

Abstract
The Josephin domain plays an important role in the cellular functions of ataxin-3, the protein responsible for the neurodegenerative Machado–Joseph disease. We have determined the solution structure of Josephin and shown that it belongs to the family of papain-like cysteine proteases, sharing the highest degree of structural similarity with bacterial staphopain. A currently unique structural feature of Josephin is a flexible helical hairpin formed by a 32-residue insertion, which could determine substrate specificity. By using the Josephin structure and the availability of NMR chemical shift assignments, we have mapped the enzyme active site by using the typical cysteine protease inhibitors, transepoxysuccinyl-l-eucylamido-4-guanidino-butane (E-64) and [l-3-trans-(propylcarbamyl)oxirane-2-carbonyl]-l-isoleucyl-l-proline (CA-074). We also demonstrate that the specific interaction of Josephin with the ubiquitin-like domain of the ubiquitin- and proteasome-binding factor HHR23B involves complementary exposed hydrophobic surfaces. The structural similarity with other deubiquitinating enzymes suggests a model for the proteolytic enzymatic activity of ataxin-3.
Keywords: cysteine protease, NMR, polyglutamine disease, SCA3, ubiquitin
Machado–Joseph disease, also known as spinocerebellar ataxia type 3 (SCA3), is an autosomal dominant neurodegenerative disease clinically characterized by cerebellar ataxia (1). As in a larger family of neurological diseases, Machado–Joseph disease is caused by abnormal expansion of a polyglutamine (polyQ) tract in the causative protein, which for SCA3 is ataxin-3. It is widely accepted that when the expansion exceeds a threshold of ≈52 glutamines, ataxin-3 undergoes misfolding and triggers formation of intranuclear aggregates, with consequent cell death (2). On the contrary, it remains unclear what the cellular role of the nonexpanded protein is, even though this knowledge may provide important insights that could help to design a therapeutic strategy. The most accredited hypothesis, strongly supported by independent lines of evidence, is that ataxin-3 is involved in the ubiquitin (Ub) proteasome pathway; ataxin-3 is known to bind polyUb chains containing four or more subunits, most likely through the Ub interacting motifs (UIM) predicted along its sequence (3–5). Ataxin-3 is also known to interact with the N-terminal Ub-like (Ubl) domain of the homologous human Ub- and proteasome-binding factors HHR23A and -B, which are involved in translocating proteolytic substrates to the proteasome, and with the valosin-containing protein (VCP/p97) (6, 7). Finally, ataxin-3 has been shown to cleave substrates of Ub proteases and to bind the specific Ub protease inhibitor, Ub-aldehyde (5).
Another hypothesis is that, independently from a role in surveillance pathways, ataxin-3 acts as a transcriptional repressor through a multiple mechanism that would involve inhibition of histone acetylation by means of histone and histone acetyltransferase binding (8). Although this possibility is particularly attractive because a role in gene regulation also has been suggested for other proteins involved in polyQ diseases (9), thus implying a common mechanism for these pathologies, no direct evidence has been produced so far to support it.
More information is therefore needed to clarify the cellular role of ataxin-3. A powerful approach to achieve it may be to identify the structural motifs involved in the interactions formed by ataxin-3 with other partners and to characterize further their structure and function(s). As a first step toward this aim, we have recently established the domain architecture of ataxin-3 and shown that it consists of an N-terminal globular domain with significant helical content, which spans the Josephin motif, and a flexible C-terminal tail containing up to three UIMs and the polyQ tract (10). Both our analysis and other indirect evidence strongly suggest that Josephin is an important functional region of ataxin-3 that plays a role both in the normal and the pathological functions of the protein. This hypothesis is, for instance, suggested by the strong conservation of the ataxin-3 N terminus throughout all known sequences, whereas the C terminus is highly divergent. Josephin also was reported to be sufficient to bind several of the ataxin-3 partners, among which are histones (8) and the two HHR23A and -B factors (6, 7). A possible function of Josephin was predicted by biocomputing methods, which suggested that Josephin has a cysteine (Cys) protease fold and is therefore the region responsible for the Ub protease activity of the full-length protein (11). This suggestion is supported by the observation that the enzymatic activity of ataxin-3 can be inhibited by mutating the only Cys of Josephin predicted to be part of the active site (5, 12). More recently, Josephin also was implicated in ataxin-3 misfolding and therefore in SCA3 pathology: biophysical studies of the thermodynamic stability of Josephin indicate that the domain has an intrinsic tendency to aggregate and to form temperature-induced fibrils similar to those formed by expanded ataxin-3 (13).
We present here the solution structure of the Josephin domain as determined by NMR techniques. We prove conclusively that, despite the low sequence identity (≤16%), Josephin belongs to the papain superfamily of Cys proteases, with the expected Cys, His, and Asn catalytic triad in a structurally conserved active site. However, Josephin has a distinct fold, most similar to that of bacterial staphopain (14) and of the Pseudomonas avirulence protease AVRPPH3 (15). The Josephin structure suggests regions potentially important for protein–protein interactions and therefore for substrate specificity. We have explored in detail the interaction between Josephin and human HHR23B, which we have mapped onto the respective structures by NMR chemical shift perturbation. This information then was used to dock the two proteins and provide a model of their complex. We also studied the ability of standard Cys protease inhibitors to inhibit Josephin. Taken together, our data provide the bases for further prediction of functionally important residues that may be tested by site-directed mutagenesis and biochemical approaches.
