emission by the probability of the process emitting the same output randomly,
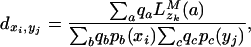 |
[2] |
where qa denotes the equilibrium frequency of character a.
Emission in states X and Y, i.e., during the creation of gaps, is defined by the equilibrium frequencies of the possible characters at the child site and their conditional probabilities given the subtree below that child. Because of the normalization step (see Eq. 2), however, we can ignore the computation and define gap scores dxi,- = d-,yj = 1.
Standard Progressive Alignment. Two sequences are aligned pairwise by searching for the most probable state path, also called the Viterbi path (15), through the HMM (see below). Because of our description of sequence sites as vectors and the normalization of the match scores (Eq. 2), we can extend the pairwise alignment to progressive multiple alignment by defining the probability vector pa(zk) for parent site zk as the conditional probabilities of characters a given the child sites in the pairwise alignment. Given Eq. 1 and defining 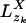 for the single child xi as
for the single child xi as
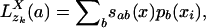 |
[3] |
(and similarly 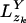 for the single child yj), then pa(zk) =
for the single child yj), then pa(zk) = 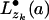 where • denotes M, X, or Y depending on which is the most probable character-matching event.
where • denotes M, X, or Y depending on which is the most probable character-matching event.
If a site is inferred as an insertion and skipped over (see below), we can either set pa(zk) = 0 or copy pa(zk) from the inserted child. Setting these probabilities to zero disallows a site inferred as an insertion from being matched against other sites at any later stage and forces the algorithm to skip over the site in all subsequent alignments (i.e., “insertions open forever”). Copying probabilities from the inserted child means that a site inferred as an insertion is permitted to be matched against other sites at a later stage (i.e., “insertions may be closed”). Biologically, the first approach seems a natural choice, but it has its drawbacks if the alignment order is incorrect or if some of the sequences are incomplete (see Results and Discussion).
Given all of the sites on the alignment path, the ancestral sequence is fully defined and can be aligned with another sequence. Note that ancestral sequences are technically not treated differently from extant ones.
Progressive Alignment of Sequences with Insertion. Our approach is based on a simple idea that a preexisting gap can be skipped over, and the subsolution preceding the gap extended as if that substring did not exist. The method requires keeping track of sequence sites that were within a gap in previous alignments (specifically, in the sequences to be aligned and in their child sequences; see below) and can then be seen as a checking of backward pointers that indicate the start sites of preexisting gaps within the alignment matrix rows and columns (Fig. 1). Applied to Gotoh's original algorithm requiring O(3L2) space for a pairwise alignment, the simple pointer approach would require four additional binary vectors [i.e., O(4L) bits memory] to flag the gapped sequence sites and practically no additional time. A computationally more attractive implementation uses a set of four extra matrices [i.e., an additional O(4L2) space] to store the subsolutions before preexisting gaps and then may restore and extend these solutions from the gap end points (see Fig. 1 A and B Bottom).
Insertions in different lineages of an evolutionary tree are independent. Therefore, in a pairwise alignment, insertions in two descendant sequences can be arranged to appear in an arbitrary order. Here, we apply the ordering that a preexisting gap in one sequence always comes before a new gap created in the other sequence (Fig. 1 A). This ordering ensures that a preexisting insertion cannot interrupt a new insertion, thus defining a single optimal solution for each skipped gap, and results in a saving in memory requirement for our modification of Gotoh's algorithm, reducing what would otherwise be an O(6L2) additional requirement to O(4L2).
In practice, quadratic space complexity is too high, and use of a linear-space algorithm is required. Our affine gap cost alignment's total O(7L2) space can be reduced to O(17L) by using Hirschberg's algorithm [based on O(9L) and O(8L) implementations of Gotoh's algorithm and our modification, respectively]. For clarity, however, we will describe our algorithm in its O(7L2) form.
Alignment Recursion. In pairwise alignment, a recursive computation defines matrices vX, vY, and vM as the probability of obtaining the alignment x1... xi:y1... yj by the extension of subalignment x1... xi-1:y1... yj or x1... xi:y1... yj-1 by a gap, or x1... xi-1:y1... yj-1 by a match, respectively; in the HMM, these computations correspond to the moves into states X, Y, and M. Our method requires keeping track of the values in these matrices before preexisting gaps, but because of the ordering of insertions, we only need to store the values of vX and vM before a gap in sequence x and the values of vY and vM before a gap in sequence y. We call these matrices tX and tM, and uY and uM, respectively. The values in the storage matrices are copied along the preexisting gap that is to be skipped over, and moves out of these matrices are only allowed back to vX, vY, and vM with one exception: in the case that a preexisting gap in the first sequence is followed by another preexisting gap in the second sequence, and the gaps are preceded and followed by a match, moves between tM and uM are possible (Fig. 1B); in all other cases, the preexisting gaps can be arranged so that they are separated by a matching pair or a new gap. Moves back to the matrices vX, vY, and vM can only be taken in positions where a preexisting gap in the corresponding sequence ends.
