

Abstract
In the methanogenic archaeon Methanosarcina barkeri Fusaro, the N5-methyl-tetrahydrosarcinapterin (CH3-H4SPT):coenzyme M (CoM) methyltransferase, encoded by the mtr operon, catalyzes the energy-conserving (sodium-pumping) methyl transfer from CH3-H4SPT to CoM during growth on H2/CO2 or acetate. However, in the disproportionation of C-1 compounds, such as methanol, to methane and carbon dioxide, it catalyzes the reverse, endergonic transfer from methyl-CoM to H4SPT, which is driven by sodium uptake. It has been proposed that a bypass for this energy-consuming reaction may occur via a direct methyl transfer from methanol to H4SPT. To test this, an mtr deletion mutant was constructed and characterized in M. barkeri Fusaro. The mutant is unable to grow on methanol, acetate or H2/CO2, but can grow on methanol with H2/CO2 and, surprisingly, methanol with acetate. 13C labeling experiments show that growth on acetate with methanol involves a previously unknown methanogenic pathway, in which oxidation of acetate to a mixture of CO2 and formic acid is coupled to methanol reduction. Interestingly, although the mutant is unable to grow on methanol alone, it remains capable of producing methane from this substrate. Thus, the proposed Mtr bypass does exist, but is unable to support growth of the organism.
Keywords: methyltransferase, mutant
Methanogenic archaea are a diverse group of anaerobic organisms that obtain energy for growth by converting a limited number of substrates to methane (1). They are found in a variety of anaerobic environments, including freshwater and marine sediments, marshes, swamps, and the gastrointestinal tracts of animals, and are responsible for essentially all of the biologically produced methane on Earth (2, 3). Each year, ≈1014 g of biologically produced methane are released into the atmosphere, where it acts as a potent greenhouse gas contributing to global warming (4). However, it is estimated that 90-99% of the methane produced is oxidized by methanotrophic bacteria. Thus, ≈1015 to 1016 g of methane are produced each year, demonstrating the critical role methanogens play in the global carbon cycle (4, 5).
Extensive biochemical studies have led to the four proposed pathways of methanogenesis, of which most methanogens can use only one (Fig. 1). The CO2 reduction pathway involves the reduction of carbon dioxide to methane with hydrogen gas as the electron donor (6). The methyl reduction pathway also uses hydrogen gas as an electron donor, but reduces methanol to methane after transfer of the methyl group to coenzyme M (CoM) (7). The acetoclastic pathway occurs through the dismutation of acetate, where acetate is first activated to acetyl-CoA (6). The carbonyl group is then oxidized to carbon dioxide, whereas the methyl moiety is transferred to tetrahydrosarcinapterin (H4SPT) and subsequently reduced to methane (6). Finally, the methylotrophic pathway involves the disproportionation of C-1 compounds, such as methanol and methylamines, to carbon dioxide and methane (8). One molecule of substrate must be oxidized to produce the reducing equivalents needed to reduce three molecules to methane (8).
Fig. 1.

Four overlapping methanogenic pathways found in M. barkeri. Many methanogens reduce CO2 to methane by using electrons derived from the oxidation of H2 (hydrogenotrophic pathway, shown in red in A). Alternatively, acetate can be split into a methyl group and an enzyme-bound carbonyl moiety. The latter is oxidized to CO2 to provide the electrons required for reduction of the methyl group to methane (aceticlastic pathway, shown in blue in B). C-1 compounds such as methanol or methyl-amines can also be disproportionated to CO2 and methane. In this pathway, one molecule of the C-1 compound is oxidized to provide electrons for reduction of three additional molecules to methane (methylotrophic pathway, shown in green in C). Finally, C-1 compounds can be reduced by using electrons derived from hydrogen oxidation (methyl reduction pathway, shown in orange in D). Steps not required by each pathway are shaded gray. The step catalyzed by the Mtr protein is indicated: note that this enzyme is predicted to be required for all pathways except the methyl-reduction pathway. CHO-MF, formyl-methanofuran; CHO-H4SPT, formyl-tetrahydrosarcinapterin; CH≡H4SPT, methenyl-tetrahydrosarcinapterin; 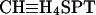 , methylene-tetrahydrosarcinapterin; CH3-H4SPT, methyl-tetrahydrosarcinapterin; CH3-CoM, methyl-coenzyme M; CoM, coenzyme M; CoB, coenzyme B; CoM-CoB, mixed disulfide of CoM and CoB; Mph/MphH2, oxidized and reduced methanophenazine; F420/F420H2, oxidized and reduced Factor 420; Fd(ox)/Fd(red), oxidized and reduced ferredoxin; Ac, acetate; Ac-Pi, acetyl-phosphate; Ac-CoA, acetyl-CoA; Ech, ferredoxin-dependent hydrogenase; Frh, F420-dependent hydrogenase; Vho, methanophenazine-dependent hydrogenase; Fpo, F420 dehydrogenase.