Materials and Methods
Sample Production. The samples of Josephin and its mutant were expressed in Escherichia coli BL21(DE3) and purified as described in ref. 16. Labeled protein was obtained by growing E. coli in synthetic medium containing 15NH4Cl and 15NH4Cl/13C-glucose as the sole source of nitrogen and nitrogen/carbon respectively. Full-length HHR23B and the isolated Ubl domain were expressed by using the pET-24d bacterial expression vector (a gift from Rick Wood, Imperial Cancer Research Fund, South Mimms, U.K.) in a RecA-deficient strain of BL21(DE3) following standard protocols. Recombinant proteins were purified by Ni-NTA affinity chromatography (Qiagen, Valencia, CA). The proteins were eluted into 20 mM Tris (pH 8.0), 300 mM NaCl, β-mercaptoethanol, and 100 mM imidazole before dialysis into the final buffer (10 mM phosphate at pH 6.0).
NMR Spectroscopy. A description of the procedures and experiments used to obtain essentially full 1H, 15N, and 13C assignment is provided in ref. 16. Spectra were processed with nmrpipe and nmrdraw (17) and analyzed by using xeasy (18). 3D 1H-15N/13C NOESY-HSQC spectra with mixing times of 100 ms and acquired at 25°C on Varian INOVA 600 and 800 spectrometers provided the distance restraints used in the final structure calculations. Potential H-bonds were identified by the persistence of amide resonances in HSQC spectra recorded 12 h after dissolving in D2O the freeze-dried sample. The intrinsic tendency of Josephin to aggregate, further enhanced by the confinement effect in liquid crystalline media (19), made measurements of residual dipolar couplings very difficult. After several attempts, 38 residual dipolar couplings were nonetheless obtained in polyacrylamide gels (20). 15NT1,T2, and heteronuclear nuclear Overhauser effect (NOE) data were recorded both at 600 and 800 MHz by using standard sequences (21, 22). The overall rotational correlation time τc was estimated from the average T1/T2 ratio, as calculated after excluding residues with T1/T2 values greater than one standard deviation from the mean.
Structure Calculations. The aria program (Version 1.2) (23) was used for the calculations starting from an extended structure with random side-chain conformations. All NOE cross-peaks from 15N- and 13C-NOESY-HSQC spectra and chemical shift assignments were provided as input, as well as angular and H-bond information. A rotational correlation time of 11.75 ns, computed for the Josephin domain, was used to compute the relaxation matrix. The standard aria protocol was used (23). Eight cycles of restraint assignment were carried out, each followed by simulated annealing following the standard cns protocol (24), except that the number of steps was doubled to improve convergence. At each of the eight iterations, 20 structures were calculated. Seven structures among them were kept, and the top 10 structures were used for evaluation. The final ensemble of NMR structures was refined in a shell of water molecules. Structural quality was assessed by procheck (25) and whatif (26).
The final atomic coordinates are available from the Protein Data Bank (PDB; ID code 1yzb).
Binding Assays. 15N-labeled Josephin solutions (0.5 mM) in 20 mM HCl·Tris (pH 6.5) were titrated with 1 M stock solutions of CaCl2 or MgSO4 to reach 1:20 protein:cation ratios. Similarly, 0.5 mM Josephin solutions were titrated with concentrated stock solutions of transepoxysuccinyl-l-eucylamido-4-guanidinobutane (E-64) or [l-3-trans-(propylcarbamyl)oxirane-2-carbonyl]-l-isoleucyl-l-proline (CA-074) to reach a protein:inhibitor ratio of 1:50 and 1:3, respectively. E-64 and CA-074 were dissolved in water and DMSO, respectively. Josephin was independently titrated against matching amounts of DMSO to discriminate the effect of the solvent from that of the inhibitor.
15N-labeled Josephin and its mutant (0.16 mM) in 10 mM phosphate at pH 6.0 were individually titrated with stepwise additions (5 μl) of a 0.6 mM stock solution of unlabeled HHR23B up to a 1:4.3 ratio. Vice versa, the 15N-labeled Ubl domain of HHR23B was titrated with unlabeled Josephin. The KD was estimated from nonlinear fit of the chemical shift variations as a function of the HHR23B concentration, assuming a binary interaction between the two components. Cross-saturation experiments (27) were performed at 800 MHz on perdeuterated 15N Josephin domain in the presence of HHR23B. Two independent experiments were performed with adiabatic decoupling sequences centered inside and outside the HHR23B aliphatic proton resonances.
Enzymatic Activity Assays. Expression and purification of GST-Ub52 using the pGEX-Ub52 vector kindly provided by Roger Everett (MRC Virology Unit, Glasgow) was achieved according to the protocol described in ref. 28. The sample was dialyzed against 20 mM Tris·HCl (pH 8.0) and 10 mM DTT, concentrated to 10 μM, and incubated with Josephin at different temperatures (in the range 25–37°C) at a 1:5 ratio. Cleaved and uncleaved substrates were resolved by SDS/PAGE and Coomassie staining after different incubation times (1, 3, 6, and 24 h). The experiments were repeated, preincubating Josephin with 520 μM E-64 or CA-074 for 1 h in the absence and presence of Ca2+ (1:10 substrate:ion ratio). GST-Ub52 then was added to each sample. To assess the effect of the solvents used to dissolve the inhibitors, Josephin and GST-Ub52 were preincubated in solutions containing the appropriate volumes of water or DMSO. Each series of experiments was repeated three times to test for consistency.