To allow long deletions to overlap insertions, we keep track of the gaps in the sequences to be aligned and also the overlapping gaps in their child sequences. Because we only need to consider sites that are currently flagged as gaps (deletions in grandchildren and their descendants need no attention), and a gap implies the absence of one of the child sites and all its descendants, our recording requires just two binary vectors for each of the two sequences (Fig. 1C). Consider the alignment of sequence z (with child sequences x and y) to its sister sequence, and imagine that the child sequence x contains an inserted site xi and that the fragment (yj-lyj-l+1... yj+m-1yj+m) represents a deletion in the lineage from z to y that includes the hypothetical sites yj and yj+1 homologous to xi-1 and xi+1, respectively. In the alignment of the parent z, we allow for gaps to be skipped over (i.e., unpenalized moves to and from the storage matrices) at the boundaries of both preexisting gaps, i.e., at sites zk that are parents either to sites yj-l and yj+m (the actual gap boundaries in z) or to site xi (the gap boundary in its child). (In Fig. 1C, these positions would correspond to the boundaries of single- and double-flagged sites, respectively.) For simplicity, we assume that sequence similarity alone ensures that the boundaries of the same gap are used for the moves to and from a storage matrix; an alternative approach would separate the skipping of gaps in child and grandchild sequences, requiring more storage matrices and increasing the additional space required by our method from O(4L2) to O(8L2).
The dynamic programming recursion for our method is given in Algorithm 1. For clarity, the steps novel to the method are boxed in gray; if these steps are ignored, the algorithm reduces to the standard one.
Algorithm 1.

Progressive alignment of sequences with insertions. A boolean variable 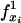 is true if site xi is within a gap, otherwise false;
is true if site xi is within a gap, otherwise false; 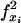 is true if site xi and one of its child sites are within a gap, otherwise false. Their values are reversed by the! operator. Conditions in parentheses are true when the variables f• they contain either all represent
is true if site xi and one of its child sites are within a gap, otherwise false. Their values are reversed by the! operator. Conditions in parentheses are true when the variables f• they contain either all represent  or all represent
or all represent  . We use ⋆ to indicate indices that range over all permitted values (X, Y, M for v⋆; X, M for t⋆; Y, M for u⋆), and vE denotes the score of the full alignment (for clarity, the possibility of preexisting gaps at the end of sequences is ignored).
. We use ⋆ to indicate indices that range over all permitted values (X, Y, M for v⋆; X, M for t⋆; Y, M for u⋆), and vE denotes the score of the full alignment (for clarity, the possibility of preexisting gaps at the end of sequences is ignored).
Results
We illustrate the performance of our method with alignments of real biological sequences, with a full description of the analyses and results in Supporting Text and Figs. 3–15, which are published as supporting information on the PNAS web site.
Analyses. In the first example, we studied 20 primate mitochondrial D-loop sequences with the aim of assessing the effects of (i) the correction for insertion sites [disabled (-) vs. enabled (+)]; (ii) the modeling of the substitution process [JC (16) vs. HKY (20)]; and, when the correction was enabled, (iii) the different ways of handling insertions [insertions may be closed (+) vs. insertions open forever (+F)]. As a reference, we aligned the same data set by using a traditional progressive algorithm [clustalw (9)]. The resulting alignments demonstrate that when the correction is enabled, variation in sequence length is preferentially explained by single insertion events instead of multiple independent deletions (Fig. 2). In contrast to heuristics that encourage any gap to appear at the same position as an existing gap (Fig. 2 A), our method tends to create gaps that are consistent with the phylogeny, i.e., are most parsimoniously explained by a single insertion or deletion event (Fig. 2 C and D). This feature is even more pronounced when the insertions are forced to stay open (Fig. 2E). In this case, all indel events are strictly consistent with the phylogeny but the algorithm is also more sensitive to missing information (see Discussion). When the traditional method is used (Fig. 2 A), the inconsistency of the indel events with the phylogeny suggests that some of the indel “hot spots” may be artifacts of its heuristic correction. Our method considers indels as phylogenetic information, and is naturally sensitive to the order in which the progressive alignments are performed. If sequences are added in an incorrect order (i.e., the guide tree is incorrect), the algorithm may attempt to explain the inconsistency with additional insertions.