, methylene-tetrahydrosarcinapterin; CH3-H4SPT, methyl-tetrahydrosarcinapterin; CH3-CoM, methyl-coenzyme M; CoM, coenzyme M; CoB, coenzyme B; CoM-CoB, mixed disulfide of CoM and CoB; Mph/MphH2, oxidized and reduced methanophenazine; F420/F420H2, oxidized and reduced Factor 420; Fd(ox)/Fd(red), oxidized and reduced ferredoxin; Ac, acetate; Ac-Pi, acetyl-phosphate; Ac-CoA, acetyl-CoA; Ech, ferredoxin-dependent hydrogenase; Frh, F420-dependent hydrogenase; Vho, methanophenazine-dependent hydrogenase; Fpo, F420 dehydrogenase.
A key step in three of the four pathways is the methyl transfer from methyl-H4SPT to CoM. In both the CO2-reduction pathway and the acetoclastic pathway, this transfer occurs concomitantly with the extrusion of sodium ions to generate an ion motive force, in a reaction catalyzed by the enzyme N5-methyl-H4SPT:CoM methyltransferase (Mtr) (9). This membrane-bound, eight-subunit enzyme, encoded by the mtrECDBAFGH operon, also catalyzes the reverse, endergonic methyl transfer from methyl-CoM to H4SPT in the methylotrophic pathway (3). Mtr is able to drive this unfavorable methyl transfer by the consumption of the sodium ion gradient (10, 11). It has been proposed that a direct methyl transfer from methanol to H4SPT may occur to bypass this energy-consuming step, but this has yet to be shown experimentally (12).
To address the issue of an Mtr bypass and to further investigate the physiological role of Mtr, we constructed and characterized an mtr deletion mutant in Methanosarcina barkeri Fusaro. The Δmtr mutant was not viable on methanol, acetate, or H2/CO2, but was able to use combinations of methanol plus H2/CO2 or methanol plus acetate. Although the mutant was unable to grow on methanol alone, cell suspensions were able to convert methanol to carbon dioxide and methane, demonstrating that a bypass of Mtr is possible.
Materials and Methods
Strains, Media, and Growth Conditions. M. barkeri Fusaro (DSM 804) was grown at 37°C in high salt (HS) broth medium (13) or on agar-solidified medium as described (14). HS medium was supplemented as appropriate with 125 mM methanol, 40 mM sodium acetate, or H2/CO2 (80/20) mix at 150 kPa over ambient pressure. Puromycin was added at 2 μg/ml for selection of the pac gene (15). 8-aza-2,6-diaminopurine (8-ADP) was added at 20 μg/ml for selection of hpt disruption (15).
DNA Methods and Plasmid Construction. Standard methods were used for plasmid DNA isolation and manipulation (16). Genomic DNA isolation and DNA hybridization were as described (13, 14, 17). All plasmid constructions are described in Table 4, which is published as supporting information on the PNAS web site. Complete DNA sequences of all plasmids used are available upon request.
Construction and Complementation of M. barkeri Δmtr. Liposome-mediated transformation and homologous recombination-mediated gene replacement were used to construct the M. barkeri Δmtr::pac-ori-aph mutant (hereafter designated Δmtr) (14, 17). M. barkeri Fusaro was transformed with NotI-cut pPW7, and transformants were selected on HS agar containing methanol, acetate, and puromycin under H2/CO2 gas phase. Complementation of the mutation was accomplished by transforming the Δmtr mutant with SpeI-cut pPW25-3 (which carries hpt::mtrECDBAFGH) and selecting transformants on HS agar supplemented with methanol and puromycin under N2/CO2 gas phase and screening for 8-ADP resistance.
Cell Suspension Experiments. Wild-type and Δmtr cells grown on methanol plus acetate were collected in late exponential phase (OD600 = 0.6-0.7) by centrifugation at 5,000 × g for 15 min at 4°C. Cells were washed once with plain HS medium (no substrate) and resuspended in plain HS to a final cell concentration of 109 cells per ml. A total of 2 ml of the suspension were used in the assays, which were conducted under strictly anaerobic conditions in 25-ml tubes sealed with butyl rubber stoppers. Sparsomycin (25 μg/ml) was added to prevent protein synthesis, and substrates were added at the concentrations indicated in Table 2. Cells were held on ice until use, and assays were started by transferring the tubes to 37°C. For rate determinations, the cell concentration was halved. Gas phase samples were withdrawn at various time points and assayed for CH4 by gas chromatography at 225°C in a Hewlett Packard gas chromatograph (5890 Series II) equipped with a flame ionization detector. The column used was of stainless steel filled with 80/120 Carbopack B/3% SP-1500 (Supelco, Bellefonte, PA) with helium as the carrier gas. For total CH4 production, assays were incubated overnight at 37°C and then gas phase samples were withdrawn and analyzed for CH4. Total cell protein was determined by using the Bradford method (18) after an aliquot of the cells was lysed by sonication for 10 s.