Molecular Docking Calculations. Docking of Josephin with the Ubl domain of HHR23B was performed with the software haddock1.3 (29). The starting structures were model 1 of the Ubl domain (PDB ID code 1p1a) and the 10 best NMR structures of Josephin. The data obtained from chemical shift mapping and surface accessibility data of Josephin were used to define active (Tyr-27, Phe-28, Val-86, Trp-87, Gly-88, and Leu-89) and passive (Glu-26, Ser-29, Val-31, Glu-32, Lys-85, Glu-90, and Asp-168) residues according to haddock1.3 definitions. Accordingly, residues Thr-7, Leu-8, Leu-46, Ile-47, Tyr-48, Ala-49, Ile-52, Phe-69, Val-71, Met-73, and Thr-75 of HHR23B Ubl were selected as active residues, and Lys-6, Gln-9, Gln-44, Lys-45, Gly-50, Lys-51 were chosen as passive residues. Initially, 1,000 structures of the complex were generated by rigid body energy minimization. The 200 best structures in terms of the total energy were selected for a semiflexible simulated annealing followed by a refinement in explicit water solvent.
Results
Josephin Has a Mixed αβ Fold. Initially, we prepared a construct encompassing residues 1–205 on the basis of secondary structure predictions (11). The 15N-HSQC spectrum showed that the construct is folded, but progressive broadening of the resonance linewidths indicates that it has a strong tendency to aggregate within hours, making it unsuitable for structure determination (data not shown). We therefore proceeded with the better-behaved construct 1–182, whose boundaries are based on limited proteolysis experiments (10). As described in ref. 13, this construct also aggregates but more slowly, so that appreciable aggregation, at 25°C and submillimolar NMR concentrations, is only observed after ≈1 week. Structure determination was achieved for this construct. The Josephin structure has high precision and accuracy as demonstrated by the standard quality controls (see Table 1, which is published as supporting information on the PNAS web site). The rms deviation (rmsd) of the NMR bundle from the mean structure as calculated for the backbone atoms of residues 3–40 and 76–180 is 0.49 Å, indicating that the backbone conformation of Josephin is well defined (Fig. 1A).
Fig. 1.

Structure of the Josephin domain. (A) The NMR bundle (Left) and a ribbon representation (Right) of the Josephin structure. (B) The electrostatic surface potential of Josephin as calculated by molmol. The structure is displayed by using the same orientation as in A and rotated by 180° along the y axis.
The structure consists of two subdomains separated by a cleft (Fig. 1 A). The N-terminal subdomain includes four α-helices (α1, residues 14–22; α2, 31–47; α3, 56–62; α4, 78–85), of which α1 and α4 lie approximately within the plane of the β-sheet, while α2 and α3 form a long helical hairpin that appears as a thumb-like extension protruding from the core structure into solution. The C-terminal subdomain contains instead a six-stranded antiparallel β-sheet, with a β1, β6, β2, β3, β4, β5 topology. β1 is connected to β2 by two short 310-helices that flank and stabilize the conformation of the interconnecting loop. Strands β2–β5 are connected by short turns, whereas β5 is connected to β6 by a 12-aa-long α-helix (residues Tyr-147–Glu-158) located on one side of the sheet. Overall, the surface is highly negatively charged with a large patch that clusters around the α2–α3 helical hairpin (Fig. 1B). At least two exposed hydrophobic patches are also visible on the surface of the Josephin structure, one being centered around Trp-87 (see Fig. 5, which is published as supporting information on the PNAS web site).
Josephin Has a Rigid Scaffold with the Exception of a Flexible Helical Hairpin. Overall, the Josephin structure is relatively rigid as indicated by NMR relaxation experiments recorded from freshly prepared protein (see Fig. 6, which is published as supporting information on the PNAS web site). The estimated correlation time (τc) at 25°C is 11.75 ± 0.07 ns, which confirms that the protein is mainly monomeric under the conditions used. An interesting feature is the helical hairpin formed by α2 and α3, comprising two antiparallel relatively well-defined helices. Although the hairpin is well attached to the rest of the structure by several salt bridges, hydrophobic interactions, and H-bonds, the relaxation parameters indicate that this region is overall distinctly more flexible than the rest of the protein, with extensive fast local motions that deviate from those expected for a rigid body. It exhibits short T1, long T2, and small NOE values, suggesting the presence of internal motions on the nano- and picosecond timescale. These dynamical properties also are reflected by the average backbone rmsd values of the structural ensemble, which are appreciably higher in the hairpin than in the rest of the molecule. The exposed hairpin therefore appears to behave as an exposed waving hand, which could be available for interactions with other molecules.