Fig. 2.

The guide tree used for the D-loop alignment (Left) and the central part of the alignment (Right) as inferred by clustalw (A), JC- (B), JC+ (C), HKY+ (D), and HKY+F (E). In A, the gaps are gathered at the same sites but are inconsistent with the phylogeny, and the outgroup sequence (top) is clearly overmatched. In comparison with B, the new correction in C preferentially infers insertions instead of multiple deletions. As shown by C and D, the matching of sites depends on the substitution model used. In E, the insertions-open-forever rule prevents the distant outgroup being matched to sites earlier inferred as insertions and creates indels that are strictly consistent with the phylogeny. Blue, green, orange, and red represent the bases A, C, G, and T, respectively, and gray is used for sites where the true base is unknown. Up (blue) and down (red) triangles above the alignments show the inferred deletion and insertion events, respectively; at sites marked with diamonds (green), the state of the root sequence is unknown. See text for further details.
Our approach allows for the use of any substitution model for character evolution and can thus take into account, e.g., unequal base frequencies and transition/transversion bias (17). The alignments generated by using different models (JC vs. HKY; Fig. 2 C and D) reveal that the result obtained depends on the parameter values chosen, suggesting that subsequent analyses (e.g., phylogenetic inference) that estimate the same parameters may depend on the initial choices made for the sequence alignment.
The benefits of our approach are more obvious in our second example, the alignment of CAV2 region genomic sequences of 5.2–7.0 kilobases in length from chicken and 14 mammals. This data set contains many (often rather long) insertion elements only present in one or a few sequences. The alignments produced with a traditional progressive algorithm are clearly overmatched and too compact, causing a misalignment of an exon; in contrast, our approach correctly aligns the exons in all sequences. When the inferred insertions are forced to be skipped over (+F option), the resulting alignment is very gappy (and aesthetically rather unpleasing) but has the long insertions, each evolutionarily unrelated, correctly placed in single (or only few) sequences.
Software. We have implemented the method described here (in linear memory form) as a part of our software package for multiple sequence alignment. Source code is distributed under the General Public License and can be obtained, along with the corresponding data sets and alignments, upon request from A.L.
Discussion
We have developed an algorithm for progressive multiple alignment that distinguishes insertions from deletions and thus avoids penalizing a single insertion event multiple times. With the proper handling of insertion events, our method can avoid the typical overmatching of sequence sites and should produce evolutionarily more correct alignments. Unlike traditional progressive methods, the method also provides realistic estimates of the process that generated the sequences: although the reconstructed sequences contain inserted fragments and get longer in the progressive steps, the recording of the skipped-over gaps permits us to distinguish the inserted regions and to infer realistic ancestral sequences. Obviously, our algorithm does not solve all of the problems that heuristic progressive alignment methods may suffer from; for example, if an alignment of two extant sequences is globally nonoptimal, improved alignments at internal nodes are not able to correct the early error.
We have implemented the correction for insertion sites with a phylogenetic scoring function that can be based on common evolutionary substitution models and takes into account the descendants and their phylogeny for the two sites to be matched. As the score depends on the characters in the descendants, the approach is robust to locally ambiguous or erroneous alignments and, in common with methods aligning alignments, benefits from the information gathered during the progressive alignment steps. Furthermore, the normalization that we perform for the match and gap scores ensures that nonhomologous sequences are much less likely to be erroneously matched. Note that our approach does not contain hidden heuristics: although the gap opening probability is dependent on the evolutionary distance and the substitution model can be arbitrarily complex, the algorithm is purely based on pairwise alignments. The clarity of the approach and the description of the algorithm as a HMM permits us to extend the model easily, e.g., to take into account multiple evolutionary processes, and to use standard tool sets to estimate model parameters or compute posterior probabilities for the obtained solutions.
Within our algorithm, sites that have been inferred as insertions can be forced to be skipped over in all subsequent alignments. Alternatively, matching characters against these sites may be allowed at a later stage. If the phylogeny, and thus the order of alignments, is known to be correct, it may be beneficial to use the phylogenetic information in the patterns of indels by applying the “insertions open forever” rule. This rule may be particularly valuable when sequence similarity is low. However, the dilemma of multiple alignment is that the correct phylogeny is seldom known before the alignment is performed, and a fully correct alignment guide tree cannot generally be expected. By allowing for characters to be matched against skipped-over sites (“insertions may be closed”), the algorithm is less vulnerable to incorrect alignment order. This approach also better handles truncated sequences and missing information because when the strict insertions-open-forever rule is applied for the alignment of incomplete sequences, the sites that are not missing can be inferred as insertions and the resulting alignment can be spread out by many gaps.