Table 2. Methane production by cell suspensions of M. barkeri strains.
|
M. barkeri
|
M. barkeri Δmtr
|
|||
|---|---|---|---|---|
| Substrate | CH4 produced, μmol | Rate of CH4 production, nmol·min−1·mg−1 | CH4 produced, μmol | Rate of CH4 production, nmol·min−1·mg−1 |
| N2/CO2 | <1 | <1 | <1 | <1 |
| H2/CO2 | 308 ± 55 | 221 ± 37 | <1 | <1 |
| Me | 350 ± 18 | 328 ± 26 | 109 ± 5 | 9 ± 4 |
| Me + H2/CO2 | 691 ± 39 | 731 ± 36 | 442 ± 46 | 422 ± 41 |
| Me + Ac | 356 ± 6 | 309 ± 6 | 491 ± 13 | 171 ± 20 |
Assays contained 500 μmol methanol and/or 500 μmol acetate and were conducted as described in Materials and Methods; gas phase was either N2/CO2 (80%/20%) or H2/CO2 (80%/20%) at 250 kPa.
Labeled Cell Suspension Experiments. Cell suspension assays were performed as described above except HS Pipes was used as wash and assay buffer: 50 mM Pipes, pH 6.8/400 mM NaCl/13 mM KCl/54 mM MgCl2/2 mM CaCl2/2.8 mM cysteine/0.4 mM Na2S. Labeled substrates were added at the concentrations indicated in Table 4. Gas phase samples were removed after overnight incubation at 37°C and assayed for 13CH4 and 13CO2 by gas chromatography-mass spectrometry. This was done at 50°C in an HP6890 gas chromatography system equipped with an HP5973 mass selective detector. A Carbon-Plot capillary column (30 m, 0.32-mm inner diameter; Agilent Technologies, Colorado Springs, CO) was used at 1.3 ml/min flow rate of helium. The supernatant of the cell suspensions was analyzed by 13C-NMR as in (19). Yeast formate dehydrogenase (Sigma) was used to measure formate levels in the supernatants by following the reduction of NAD to NADH at 340 nm (20).
Preparation of Cell Extracts. Wild-type and Δmtr cultures were grown in methanol plus acetate to late exponential phase (OD600 = 0.6-0.7). Cells were harvested by centrifugation at 5,000 × g for 15 min at 4°C. Cells were washed once in anaerobic HS Mops: 50 mM Mops, pH 7.0/400 mM NaCl/13 mM KCl/54 mM MgCl2/2 mM CaCl2. Cells were lysed by sonication (1 × 10 s) after resuspension in anaerobic 50 mM Mops (pH 7.0) with a few crystals of DNase I. Intact cells and debris were removed by centrifugation at 16,000 × g for 2 min. Extracts were transferred to fresh vials and kept on ice for up to 6 h.
Methyltransferase Assays. Methyltransferase activity was determined by measuring the formation of methyl-CoM from formaldehyde, H2 and coenzyme M by wild-type and Δmtr extracts. Methanol-free formaldehyde was prepared from paraformaldehyde (Sigma). A 400 mM paraformaldehyde stock solution was prepared in deionized water, heated to 65°C for 1 h, cooled to room temperature, and stored at 4°C for up to 1 week. One-milliliter assays were done in triplicate in sealed 10-ml anaerobic vials containing crude extract (2 mg of protein), 8 mM formaldehyde, 1 mM NaCl, 2.5 mM ATP, 1.5 mM CoM, 5 mM BES, and 3.2 mM Ti (III) citrate in 50 mM Mops (pH 7.0) under a 100% H2 gas phase. Assays were incubated at 37°C, and 30-μl samples were removed at various time points, added to 700 μl of 0.5 mM 5,5-dithio-bis(2-nitrobenzoic acid), and the loss of absorbance was measured at 415 nm. Methanol:CoM methyl transferase activity was tested in the extracts as above except the assay contained crude extract (2 mg of protein), 1.5 mM CoM, 3.2 mM BES, 10 mM ATP, 20 mM MgCl2, 100 mM methanol, and 3.2 mM Ti (III) citrate in 50 mM Mops (pH 7.0).