Josephin Is a Cys Protease. A structural comparison with the PDB (30) shows that the Josephin architecture bears a clear similarity with members of the papain-like Cys protease family, which includes papain, cathepsin, staphopain, and different Ub hydrolases, in agreement with previous fold predictions (11) (Fig. 2). The secondary structure elements of Josephin that have a similar organization in Cys proteases include the central antiparallel β-sheet with the β1, β6, β2, β3, β4, β5 topology, the α5 helix, and the cleft between the two subdomains, in which the active site sits. Of the residues in the catalytic triad, the nucleophilic Cys-14, demonstrated by mutagenesis to be critical for Ub protease activity of ataxin-3 (5), is part of the buried α1. His-119 and Asp-134 are respectively located at the N and C termini of β3 and β4, whereas Gln-9, present in the extended N terminus, is structurally equivalent to Gln-19 in papain, which participates in the formation of a catalytically active structure known as the “oxy-anion hole” (31).
Fig. 2.

Comparison between the Josephin structure with those of the Cys proteases staphopain (PDB ID code 1cv8), avirulence protein (PDB ID code 1ukf), papain (PDB ID code 1pe6), YUH1 (PDB ID code 1cmx), and UCH-L3 (PDB ID code 1uch). The side chains of the catalytic triad are shown on each structure.
The two closest matches according to dali (32) are the crystal structures of staphopain from Staphylococcus aureus (14) and the Pseudomonas avirulence protease AVRPPH3 (15), even though the overall sequence identities are well below the level regarded as significant (16% and 11%). The two structures superpose to Josephin with Z-scores of 6.7 and 6.3 and rmsds of 3.5 and 3.4 Å over 113 and 121 equivalent Cα atoms, respectively. However, despite the higher score, staphopain has longer strands and additional secondary structure elements that make it topologically less close to Josephin than AVRPPH3. This structure shares with Josephin the same topology and a similar arrangement to the all-helical N terminus (corresponding to the α1–α4 helices in Josephin), although it is shorter and its orientation relative to the rest of the structure is distinctly different. Papain (33) also shares significantly high structural similarity (Z-score 5.3 and rmsd 4.2 Å, superposing 121 equivalent Cα atoms). In comparison, the structures of the two deubiquinating enzymes, human UCH-L3 (34) and yeast YUH1 (35), used for previous comparative modeling (36), show smaller rmsds but over fewer residues (2.8 and 2.7 Å over 82 and 91 residues, respectively).
Josephin Is Not a Ca2+-Activated Protease. The papain superfamily of Cys proteases comprises, in addition to the papain and the bleomycin hydrolase groups, calpains (37). These are Ca2+-dependent Cys proteases that coordinate the ion with high specificity through conserved negatively charged residues and are activated through a Ca2+-induced conformational change (38). Because Josephin is also a highly acidic protein, we tested its ability to coordinate Ca2+. Titration of Josephin with Ca2+ shows a clear influence of the cation on the amide resonances of Cys-14, Asn-68, Ser-76, and Glu-118 (see Fig. 7, which is published as supporting information on the PNAS web site). However, Mg2+ induces similar effects, and no conformational changes are associated with cation binding as judged from comparison of NOESY-like experiments run on the apo- and holo-forms. We therefore conclude that Ca2+ has an unspecific effect that is not required for enzymatic activity.
Josephin Binds Protease Inhibitors Selectively. To characterize the mechanism of enzyme inhibition and map the inhibitor-binding site onto the Josephin structure, we carried out binding studies with two typical Cys protease inhibitors, E-64 and CA-074. E-64 is a well-documented, potent, and irreversible inhibitor of papain-like Cys proteases, such as papain, ficin, actinidin, and cathepsins B and L (39). Along with its derivative, CA-074, E-64 is known to recognize specifically the catalytic Cys and bind covalently to its sulfydryl group (40).
Titrations were followed by monitoring the changes induced in the Josephin HSQC spectra by progressive addition of each of the two ligands separately. When Josephin was titrated with E64 up to a protein:inhibitor ratio of 1:50, no variations were observed in the Josephin spectrum (data not shown), indicating that the inhibitor does not bind to the protein. Addition of Ca2+ to the solution made no difference. To find a rational explanation for this observation, we compared the structures of known Cys protease/E-64 complexes with that of Josephin. We observed two features that could explain why E-64 is unable to bind (see Fig. 8, which is published as supporting information on the PNAS web site): Phe-74 may cause steric hindrance, thus preventing the inhibitor from binding. Glu-118, whose side-chain packs against Phe-74, also may contribute to closing the binding groove.
In contrast, significant changes in a well-defined region of the protein were observed upon titration with CA-074, already at a 1:3 inhibitor:protein ratio (see Fig. 8). Spatially, the affected residues cluster around the catalytic site (Cys-14, His-119, and Asn-134), indicating inhibitor occupancy. Because the two inhibitors mainly differ for the long hydrophobic chain attached to the guanidinium group present in E-64, which is substituted by a shorter pyrrolidine group in CA-074, it is reasonable to assume that this part of the molecule discriminates binding. Superposition of the crystal structure of a cathepsin B/CA-074 complex (40) onto Josephin suggests that Trp-120 and Leu-136 could create a hydrophobic  pocket [as defined in Schechter and Berger (41)] able to accommodate the -l-iso-leucyl moiety of CA-074 (Fig. 3A). The
pocket [as defined in Schechter and Berger (41)] able to accommodate the -l-iso-leucyl moiety of CA-074 (Fig. 3A). The  subsite could be occupied mainly by His-6, Asn-21, and Thr-138 and contribute to stabilizing the Pro pyrrolidine ring mainly through interaction with the imidazole nitrogens of the His-6 and Asn-21 side chains. Gln-9 and Cys-14, located on the opposite side of the hydrophobic S′1 pocket across the inhibitor, could produce a hydrophilic environment that could stabilize the oxirane atom of CA-074.
