

Abstract
Sulfinamides play a crucial role in organic synthesis and pharmaceuticals. In this study, we introduce a highly effective method for the deoxygenative radical addition to N-tritylsulfinylamine, which produces sulfinamides via photoredox catalysis. This method is compatible with a diverse array of functional groups and the resulting sulfonamides were achieved in moderate to high yields. Furthermore, the synthetic applications to access various sulfur(vi)-centered functional groups highlight the practicality of this approach.
Sulfinamides play a crucial role in organic synthesis and pharmaceuticals.
Sulfinamides are important compounds widely found in drug candidates, agrochemicals, and asymmetric catalysis (Fig. 1).1–3 Chiral sulfinamides serve as versatile auxiliary to access important chiral amine building blocks. For example, Ellman's tert-butanesulfinamide is an important precursor for the synthesis of chiral amines and can also function as ligands for asymmetric catalysis.4,5 Besides, sulfinamides have been utilized as amide bioisosteres and have been extensively employed in the research and development of anti-HIV drugs.6 They are commonly featured in active pharmaceutical ingredients (APIs) and agrochemical agents.7 Importantly, the sulfinamides are universal synthetic intermediates that can be used to prepare a variety of sulfur-containing functional groups, such as sulfonamide and sulfonimidamide.8–11 Due to their wide applications, numerous synthetic methods have been reported.12,13 Previous methods have primarily focused on converting sulfur-containing functional groups, such as sulfinates,14–20 thiols,21–24 and sulfenamide,25 which, in general, have limited availability from commercial vendors. Another approach features the formation of the C–S bond through the nucleophilic attack on the S(iv) functional groups such as N-sulfinylamine (R-NSO) with carbon-centered nucleophiles (Fig. 1b).26 These nucleophiles can engage with N-sulfinylamine via both ionic and radical pathways. The ionic pathway typically involves alkyl metallic reagents27 and transition-metal-catalyzed addition of internal alkenes,28 or arylboron compounds29–31 to N-sulfinylamine (R-NSO). However, the reactive organometallic reagents are generally air- and moisture-sensitive, which limits their functional group compatibility. Many substrates are challenging to handle and are available in limited commercial supply.
Fig. 1. Motivation and synthetic design.

To address the aforementioned issues, the development of efficient approaches to synthesize sulfinamides under relatively mild conditions by coupling N-sulfinylamines with highly reactive radicals has become increasingly attractive. In 2022, Li and co-workers first disclosed photoredox alkylation of sulfinylamines with 1,4-dihydropyri-dines (DHPs) as radical precursors to generate alkyl sulfinamides.32 Similar approaches employing potassium trifluoro(organo)borates as radical precursors with Tr-NSO have also been reported.33 Meanwhile, Willis et al. and Larionov et al. use readily available carboxylic acids as alkyl radical sources to achieve radical-mediated decarboxylated sulfinamidation.34,35 Recently, Hu and co-workers introduced a general photoredox protocol that employs aliphatic C–H bonds as latent radical precursors.36 Inspired by the protocol developed by MacMillan and co-workers, which involves the deoxygenation of alcohols with N-heterocyclic carbenes (NHC) to form the corresponding carbon radicals,37–45 we speculate that alkyl radicals generated from deoxygenation of alcohols by NHC could be combined with N-sulfinylamines to yield the corresponding sulfinamide products. Following this strategy, herein, we reported our development of photoredox-catalyzed deoxygenative radical transformation of alcohols to sulfinamides, enabling a variety of alcohols to be converted to sulfonamides through deoxygenative pathway under photocatalytic condition (Fig. 1c).