Results
Isolation of an M. barkeri Δmtr Mutant. Homologous recombination-mediated gene replacement was used to isolate a mutant with a deletion of the mtrECDBAFGH operon. A plasmid, pPW7, containing a puromycin resistance cassette (pac-ori-aph) flanked by ≈1 kb of the upstream and downstream regions of the mtr operon was constructed. This plasmid was linearized and introduced into M. barkeri Fusaro. Through a double recombination event between the chromosome and the plasmid, the pac cassette replaced the mtr operon resulting in a puromycin-resistant strain. Several puromycin-resistant colonies were screened by Southern blot, and four Δmtr mutants were identified (data not shown). One mutant (Δmtr) was selected for further characterization.
To verify that the mutant lacked Mtr activity, an indirect formaldehyde assay was used to demonstrate that Δmtr extracts were unable to catalyze the methyl transfer from methyl-H4SPT to coenzyme M. Extracts were given formaldehyde, CoM, H2, and BES and the methylation of CoM was followed spectrophotometrically by a decrease in absorbance at 415 nm. Wild-type extracts were able to catalyze this transfer (27.7 ± 0.1 nmol/min/mg extract), whereas mutant extracts had essentially no activity (0.4 ± 0.2 nmol/min/mg extract), indicating that no other enzymes were present in Δmtr that could catalyze this reaction. We confirmed that the Δmtr extracts were not inactivated by O2 exposure by demonstrating wild-type activity levels of the oxygen-sensitive enzyme methanol:CoM methyltransferase, which catalyzes the transfer of a methyl group from methanol to CoM (data not shown). The lack of Mtr activity in the mutant strain was, therefore, not due to inactivation of the extract.
Growth Phenotypes of M. barkeri Δmtr. The ability of Δmtr to grow on a variety of substrates was tested (Table 1). As expected, the mutant was unable to grow on H2/CO2, acetate, or methanol, but was able to use combinations of methanol plus H2/CO2 and surprisingly, methanol plus acetate. It had been previously reported that acetate catabolism by Methanosarcina strains was repressed by methanol as well as H2/CO2 (21, 22). Only acetate-adapted cells have been shown to cometabolize methanol and acetate before switching to metabolizing methanol alone (22, 23). Because Δmtr does not grow on methanol alone, we assumed that acetate oxidation was providing the reducing equivalents necessary for methanol reduction, which, if proven, would represent an unprecedented methanogenic pathway.
Table 1. Growth of M. barkeri strains in various media.
| Doubling time, h
|
|||
|---|---|---|---|
| Substrate | Wild type | Δmtr::pac-ori-aph | Δmtr::pac-ori-aph hpt::mtrECDBAFGH |
| H2/CO2 | 7.2 ± 0.7 | NG | 9.4 ± 0.3 |
| Ac | 60.1 ± 7.9 | NG | 67.1 ± 9.8 |
| Me | 5.7 ± 0.5 | NG | 10.7 ± 0.5 |
| Me + H2/CO2 | 5.8 ± 1.9 | 5.4 ± 0.5 | 6.0 ± 0.5 |
| Me + Ac | 6.7 ± 0.6 | 13.2 ± 1.2 | 11.9 ± 0.1 |
Growth rate was measured by measuring optical density during growth in HS broth with the indicated substrates; Me, methanol; Ac, acetate. Doubling time (in hours) from at least three independent measurements is reported. Positive cultures, except acetate, typically grew within 3 days; Ac cultures grew within 4 weeks. NG, no growth after incubation for at least 6 months.
To confirm that the growth phenotypes observed for Δmtr were a result of the deletion of the mtr locus, a complementing copy of the mtr operon was placed on the chromosome and the growth phenotypes tested. This was accomplished by construction of a Δmtr strain with the wild-type mtr operon and flanking regions recombined into the hpt locus, previously shown to be a permissive site for the insertion of DNA in M. barkeri (24). The Δmtr, Δhpt::mtrECDBAFGH strain was verified by DNA hybridization and shown to grow on all substrates used by the wild type (Table 1). Therefore, the growth phenotypes observed for Δmtr are due to the deletion of the mtr operon.
Methane Production by M. barkeri Δmtr Cell Suspensions. To examine whether the lack of growth of Δmtr on H2/CO2, acetate or methanol was due to a block in methanogenesis, we quantified the amount of methane produced from various substrates by resting cell suspensions (Table 2). Methane was not produced by Δmtr cells when H2/CO2 alone or acetate alone were supplied as substrates. However, when given methanol alone, ≈20% of the substrate was converted to methane. In this situation, methanol oxidation was the only source of reducing equivalents needed for the production of methane, implying that Δmtr cells must be able to oxidize methanol in the absence of Mtr to produce methane.
A significant increase in both the amount of methane produced as well as the rate of methane production by Δmtr cells was observed when acetate was given in combination with methanol, indicating that the acetate contributes to the production of methane from methanol. These data suggest that Δmtr cells oxidize acetate to produce the reducing equivalents needed to reduce methanol to methane.