subsite could be occupied mainly by His-6, Asn-21, and Thr-138 and contribute to stabilizing the Pro pyrrolidine ring mainly through interaction with the imidazole nitrogens of the His-6 and Asn-21 side chains. Gln-9 and Cys-14, located on the opposite side of the hydrophobic S′1 pocket across the inhibitor, could produce a hydrophilic environment that could stabilize the oxirane atom of CA-074.
Fig. 3.

Testing protease inhibitor binding. (A) Stereo picture of the CA-074 fit into the Josephin binding site. The complex was built by homology on the coordinates of a cathepsin/CA-074 complex (PDB ID code 1qdq) (40). The residues most affected by the titration are displayed in blue around the inhibitor, shown in red. The subsites are indicated. (B) Enzymatic assays. The gel was loaded after a 3-h incubation of GST-Ub52 with Josephin at 37°C.
Enzymatic Activity of Josephin Is Selectively Inhibited by CA-074. To support the conclusions formulated on the basis of structural considerations, we tested the role of protease inhibitors and Ca2+ on the enzymatic activity of Josephin. We first reproduced previous results by incubating Josephin with a model substrate comprising a human Ub52 fusion protein precursor linked to GST (GST-Ub52) at an enzyme:substrate molar ratio of 1:5 (12). Ub cleavage was monitored by SDS/PAGE (Fig. 3B). Under these conditions, Josephin cleaves ≈50% of the GST-Ub52 substrate after 3 h at 37°C. Substrate cleavage was almost quantitatively inhibited by CA-074, whereas it was unaffected by E-64. No influence was observed when the experiments were repeated in the presence of Ca2+.
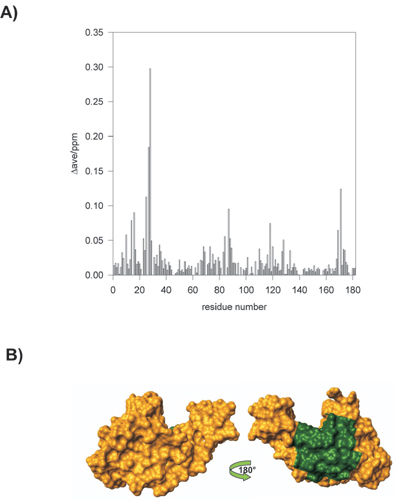
Mapping the Josephin/HHR23B Interface. The N-terminal Ubl domain of HHR23B has been shown to be necessary and sufficient for interacting with ataxin-3 (6). We used NMR chemical shift perturbation to map the interaction onto the respective 3D structures. First, 15N-labeled Josephin was titrated with unlabeled full-length HHR23B. The chemical shift variations reached a plateau at an ≈1:1 molar ratio. Curve fitting provided a KD of 12 ± 3 μM. The Josephin residues exhibiting the largest chemical shift variations upon HHR23B binding are Gly-25, Tyr-27, Phe-28, Trp-87, and Leu-89 (see Fig. 9, which is published as supporting information on the PNAS web site). They form a well-defined exposed hydrophobic patch of ≈240 Å2 and cluster around the exposed Trp-87, a residue highly conserved across species. These results were confirmed by cross-saturation experiments (28) carried out on uniformly 2H- and 15N-labeled Josephin and unlabeled HHR23B. As with Ca2+ binding, the interaction with HHR23B does not seem to involve significant changes of the backbone conformation.
To confirm a direct involvement of Trp-87 in the interaction, a Josephin mutant was prepared in which this residue was replaced by a Lys. The mutation does not disrupt the Josephin fold, as judged both from the 1D 1H and the 2D 1H-15N HSQC spectra of the mutant protein. No significant Josephin amide chemical shift variations were observed upon titration of this sample with HHR23B, indicating that Trp-87 is indeed essential for binding (data not shown).
When chemical shift perturbation was followed by 15N-labeled HHR23B Ubl titrated by unlabeled Josephin, taking advantage of the availability of the NMR spectral assignment of the Ubl domain (42), the largest effects were observed for residues Thr-7, Leu-8, Gln-9, Leu-46, Ile-47, Tyr-48, Ala-49, Ile-52, Phe-69, Val-71, and Met-73 (see Fig. 10, which is published as supporting information on the PNAS web site). These residues form an exposed hydrophobic patch of Ubl, which has all of the potential features to be complementary to the aromatic/hydrophobic patch on the Josephin surface. The residues involved are conserved in the homologous domain of HHR23A and are the same ones that participate to the interaction with the UBA domains and with the polyUb-binding site of S5a (43). We used this information to dock the complex with the haddock approach, a procedure that makes use of experimental chemical shift data to drive docking (29). A representation of the lowest energy structure of the Ubl/Josephin complex as obtained by this procedure is shown in Fig. 4A.