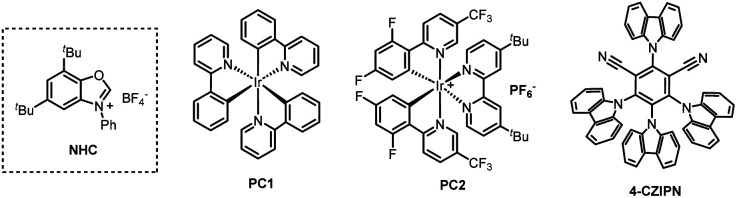
We initiated our study using (4-fluorophenyl)methanol (1) and N-sulfinyltritylamine (Tr-NSO) (2) as the model substrates (Table 1). After a series of conditions optimizations, we identified the standard conditions as following: using 2.3 equiv. of NHC (N-phenyl benzoxazolium salt) as alcohol C–O bond activating reagent, Ir(dF(CF3)ppy)2(dtbbpy)PF6 (PC2, 2 mmol%) as photocatalyst and HCOOCs as base in a mixture solution of THF/MeCN (1.5 : 1), sulfinamide 3a was isolated in 78% yield (Table 1, entry 1). The replacement of PC2 with other photocatalysts such as Ir(ppy)3, 4-CzIPN, resulted in less efficient reactions (Table 1, entries 2, 3). Alternative bases, such as Et3N or KOtBu, failed to deliver the product (Table 1, entry 4). Screening of a range of solvents revealed that the mixture of THF/MeCN is superior to single solvents or other polar solvents (Table 1, entries 5–9). The absence of a base or the use of alternative bases resulted in a suboptimal outcome, highlighting the critical role of HCOOCs in the reaction (Table 1, entries 10–12). In the absence of photocatalyst or light, the reaction was completely inhibited (Table 1, entries 13, 14).
Table 1. Optimization of reaction conditionsa.

| ||
|---|---|---|
| Entry | Variation from the standard conditions | Yieldb |
| 1 | None | 78% |
| 2 | PC1 instead of PC2 | 7% |
| 3 | 4-CZIPN | 71% |
| 4 | Et3N/KOtBu instead of pyridine | Trace |
| 5 | MTBE instead of THF | 34% |
| 6 | DCM instead of MeCN | Trace |
| 7 | DMSO instead of MeCN | 40% |
| 8 | DCE instead of MeCN | 20% |
| 9 | THF instead of MeCN | 71% |
| 10 | DIPEA instead of HCOOCs | N.D. |
| 11 | Cs2CO3 instead of HCOOCs | 42% |
| 12 | In the absence of base | Trace |
| 13 | In the absence of PC | N.D. |
| 14 | In the dark | N.D. |
| ||
Unless otherwise noted, all reactions were performed with 1 (0.25 mmol), 2 (0.10 mmol), NHC (0.23 mmol), pyridine (0.46 mmol), solvent A (1.5 mL), 15 min; then photocatalyst (0.002 mmol), base (0.2 mmol), solvent B (1.0 mL), blue LEDs at rt for 12 h.
Yield determined by 19F NMR using PhCF3 as an internal standard. N.D. = no detection.
With the optimized reaction conditions in hand, we then evaluated the substrate scope of alcohols in the construction of sulfinamides. As shown in Scheme 1, the reaction proceeded smoothly with benzyl alcohol substrates bearing an electron-donating or electron-withdrawing groups at the ortho-, meta- and para-position, yielding the corresponding sulfonamide in moderate to good yields (41–87%). Benzyl alcohols containing various functional groups, including alkyl (3b, 3c and 3l), alkoxy (3e, 3f and 3m), phenyl (3d), cyanide (3g, 3n), fluoro (3a, 3p), chloro (3i, 3q) bromo (3j, 3r) and trifluoromethyl (3k, 3o) groups, were suitable substrates for the transformation, demonstrating remarkable functional group compatibility. Polysubstituted benzyl alcohols also reacted smoothly with Tr-NSO to generate products 3s and 3t in 72% and 65% yields, respectively. Substitution at the ortho-position results in a relatively lower yield due to the steric effect, as exemplified by 3u. Notably, alcohols containing heterocycles, such as 3-thiophenemethanol and 3-furanmethanol, also yielded products in moderate yields (3v, 3x). Additionally, secondary alcohols delivered the desired product 3y in 41% yield with 1 : 1 dr. Propargyl alcohol was also smoothly converted to the desired sulfinamide product (3z). Unfortunately, simple alkyl alcohol is not suitable in the current catalytic system. Variation of the sulfinylamine reagent was also possible, with the N-t-octyl (2aa) and N-Si(i-Pr)3 (2ab) substituted reagents, both of which benefit from steric-stabilization, providing the corresponding sulfinamides in moderate yields.