Interestingly, a significant decrease in the rate of methanogenesis from methanol plus H2/CO2 was observed in the Δmtr mutant relative to the wild type. This observation suggests that, in the wild type, both CO2 and methanol reduction occur simultaneously, leading to a higher rate than is observed in the mutant, which is unable to reduce CO2.
Oxidation of 13C-Labeled Methanol by M. barkeri Δmtr Cell Suspensions. The results from the Δmtr cell suspension assays above implied that the oxidation of methanol was occurring without Mtr. To demonstrate this directly, cell suspension experiments were repeated as above, except 13C-labeled substrates were used and the gas samples were analyzed by gas chromatography-mass spectrometry (Table 3). When given labeled methanol alone, wild-type cells disproportionated methanol to labeled methane and labeled carbon dioxide in the expected 3:1 ratio. Under these same conditions, 99% of the methane and 95% of the carbon dioxide produced by Δmtr cells was also labeled, indicating that all of the methane and carbon dioxide came from the labeled methanol. This production of labeled carbon dioxide by Δmtr cells clearly demonstrated that methanol was oxidized in the absence of Mtr and, therefore, an Mtr bypass must exist.
Table 3. [13C]methane and carbon dioxide production by M. barkeri strains.
| Substrate | CH4 produced, μmol | % 13CH4 | CO2 produced, μmol | % 13CO2 | CH4:CO2 |
|---|---|---|---|---|---|
| M. barkeri Δmtr | |||||
| 13CH3OH | 72 ± 6 | 99 | 22 ± 2 | 95 | 3.2:1 |
| 13CH3OH + CH3COO− | 440 ± 6 | 98 | 130 ± 2 | 4 | 3.4:1 |
| CH3OH + 13CH3COO− | 396 ± 27 | <1 | 125 ± 6 | 36 | 3.2:1 |
| CH3OH + CH313COO− | 364 ± 11 | <1 | 124 ± 3 | 62 | 2.9:1 |
| M. barkeri | |||||
| 13CH3OH | 345 ± 17 | 99 | 116 ± 6 | 97 | 3:1 |
| 13CH3OH + CH3COO− | 378 ± 6 | 98 | 133 ± 3 | 92 | 2.8:1 |
| CH3OH + 13CH3COO− | 312 ± 11 | <1 | 126 ± 5 | 2 | 2.5:1 |
| CH3OH + CH313COO− | 322 ± 6 | <1 | 126 ± 1 | 5 | 2.6:1 |
Assays contained 500 μmol methanol and/or 500 μmol acetate and were conducted as described in Materials and Methods; gas phase was either N2/CO2 (80%/20%) or H2/CO2 (80%/20%) at 250 kPa.
It should be pointed out that methanol is commonly contaminated with formaldehyde, which is formed by the autoxidation of methanol in air. Because formaldehyde is readily oxidized by Methanosarcina cells, it seemed possible that contaminating formaldehyde, rather than methanol, was being oxidized to produce the reducing equivalents needed to reduce methanol to methane in the Δmtr cell suspension assays. The 13C-labeled methanol used in our study was received in anaerobic ampoules under N2 and was stored under anaerobic conditions to minimize the possibility of autoxidation. To rigorously exclude the possibility of formaldehyde contamination, we examined the methanol stock solution used in our cell suspension assays via 1H NMR. Formaldehyde was readily detected in controls, but absent from the stock solution used in our assays (data not shown).
Oxidation of 13C-Labeled Acetate by M. barkeri Δmtr Cell Suspensions. When wild-type cells were given labeled methanol plus unlabeled acetate, 98% of the methane and 92% of the carbon dioxide produced were labeled (Table 3). Thus, consistent with previous results (21, 22), wild-type cells primarily disproportionate the methanol, even in the presence of acetate. When Δmtr cells where given labeled methanol and unlabeled acetate, all of the methane produced was labeled, but only 4% of the carbon dioxide produced was labeled. It is clear that, although some methanol oxidation was occurring, the majority of the carbon dioxide came from the oxidation of acetate; this was demonstrated more directly when Δmtr cells were given labeled acetate and unlabeled methanol. In this case, Δmtr cells produced labeled carbon dioxide and unlabeled methane. This finding shows that, when Δmtr was grown on methanol plus acetate, the acetate was oxidized to carbon dioxide, but not reduced to methane. These data support the hypothesis that Δmtr was growing through a new methanogenic pathway in which acetate oxidation provides reducing equivalents for methanol reduction to methane.