Fig. 4.

Modeling of the interaction surfaces of Josephin with some of its molecular partners. (A) Model of the Josephin/HHR23B complex as built by haddock. Josephin is shown in blue and magenta, and HHR23B Ubl domain is shown in gold and pale green (PDB ID code 1p1a). The side chains of the residues involved in the interaction are displayed. (B) Model of YUH1 (Left, PDB ID code 1cmx) and Josephin (Right) complexes with Ub-aldehyde. The YUH1 coordinates were used as a template to model the Josephin complex by fitting the coordinates of the two proteases according to the dali superposition.
Discussion
Consequences of Our Results for the Catalytic Activity of the Josephin Domain. We have reported the 3D structure of the Josephin domain, the only independently folded unit of ataxin-3, which is thought to play an important role in the functions of both the normal and pathological forms of the protein (13). We have shown that, according to previous predictions (11), the Josephin structure has a papain-like fold. The overall structural similarity of the active site suggests that the catalytic mechanism of Josephin resembles that of other papain-like enzymes. Josephin Cys-14 and His-119 then would form a thiolate–imidazolium ion pair, while Asn-134 would be responsible for the correct orientation of the imidazolium ring of the active-site His-119. Gln-9, together with the adjacent amide of Cys-14, can form a potential oxy-anion hole, like that of papain (31).
Our data suggest that, in contrast with what is observed for other subfamilies of Cys proteases, Josephin is already in its active form: the active site is overall well exposed, and the distance between the components of the catalytic triad is, within the resolution of our structure, that expected for active Cys proteases. Apart from a partial shielding of the catalytic triad Asn by the side chain of the nearby Leu-136, there are also no secondary structure elements or loops that could in principle mask access to the catalytic site, thus suggesting that substrate binding may occur without major structural rearrangements, such as those observed, for instance, in other deubiquitinating enzymes (35, 44). Although the protein is highly negatively charged, we also can exclude a role of Ca2+ in protease activation, such as that observed for calpains, because the interactions with this cation are weak and unspecific.
To explore further the catalytic subsites, we probed Josephin with two typical protease inhibitors, chosen because of a different potential ability to fit into the catalytic subsites. We observed high specificity of recognition. Josephin is not affected by E-64 but is able to bind CA-074. Consistently, proteolytic activity is quantitatively inhibited only by CA-074. A similar specificity is observed also in other Cys proteases, such as the streptococcal endopeptidase IdeS (45). The information collected so far will help us to understand the chemical requirements necessary to design specific inhibitors of Josephin.
Structural Bases of Protein–Protein Recognition. Knowledge of the Josephin structure allowed us to investigate the structural bases of its interaction with other proteins. We studied in detail the determinants of recognition of HHR23B, which, together with the high homolog HHR23A, is the human ortholog of yeast RAD23, involved in nucleotide excision repair (43). By chemical shift mapping and cross-correlation experiments, we have demonstrated that Josephin and HHR23B form a stable complex. On the Josephin surface, this binding site involves an exposed aromatic cluster, of which Trp-87 is a key element because its mutation is sufficient to abolish binding. The patch is on the face opposite to the active site (Fig. 4A), strongly suggesting that interaction with the HHR23 proteins is well distinct from protease activity. The interaction, mapped to the N-terminal Ubl domain of HHR23B, involves a highly conserved hydrophobic patch exposed on the surface of this domain. This region, which has all of the features necessary to complement the Josephin binding site, is the same surface involved in the interaction with UBA domains, the S5a polyUb-binding site, and the proteasome (43, 46).
Another region of Josephin that is likely to mediate protein–protein interactions is the flexible α2-α3 hairpin present in the proximity of the active site. A similar but not topologically equivalent secondary structure element also may be observed in other Cys proteases, such as in the avirulence protease AVRPPH3 (15). In this structure, however, the hairpin packs much more tightly against the rest of the globular domain, whereas in Josephin the longer helices are exposed and flexible in solution. Because of its proximity to the active site, the hairpin could determine specificity and stabilize protease substrate(s) and/or inhibitors.
Excellent candidates for interacting with this region are the histones (8), because they would have the correct charge complementarity to the highly negatively charged surface of the hairpin. An even more obvious molecular partner of Josephin is Ub. Ataxin-3 has in fact been implicated with Ub at two different levels. PolyUb binding seems to involve exclusively the UIMs: ataxin-3 constructs containing only the UIM sequences are still able to interact, but mutations of UIM key residues abolish binding completely (5). Conversely, Josephin has been shown by in vitro studies to be necessary and sufficient to cleave Ub and to be inhibited by Ub-aldehyde (5). In our hands, as in previous studies (12), proteolytic cleavage occurs, although with low efficiency, also at the level of monomeric Ub (Fig. 3B). A model built by exploiting the structure similarity between Josephin and the YUH1/Ub-aldehyde complex (35) shows that the Ub monomer has the right size to fit into the cavity between α2–α3 and α1 and α4 (Fig. 4B). Being longer than the structurally homologous region of YUH1, the Josephin hairpin could contribute to the stability and the specificity of the complex with additional interactions.