Scheme 1. Substrate scope of sulfinamides.a aReactions performed on 0.2 mmol scale with alcohol (2.5 equiv.), NHC (2.3 equiv.), pyridine (4.6 equiv.), THF (2.5 mL), 15 min; then Tr-NSO (1.0 equiv.), HCOOCs (2.0 equiv.), PC2 (0.004 mmol), MeCN (2.0 mL), blue LEDs 12 h. See the ESI† for experimental details.

To showcase the practical applicability of our protocol, a gram-scale reaction was conducted under standard reaction conditions, yielding 1.05 g of 3b at a 51% yield (Scheme 2a). We established reaction conditions to convert the N-tritylsulfinamides into the S(vi) functional groups (Scheme 2b). Sulfonamides could be obtained by oxidation with m-CPBA, followed by a simple S(vi) functional groups (Scheme 2b). Sulfonamides could be obtained by oxidation with m-CPBA, followed by a simple treatment with MsOH, which removed the trityl group to yield primary sulfonamide 4. The reaction of sulfinamides with 0.5 equivalents of trichloroisocyanuric acid (TCCA), followed by the addition of an amine, delivered sulfonimidamides 5 and 6 in good yields. Subsequently, the trityl group was easily removed, resulting in free sulfoximine 7. Furthermore, treating of sulfoximine 7 with benzoyl chloride led to the formation of N-benzoyl sulfoximine (8).46N-Trifluoromethylthio sulfoximines (9) were prepared from the free sulfoximine 7via the corresponding N–Br derivative with excellent yields. Additionally, a phenyl group can also be incorporated onto the N–H of sulfoximine 7 through Chan–Lam coupling (10).47
Scheme 2. Gram scale synthesis and synthetic applications.

We envisioned achieving the C(sp3)–S cross-coupling of alcohols and N-tritylsulfinylamine via the photoredox catalysis pathway outlined in Fig. 2. Our proposed mechanism starts with the condensation of alcohol substrate 1a onto benzoxazolium salt, affording activated alcohol 11. Visible-light excitation of the photocatalyst yields an oxidizing, long lived triplet excited state. This excited state photocatalyst oxidizes the anilinic nitrogen atom, yielding aminium radical cation 12, which is readily deprotonated at the α-position, forming carbon-centered radical 13. Subsequent β-scission is thermodynamically favored, yielding and alkyl radical 14 and inert byproduct 15. This radical is then captured by N-sulfinylamine and subsequently undergoes single electron transfer (SET) with highly reducing photocatalyst, releasing cross coupling product 3a.
Fig. 2. Proposed mechanism.

In summary, we herein present an efficient protocol for the deoxygenative radical addition to N-tritylsulfinylamine, yielding sulfinamides through photoredox catalysis. This protocol is amenable to a wide range of functional groups, including primary and secondary benzyl alcohols, as well as propargyl alcohol. The sulfonamides were obtained in moderate to high yields. Synthetic applications for constructing various sulfur(vi)-centered functional groups demonstrate the utility of this protocol. Further exploration of this strategy for alkyl alcohol substrates is ongoing.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We gratefully acknowledge the funding support of the National Key R&D Program of China (2021YFF0701600), NSFC (22271053, 22471040, 21921003), and Shanghai Municipal Education Commission (20212308).