Although the data from the labeling experiments demonstrated that Δmtr cells were oxidizing acetate in the presence of methanol, the oxidation of the methyl and carbonyl groups of acetate did not occur as expected. When the carbonyl group of acetate was labeled, 62% of the carbon dioxide produced was labeled, whereas only 36% of the carbon dioxide was labeled when the label was on the methyl group of acetate. We assumed that both carbons of acetate would be oxidized to CO2; therefore, we expected 50% of the carbon dioxide to be labeled. Furthermore, we expected that 1 mole of acetate would provide enough reducing equivalents to reduce 4 moles of methanol to methane, resulting in a 4:1 ratio of methane to carbon dioxide. However, the observed ratio was lower, indicating that Δmtr cells may be producing some other metabolic product in this new pathway. Because we had previously shown that Methanosarcina acetivorans produces formate when grown on carbon monoxide (19), we hypothesized that the missing product might be formate. To test this possibility, we assayed formate production in the cell suspension supernatants using formate dehydrogenase. Supernatants from Δmtr cells given methanol plus methyl-labeled acetate converted ≈23% of the methyl groups to formate, whereas no formate was detected in supernatants of wild-type cells. 13C NMR analysis of these supernatants confirmed that the formate produced by Δmtr cells is derived exclusively from the methyl group of acetate and also verified that no other 13C-labeled products were present in the supernatant (Fig. 2), whereas GC-MS demonstrated that no other labeled products were present in the gas phase (data not shown).
Fig. 2.

13C-NMR analysis of supernatants from metabolism of methanol plus acetate by M. barkeri Δmtr. Supernatants from cell suspension assays were examined by 13C NMR as described. Spectrum A, cell suspension assay buffer with unlabeled methanol and acetate. Spectrum B, cell suspension supernatant from nongrowing Δmtr cells incubated in the presence of methanol plus [1-13C]acetate. Spectrum C, cell suspension supernatant from nongrowing Δmtr cells incubated in the presence of methanol plus [2-13C]acetate.
Discussion
The sodium-pumping methyl-H4SPT:CoM methyltransferase, Mtr, plays a critical role in the production and consumption of ion-motive force in methanogenic archaea. Nevertheless, we demonstrate here that Mtr is dispensable in M. barkeri under a variety of conditions.
The extremely low energy yields available from methanogenesis have led to speculation that a bypass of the energy-consuming Mtr step may function during methylotrophic methanogenesis. Some authors have even reported unpublished biochemical data in support of this hypothesis (12). In this study, we clearly demonstrate that the proposed bypass exists, although it is incapable of supporting growth. Why this bypass pathway cannot support growth remains unclear. It is possible that the rate of methane production from methanol is too slow to allow growth in the mutant (≈20-fold slower than the rate observed with methanol plus acetate, Table 2). However, in most microorganisms, slowly used substrates simply result in slower growth rates. Moreover, bypassing the energy-requiring methyl-transfer step would presumably lead to increased ATP yields and correspondingly higher growth yields. A more compelling argument is that there is simply insufficient energy available from methanol disproportionation to support extra ATP generation.
In the methylotrophic pathway, energy is conserved during electron transport from hydrogen to the CoB-S-S-CoM disulfide through the methanophenazine-dependent hydrogenase (Vho) and heterodisulfide reductase (Hdr), and during electron transport from reduced F420 to the CoB-S-S-CoM disulfide through the F420-dehydrogenase (Fpo) and heterodisulfide reductase (Hdr) (see Fig. 1). Based on the presumed stoichiometries of ion translocation during electron transport (4 H+/H2 and 4 H+/F420; ref. 25) and ATP synthesis (4 H+/ATP; ref. 26), three ATP are expected to be produced from these steps. The standard free energy available for disproportionation of four methanol to three methane and CO2 is -315 kJ/mol, which could support the synthesis of at most 7 ATP, using a value of ≈45 kJ per ATP and assuming 100% efficient energy conservation (27). Because observed efficiencies actually range from 20% to 80% (27), only 1.4 to 5.6 ATP could realistically be produced via this pathway, which agrees well with value predicted from knowledge of the electron transport chain. However, ion-motive force is also generated by the Ech hydrogenase and consumed by Mtr. Although the number of ions translocated at these steps has not yet been determined, thermodynamic constraints for each reaction (≈20 kJ/mol, ref. 28) suggest that at most one or two scalar ions could be produced/consumed at the Ech and Mtr steps. Thus, in the Mtr bypass strain, an extra 0.25-0.5 ATP would be conserved per four methanol molecules consumed. If the thermodynamic limits of ATP generation prevent additional energy conservation (and therefore growth) via an Mtr bypass, then the overall energy conservation of methylotrophic methanogenesis cannot be more than three ATP per reaction (1 ATP per mol CH4 produced). If true, the thermodynamic efficiency of the methylotrophic pathway is very finely balanced: as little as 1/16 of an ATP per mol CH4 tips the balance beyond the limits of growth.