Although explaining well the inhibition by Ub-aldehyde, this hypothesis does not, however, clarify the relationship between protease activity, which also occurs at the level of the Ub monomer, and polyUb binding, which requires chains of four or more monomers. A working hypothesis that could explain this relationship is that the UIMs of ataxin-3 are necessary to bind polyUb chains and anchor them to ataxin-3. They would have evolved to be tuned to recognize only polyUb chains of four or more subunits, because this length is the minimal one required to target protein substrates to the proteasome. Once anchored, the region between the UIMs and Josephin, mostly flexible, could act as a folding arm that would bring the Ub chain into the active site. The efficiency of proteolytic cleavage that is low with the monomer would increase thanks to the anchoring. This mechanism may in the cell help to select between ubiquitinated substrates and confer specificity to the ataxin-3 function.
In conclusion, we believe that the structure of Josephin constitutes the first step to a more thorough characterization of the cellular role of ataxin-3 and provides insights that can then be probed experimentally.
Supplementary Material
Acknowledgments
We thank the NMR Centre of Mill Hill for technical support and John McCormick for protein production. This work was supported by the European Integrated Project on Spinocerebellar Ataxias (EUROSCA) grant and by the Cancer Research UK (P.P.K. and N.Q.M.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: SCA3, spinocerebellar ataxia type 3; Ub, ubiquitin; Ubl, Ub-like; UIM, Ub interacting motif; NOE, nuclear Overhauser effect; PDB, Protein Data Bank; rmsd, rms deviation; E-64, transepoxysuccinyl-l-eucylamido-4-guanidino-butane; CA-074, [l-3-trans-(propylcarbamyl)oxirane-2-carbonyl]-l-isoleucyl-l-proline.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 1yzb).
References
- 1.Kawagushi, Y., Okamoto, T., Taniwaki, M., Aizawa, M., Inoue, M., Katayama, S., Kawakami, H., Nakamura, S., Nishimura, M., Akigushi, I., et al. (1994) Nat. Genet. 8, 221-228. [DOI] [PubMed] [Google Scholar]
- 2.Taylor, J. P., Hardy, J. & Fischbeck, K. H. (2002) Science 296, 1991-1995. [DOI] [PubMed] [Google Scholar]
- 3.Chai, Y., Berke, S. S., Cohen, R. E. & Paulson, H. L. (2004) J. Biol. Chem. 279, 3605-3611. [DOI] [PubMed] [Google Scholar]
- 4.Donaldson, K. M., Li, W., Ching, K. A., Batalov, S., Tsai, C. C. & Joazeiro, C. A. (2003) Proc. Natl. Acad. Sci. USA 100, 8892-8897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burnett, P., Li, F. & Pittman, R. N. (2003) Hum. Mol. Genet. 12, 3195-3205. [DOI] [PubMed] [Google Scholar]
- 6.Wang, G., Sawai, N., Kotliarova, S., Kanazawa, I. & Nukina, N. (2000) Hum. Mol. Genet. 9, 1795-1803. [DOI] [PubMed] [Google Scholar]
- 7.Doss-Pepe, E. W., Stenroos, E. S., Johnson, W. G. & Madura, K. (2003) Mol. Cell. Biol. 23, 6469-6483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li, F., MacFarlan, T., Pittman, R. N. & Chakvarti, D. (2002) J. Biol. Chem. 277, 45004-45012. [DOI] [PubMed] [Google Scholar]
- 9.Neri, C. (2001) Trends Mol. Med. 7, 283-284. [DOI] [PubMed] [Google Scholar]
- 10.Masino, L., Musi, V., Menon, R. P., Fusi, P., Kelly, G., Frenkiel, T. A., Trottier, Y. & Pastore, A. (2003) FEBS Lett. 549, 21-25. [DOI] [PubMed] [Google Scholar]
- 11.Scheel, H., Tomiuk, S. & Hofmann, K. (2003) Hum. Mol. Genet. 12, 2845-2852. [DOI] [PubMed] [Google Scholar]
- 12.Chow, M. K. M., Mackay, J. P., Whisstock, J. C., Scanlon, M. J. & Bottomley, S. P. (2004) Biochem. Biophys. Res. Commun. 322, 387-394. [DOI] [PubMed] [Google Scholar]
- 13.Masino, L., Nicastro, G., Menon, R. P., Dal Piaz, F., Calder, L. & Pastore, A. (2004) J. Mol. Biol. 344, 1021-1035. [DOI] [PubMed] [Google Scholar]
- 14.Hofmann, B., Schomburg, D. & Hecht, H. J. (1993) Acta Crystallogr. A 49, Suppl., 102. [Google Scholar]
- 15.Zhu, N., Shao, F., Innes, R. W., Dixon, J. E. & Xu, Z. (2004) Proc. Natl. Acad. Sci. USA 101, 302-309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nicastro, G., Masino, L., Frenkiel, T. A., Kelly, G., McCormick, J., Menon, R. P. & Pastore, A. (2004) J. Biomol. NMR 30, 457-458. [DOI] [PubMed] [Google Scholar]