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra00158g
Notes and references
- Petkowski J. J. Bains W. Seager S. J. Nat. Prod. 2018;81:423–446. doi: 10.1021/acs.jnatprod.7b00921. [DOI] [PubMed] [Google Scholar]
- Apaydın S. Török M. Bioorg. Med. Chem. Lett. 2019;29:2042–2050. doi: 10.1016/j.bmcl.2019.06.041. [DOI] [PubMed] [Google Scholar]
- Li W. Zhang J. Acc. Chem. Res. 2024;57:489–513. doi: 10.1021/acs.accounts.4c00449. [DOI] [PubMed] [Google Scholar]
- Ellman J. A. Owens T. D. Tang T. P. Acc. Chem. Res. 2002;35:984–995. doi: 10.1021/ar020066u. [DOI] [PubMed] [Google Scholar]
- Robak M. T. Herbage M. A. Ellman J. A. Chem. Rev. 2010;110:3600–3740. doi: 10.1021/cr900382t. [DOI] [PubMed] [Google Scholar]
- Rönn R. Gossas T. Sabnis Y. A. Daoud H. Åkerblom E. Danielson U. H. Sandström A. Bioorg. Med. Chem. 2007;15:4057–4068. doi: 10.1016/j.bmc.2007.03.089. [DOI] [PubMed] [Google Scholar]
- Moree W. J. van Gent L. C. van der Marel G. A. Liskamp R. M. J. Tetrahedron. 1993;49:1133–1150. [Google Scholar]
- Aota Y. Kano T. Maruoka K. J. Am. Chem. Soc. 2019;141:19263–19268. doi: 10.1021/jacs.9b11298. [DOI] [PubMed] [Google Scholar]
- Greed S. Briggs E. L. Idiris F. I. M. White A. J. P. Lucking U. Bull J. A. Chem.–Eur. J. 2020;26:12533–12538. doi: 10.1002/chem.202002265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greed S. Symes O. Bull J. A. Chem. Commun. 2022;58:5387–5390. doi: 10.1039/d2cc01219g. [DOI] [PubMed] [Google Scholar]
- Tsuzuki S. Kano T. Angew. Chem., Int. Ed. 2023;62:e202300637. doi: 10.1002/anie.202300637. [DOI] [PubMed] [Google Scholar]
- Otocka S. Kwiatkowska M. Madalinska L. Kielbasinski P. Chem. Rev. 2017;117:4147–4181. doi: 10.1021/acs.chemrev.6b00517. [DOI] [PubMed] [Google Scholar]
- Wojaczyńska E. Wojaczyński J. Chem. Rev. 2020;120:4578–4611. doi: 10.1021/acs.chemrev.0c00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis F. A. Reddy R. E. Szewczyk J. M. Reddy G. V. Portonovo P. S. Zhang H. Fanelli D. Zhou P. Carroll P. J. J. Org. Chem. 1997;62:2555–2563. doi: 10.1021/jo970077e. [DOI] [PubMed] [Google Scholar]
- Billard T. Greiner A. Langlois B. R. Tetrahedron. 1999;55:7243–7250. [Google Scholar]
- Han Z. Krishnamurthy D. Grover P. Fang Q. K. Su X. Wilkinson H. S. Lu Z.-H. Magiera D. Senanayake C. H. Tetrahedron. 2005;61:6386–6408. [Google Scholar]
- Zhu R.-H. Shi X.-X. Tetrahedron: Asymmetry. 2011;22:387–393. [Google Scholar]
- Li B. Hu J. Liao M. Xiong Q. Zhang Y. Chi Y. R. Zhang X. Wu X. J. Am. Chem. Soc. 2024;146:25350–25360. doi: 10.1021/jacs.4c10486. [DOI] [PubMed] [Google Scholar]
- Liao M. Liu Y. Long H. Xiong Q. Lv X. Luo Z. Wu X. Chi Y. R. Chem. 2024;10:1541–1552. [Google Scholar]
- Wei T. Wang H.-L. Tian Y. Xie M.-S. Guo H.-M. Nat. Chem. 2024;16:1301–1311. doi: 10.1038/s41557-024-01522-z. [DOI] [PubMed] [Google Scholar]
- Chatterjee S. Makai S. Morandi B. Angew. Chem., Int. Ed. 2021;60:758–765. doi: 10.1002/anie.202011138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Z. Zou J. Mou C. Jin Z. Ren S.-C. Chi Y. R. Org. Lett. 2022;24:8907–8913. doi: 10.1021/acs.orglett.2c03770. [DOI] [PubMed] [Google Scholar]
- Feng C.-W. Wang D.-Y. Lu H.-L. Xi Z.-W. Shen Y.-M. Cao J. Org. Lett. 2022;24:4485–4489. doi: 10.1021/acs.orglett.2c01824. [DOI] [PubMed] [Google Scholar]
- Negrellos A. Rice A. M. Dos Santos P. C. King S. B. ACS Chem. Biol. 2023;18:2524–2534. doi: 10.1021/acschembio.3c00526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L.-j. Chen S.-s. Li G.-x. Zhu J. Wang Q.-w. Tang Z. ACS Catal. 2019;9:1525–1530. [Google Scholar]