The biochemical nature of the Mtr bypass also remains unclear, but data from other mutants suggests that it requires at least some of the steps of the standard methylotrophic pathway. Mutants lacking the methenyl-H4SPT cyclohydrolase (Mch), which catalyzes the conversion of methenyl-H4SPT to formyl-H4SPT, are unable to produce methane from methanol in cell suspensions (29). If the Mtr bypass did not use at least part of the standard pathway, then the Mch mutant would also remain capable of methanol-dependent methanogenesis via the bypass pathway. Therefore, the bypass must join the standard pathway somewhere between methenyl-H4SPT and methyl-H4SPT (see Fig. 1). As discussed above, direct methyl transfer from methanol to H4SPT could account for this observation (12). One possible candidate enzyme for this reaction is encoded by the mtxXAH operon. The MtxA and the MtxH proteins have been shown to contain high sequence similarity to MtrA and MtrH, respectively (30). Northern blot analysis has shown that the three genes form a transcriptional unit whose expression is highest in cells grown on methylated compounds (30). Alternatively, a methanol dehydrogenase could oxidize methanol to formaldehyde, which would then spontaneously react with H4SPT to form methylene-H4SPT. Although previous attempts to demonstrate methanol dehydrogenase activity in M. barkeri did not succeed (12), several putative alcohol dehydrogenases, which might catalyze this reaction, are annotated in the M. barkeri genome (http://genome.jgi-psf.org/draft_microbes/metba/metba.home.html).
The Δmtr mutant was able to use a combination of methanol plus acetate via a previously unrecognized pathway in which the oxidation of acetate provides the reducing equivalents needed to reduce methanol to methane. Given the abundance of acetate in anaerobic environments (2), it seems possible that this pathway may play a significant role in nature, particularly in low-sodium environments where the activity of sodium-dependent enzymes like Mtr would be reduced. The Mtr-independent methanol-acetate pathway is somewhat reminiscent of the methanol-H2 pathway of Methanosphaera stadtmaniae, which is also incapable of methanol oxidation due to the absence of formylmethanofuran dehydrogenase.
Careful examination of the methanol-acetate pathway emphasizes the metabolic flexibility of M. barkeri. Although acetate-adapted Methanosarcina strains can metabolize methanol and acetate simultaneously, acetate catabolism is rapidly down-regulated by continued cultivation in the presence of methanol (22, 23). Our results indicate that the Δmtr mutant is able to recognize the need for continued acetate oxidation and override this down-regulation. Clearly, the mtr mutation forces the organism to adapt its metabolism to use the available substrates for growth. The production of formate when growing on methanol plus acetate was also surprising. Our results indicate that the formate produced by Δmtr cells is derived exclusively from the methyl group of acetate, and thus is likely to arise from one of the intermediates of the C-1 oxidation pathway. We believe the most likely source to be hydrolysis of either formyl-H4SPT or formylmethanofuran. A similar reaction is found in the methylotrophic bacterium Methylobacterium extorquens AM1, where the Ftr complex catalyzes the hydrolysis of formylmethanofuran to produce formate (31). Our labeling data indicate that at least some of the methyl groups are oxidized completely to CO2; thus, it is unclear why formate is produced. Possibly, methyl group oxidation is kinetically limited resulting in an accumulation of C-1 intermediates. To relieve this backup and release cofactors for continued metabolism, the mutant is forced to hydrolyze some of the formyl groups producing formate.
Our characterization of the Δmtr mutant demonstrates the effectiveness of using a genetic approach to examine the methanogenic process. By examining the phenotypic consequences of the loss of Mtr in vivo, the presence of several unsuspected biochemical pathways, including a previously unsuspected methanogenic pathway, was revealed. These studies highlight the flexibility of the Methanosarcina genus and support the notion that this generalist organism competes with more specialized methanogens via its ability to adapt its metabolism to changing conditions (2).
Supplementary Material
Acknowledgments
We thank Andrew Eliot, Adam Guss, Gargi Kulkarni, Arpita Bose, and Donna Kridelbaugh for critical reading of the manuscript. We also thank K. S. Suslick (Department of Chemistry, University of Illinois) for providing the GC/MS equipment and A. Eliot and V. Mainz for help with the NMR studies. This work was supported by National Science Foundation Grant MCB 0212466 (to W.W.M.) and by a National Science Foundation Predoctoral Fellowship (to P.V.W.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: CoM, coenzyme M; H4SPT, tetrahydrosarcinapterin; HS, high salt.
References
- 1.Deppenmeier, U., Muller, V. & Gottschalk, G. (1996) Arch. Microbiol. 165, 149-163. [Google Scholar]
- 2.Zinder, S. H. (1993) in Methanogenesis: Ecology, Physiology, Biochemistry and Genetics, ed. Ferry, J. G. (Chapman & Hall, New York), pp. 128-206.