- 17.Delaglio, F., Grzesiek, S., Vuister, G. W., Pfeifer, F. & Bax, A. (1995) J. Biomol. NMR 3, 277-293. [DOI] [PubMed] [Google Scholar]
- 18.Bartels, C., Xia, T., Gunther, P., Billeter, M. & Wuthrich, K. (1995) J. Biomol. NMR 5, 1-10. [DOI] [PubMed] [Google Scholar]
- 19.Munishkina, L. A., Cooper, E. M., Uversky, V. N. & Fink, A. L. (2004) J. Mol. Recognit. 17, 456-464. [DOI] [PubMed] [Google Scholar]
- 20.Sass, H. J., Musco, G., Stahl, S. J., Wingfield, P. T. & Grzesiek, S. (2000) J. Biomol. NMR 18, 303-309. [DOI] [PubMed] [Google Scholar]
- 21.Kay, L. E., Torchia, D. A. & Bax, A. (1989) Biochemistry 28, 8972-8979. [DOI] [PubMed] [Google Scholar]
- 22.Boyd, J., Hommel, U. & Campbell, I. D. (1990) Chem. Phys. Lett. 175, 477-482. [Google Scholar]
- 23.Nilges, M. (1997) Folding Des. 2, 53-57. [Google Scholar]
- 24.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Unstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., et al. (1998) Acta Crystallogr. D 54, 905-921. [DOI] [PubMed] [Google Scholar]
- 25.Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993) J. Appl. Crystallogr. 26, 283-289. [Google Scholar]
- 26.Vriend, G. (1990) J. Mol. Graphics 8, 52-56. [DOI] [PubMed] [Google Scholar]
- 27.Takahashi, H., Nakanishi, T., Kami, K., Arata, Y. & Shimada, I. (2000) Nat. Struct. Biol. 7, 220-223. [DOI] [PubMed] [Google Scholar]
- 28.Halowaty, M. N. Sheng, T., Nguyen, T., Arrowsmith, C. & Frappier, L. (2003) J. Biol. Chem. 278, 47753-47761. [DOI] [PubMed] [Google Scholar]
- 29.Dominguez, C., Boelens, R. & Bonvin, A. M. (2003) J. Am. Chem. Soc. 125, 1731-1737. [DOI] [PubMed] [Google Scholar]
- 30.Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissing, H., Shindyalov, I. N. & Bourne, P. E. (2000) Nucleic Acids Res. 28, 235-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Drenth, J., Kalk, K. H. & Swen, H. M. (1976) Biochemistry 15, 3731-3738. [DOI] [PubMed] [Google Scholar]
- 32.Holm, L. & Sander, C. (1993) J. Mol. Biol. 233, 123-138. [DOI] [PubMed] [Google Scholar]
- 33.Yamamoto, D., Matsumoto, H., Ohishi, H., Ishida, T., Inoue, M., Kitamura, K. & Nizuno, H. (1991) J. Biol. Chem. 266, 14771-14776. [DOI] [PubMed] [Google Scholar]
- 34.Johnston, S. C., Larsen, C. N., Coo, W. J., Wilkinson, K. D. & Hill, C. P. (1997) EMBO J. 16, 3787-3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnston, S. C., Riddle, S. M., Cohen, R. E. & Hill, C. P. (1999) EMBO J. 18, 3877-3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Albrecht, M., Golatta, M., Wullner, U. & Lengauer, T. (2004) Eur. J. Biochem. 271, 3155-3170. [DOI] [PubMed] [Google Scholar]
- 37.Berti, P. J. & Storer, A. C. (1995) J. Mol. Biol. 246, 273-283. [DOI] [PubMed] [Google Scholar]
- 38.Moldoveanu, T., Hosfield, C. M., Lim, D., Elce, J. S., Jia, Z. & Davies, P. L. (2002) Cell 108, 649-660. [DOI] [PubMed] [Google Scholar]
- 39.Otto, H. H. & Schirmeister T. (1997) Chem. Rev. 97, 133-171. [DOI] [PubMed] [Google Scholar]
- 40.Yamamoto, A., Tomoo, K., Matsugi, K., Hara, T., In, Y., Murata, M., Kitamura, K. & Ishida, T. (2002) Biophys. Biochem. Acta 1597, 244-251. [DOI] [PubMed] [Google Scholar]
- 41.Schechter, J. & Berger, A. (1967) Biochem. Biophys. Res. Commun. 27, 157-162. [DOI] [PubMed] [Google Scholar]
- 42.Walters, K. J., Jleijnen, M. F., Goh, A. M., Wagner, G. & Howley, P. M. (2002) Biochemistry 41, 1767-1777. [DOI] [PubMed] [Google Scholar]
- 43.Ryu, K. S., Lee, K. J., Bae, S. H., Kim, B. K., Kim, K. A. & Choi, B. S. (2003) J. Biol. Chem., 278, 36621-36627. [DOI] [PubMed] [Google Scholar]
- 44.Hu, M., Li, P., Li, M., Li, W., Yao, T., Wu, J. W., Gu, W., Cohen, R. E. & Shi, Y. (2002) Cell 111, 1041-1054. [DOI] [PubMed] [Google Scholar]
- 45.Wenig, K., Chatwell, L., von Pawel-Rammingen, U., Bjorck, L., Huber, R. & Sondermann, P. (2004) Proc. Natl. Acad. Sci. USA 101, 17371-17376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fujiwara, K., Tenno, T., Sugasawa, K., Jee, J. G., Ohki, I., Kojima, C. & Tochio, H. (2004) J. Biol. Chem. 279, 4760-4767. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}