- Davies T. Q. Willis M. C. Chem.–Eur. J. 2021;27:8918–8927. doi: 10.1002/chem.202100321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding M. Zhang Z.-X. Davies T. Q. Willis M. C. Org. Lett. 2022;24:1711–1715. doi: 10.1021/acs.orglett.2c00347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayeh L. Le P. Q. Tambar U. K. Nature. 2017;547:196–200. doi: 10.1038/nature22805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo P. K. T. Willis M. C. J. Am. Chem. Soc. 2021;143:15576–15581. doi: 10.1021/jacs.1c08052. [DOI] [PubMed] [Google Scholar]
- Shi Y. Yuan Y. Li J. Yang J. Zhang J. J. Am. Chem. Soc. 2024;146:17580–17586. doi: 10.1021/jacs.4c03473. [DOI] [PubMed] [Google Scholar]
- Xi L. Fang X. Wang M. Shi Z. J. Am. Chem. Soc. 2024;146:17587–17594. doi: 10.1021/jacs.4c04050. [DOI] [PubMed] [Google Scholar]
- Li L. Zhang S.-q. Chen Y. Cui X. Zhao G. Tang Z. Li G.-x. ACS Catal. 2022;12:15334–15340. [Google Scholar]
- Yan M. Wang S.-f. Zhang Y.-p. Zhao J.-z. Tang Z. Li G.-x. Org. Biomol. Chem. 2024;22:348–352. doi: 10.1039/d3ob01782f. [DOI] [PubMed] [Google Scholar]
- Andrews J. A. Kalepu J. Palmer C. F. Poole D. L. Christensen K. E. Willis M. C. J. Am. Chem. Soc. 2023;145:21623–21629. doi: 10.1021/jacs.3c07974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang H. T. Porey A. Nand S. Trevino R. Manning-Lorino P. Hughes W. B. Fremin S. O. Thompson W. T. Dhakal S. K. Arman H. D. Larionov O. V. Chem. Sci. 2023;14:13384–13391. doi: 10.1039/d3sc04727j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia G.-D. Li R. Zhang L. Wei Y. Hu X.-Q. Org. Lett. 2024;26:3703–3708. doi: 10.1021/acs.orglett.4c00612. [DOI] [PubMed] [Google Scholar]
- Dong Z. MacMillan D. W. C. Nature. 2021;598:451–456. doi: 10.1038/s41586-021-03920-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intermaggio N. E. Millet A. Davis D. L. MacMillan D. W. C. J. Am. Chem. Soc. 2022;144:11961–11968. doi: 10.1021/jacs.2c04807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R. Intermaggio N. E. Xie J. Rossi-Ashton J. A. Gould C. A. Martin R. T. Alcázar J. MacMillan D. W. C. Science. 2024;383:1350–1357. doi: 10.1126/science.adl5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D.-P. Zhang X.-S. Liu S. Hu X.-G. Nat. Commun. 2024;15:3401. doi: 10.1038/s41467-024-47711-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long A. Oswood C. J. Kelly C. B. Bryan M. C. MacMillan D. W. C. Nature. 2024;628:326–332. doi: 10.1038/s41586-024-07181-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao E. Prieto Kullmer C. N. Sakai H. A. MacMillan D. W. C. J. Am. Chem. Soc. 2024;146:5067–5073. doi: 10.1021/jacs.3c14460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S. Li Z. Wang L. Zeng T. Hu Q. Zhu J. Org. Lett. 2024;26:264–268. doi: 10.1021/acs.orglett.3c03857. [DOI] [PubMed] [Google Scholar]
- Wang J. Z. Lyon W. L. MacMillan D. W. C. Nature. 2024;628:104–109. doi: 10.1038/s41586-024-07165-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.-L. Gao Y.-Z. Cai S.-H. Yu H. Shen S.-J. Ping Q. Yang Z.-P. Nat. Commun. 2024;15:2733. doi: 10.1038/s41467-024-46713-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho G. Y. Bolm C. Org. Lett. 2005;7:1351–1354. doi: 10.1021/ol050176b. [DOI] [PubMed] [Google Scholar]
- Vijayan A. Rao D. N. Radhakrishnan K. V. Lam P. Y. S. Das P. Synthesis. 2020;53:805–847. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting this article have been included as part of the ESI.†