- 3.Gottschalk, G. & Thauer, R. K. (2001) Biochim. Biophys. Acta 1505, 28-36. [DOI] [PubMed] [Google Scholar]
- 4.Thauer, R. K. (1998) Microbiology 144, 2377-2406. [DOI] [PubMed] [Google Scholar]
- 5.Galagan, J. E., Nusbaum, C., Roy, A., Endrizzi, M. G., Macdonald, P., FitzHugh, W., Calvo, S., Engels, R., Smirnov, S., Atnoor, D., et al. (2002) Genome. Res. 12, 532-542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weimer, P. J. & Zeikus, J. G. (1978) Arch. Microbiol. 119, 49-57. [DOI] [PubMed] [Google Scholar]
- 7.Muller, V., Blaut, M. & Gottschalk, G. (1987) Eur. J. Biochem. 162, 461-466. [DOI] [PubMed] [Google Scholar]
- 8.Deppenmeier, U., Lienard, T. & Gottschalk, G. (1999) FEBS Lett. 457, 291-297. [DOI] [PubMed] [Google Scholar]
- 9.Becher, B., Muller, V. & Gottschalk, G. (1992) J. Bacteriol. 174, 7656-7660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gartner, P., Weiss, D. S., Harms, U. & Thauer, R. K. (1994) Eur. J. Biochem. 226, 465-472. [DOI] [PubMed] [Google Scholar]
- 11.Harms, U., Weiss, D. S., Gartner, P., Linder, D. & Thauer, R. K. (1995) Eur. J. Biochem. 228, 640-648. [DOI] [PubMed] [Google Scholar]
- 12.Keltjens, J. T. & Vogels, G. D. (1993) in Methanogenesis: Ecology, Physiology, Biochemistry and Genetics, ed. Ferry, J. G. (Chapman & Hall, New York), pp. 253-303.
- 13.Metcalf, W. W., Zhang, J. K., Shi, X. & Wolfe, R. S. (1996) J. Bacteriol. 178, 5797-5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boccazzi, P., Zhang, J. K. & Metcalf, W. W. (2000) J. Bacteriol. 182, 2611-2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pritchett, M. A., Zhang, J. K. & Metcalf, W. W. (2004) Appl. Environ. Microbiol. 70, 1425-1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ausubel, F. M., Brent, R., Kingston, R. E., Moore, D. D., Seidman, J. G., Smith, J. A. & Struhl, K. (1992) Current Protocols in Molecular Biology (Wiley, New York).
- 17.Zhang, J. K., White, A. K., Kuettner, H. C., Boccazzi, P. & Metcalf, W. W. (2002) J. Bacteriol. 184, 1449-1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bradford, M. M. (1976) Anal. Biochem. 72, 248-254. [DOI] [PubMed] [Google Scholar]
- 19.Rother, M. & Metcalf, W. W. (2004) Proc. Natl. Acad. Sci. USA 101, 16929-16934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hopner, T. & Knappe, J. (1974) in Methods of Enzymatic Analysis, ed. Bergmeyer, H. U. (Verlag Chemie, Weinheim), Vol. 3, pp. 1551-1555. [Google Scholar]
- 21.Zinder, S. H. & Elias, A. F. (1985) J. Bacteriol. 163, 317-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith, M. R. & Mah, R. A. (1978) Appl. Environ. Microbiol. 36, 870-879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krzycki, J. A., Wolkin, R. H. & Zeikus, J. G. (1982) J. Bacteriol. 149, 247-254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meuer, J., Kuettner, H. C., Zhang, J. K., Hedderich, R. & Metcalf, W. W. (2002) Proc. Natl. Acad. Sci. USA 99, 5632-5637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deppenmeier, U. (2004) J. Bioenerg. Biomembr. 36, 55-64. [DOI] [PubMed] [Google Scholar]
- 26.Muller, V. (2004) J. Bioenerg. Biomembr. 36, 115-125. [DOI] [PubMed] [Google Scholar]
- 27.Thauer, R. K., Jungermann, K. & Decker, K. (1977) Bacteriol. Rev. 41, 100-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keltjens, J. T. & van der Drift, C. (1986) FEMS Microbiol. Rev. 39, 259-303. [Google Scholar]
- 29.Guss, A. M., Mukhopadhyay, B., Zhang, J. K. & Metcalf, W. W. (2005) Mol. Microbiol. 55, 1671-1680. [DOI] [PubMed] [Google Scholar]
- 30.Harms, U. & Thauer, R. K. (1997) Eur. J. Biochem. 250, 783-788. [DOI] [PubMed] [Google Scholar]
- 31.Pomper, B. K., Saurel, O., Milon, A. & Vorholt, J. A. (2002) FEBS Lett. 523, 133-137. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.