By skipping gaps as a whole, our algorithm correctly handles insertions at the same site in sister subtrees, in a subtree and its sister edge, and in two sister edges; it may fail, however, in cases where a deletion in one edge involves a site adjacent to an insertion in its sister edge. Simultaneous insertion and deletion ending at the same site should be rather unlikely and, given a reasonable level of sequence similarity, the subsequent alignment is able to correct the error (although a single insertion now gets penalized twice); multiple events at the same site can be further reduced by a denser sampling of sequences. These insertions are correctly taken care of by our algorithm, but because the number of insertions rises with the number of sequences, they are increasingly problematic to traditional progressive alignment methods. The heuristic adjustment of gap penalties (9) helps to gather gaps in the same alignment columns, but without a clear distinction between insertion and deletion events, commonly used software packages have an inherent tendency to overmatch the sequence sites and to produce overly compact alignments. They not only fail to see alignment gaps as phylogenetic information but, more seriously, may drastically underestimate the number of insertion events and infer alignments that are “too good to be true.”
Supplementary Material
Acknowledgments
We thank Simon Whelan for many inspiring discussion and two anonymous reviewers for their constructive comments on an earlier version of the manuscript. N.G. is supported by the Wellcome Trust.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: HMM, hidden Markov model; indel, insertion/deletion.
See Commentary on page 10411.
References
- 1.Levenshtein, V. I. (1966) Sov. Phys. Dokl. 10, 707-710. [Google Scholar]
- 2.Needleman, S. B. & Wunsch, C. D. (1970) J. Mol. Biol. 48, 443-453. [DOI] [PubMed] [Google Scholar]
- 3.Sankoff, D. (2000) Bioinformatics 16, 41-47. [DOI] [PubMed] [Google Scholar]
- 4.Gotoh, O. (1982) J. Mol. Biol. 162, 705-708. [DOI] [PubMed] [Google Scholar]
- 5.Hirschberg, D. S. (1975) Commun. Assoc. Comput. Mach. 18, 341-343. [Google Scholar]
- 6.Myers, E. W. & Miller, W. (1988) Comput. Appl. Biosci. 4, 11-17. [DOI] [PubMed] [Google Scholar]
- 7.Sankoff, D. (1975) SIAM J. Appl. Math. 28, 35-42. [Google Scholar]
- 8.Schuler, G. D., Altschul, S. F. & Lipman, D. J. (1991) Proteins 9, 180-190. [DOI] [PubMed] [Google Scholar]
- 9.Thompson, J. D., Higgins, D. G. & Gibson, T. J. (1994) Nucleic Acids Res. 22, 4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eddy, S. R. (1998) Bioinformatics 14, 755-763. [DOI] [PubMed] [Google Scholar]
- 11.Hogeweg, P. & Hesper, B. (1984) J. Mol. Evol. 20, 175-186. [DOI] [PubMed] [Google Scholar]
- 12.Katoh, K., Misawa, K., Kuma, K. & Miyata, T. (2002) Nucleic Acids Res. 30, 3059-3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edgar, R. C. (2004) BMC Bioinformatics 5, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hein, J. (1989) Mol. Biol. Evol. 6, 649-668. [DOI] [PubMed] [Google Scholar]
- 15.Durbin, R., Eddy, S., Krogh, A. & Mitchison, G. (1998) Biological Sequence Analysis (Cambridge Univ. Press, Cambridge, U.K.).
- 16.Gonnet, G. H. & Benner, S. A. (1996) in Algorithm Theory: SWAT'96, eds. Karlsson, R. & Lingas, A. (Springer, Berlin) pp. 380-391.
- 17.Whelan, S., Liò, P. & Goldman, N. (2001) Trends Genet. 17, 262-272. [DOI] [PubMed] [Google Scholar]
- 18.Felsenstein, J. (1981) J. Mol. Evol. 17, 368-376. [DOI] [PubMed] [Google Scholar]
- 19.Jukes, T. H. & Cantor, C. (1969) in Mammalian Protein Metabolism, ed. Munro, H. N. (Academic, New York), pp. 21-132.
- 20.Hasegawa, M., Kishino, H. & Yano, T. (1985) J. Mol. Evol. 22, 160-174. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.