

Abstract
Cell Painting is an image-based assay that offers valuable insights into drug mechanisms of action and off-target effects. However, traditional feature extraction tools such as CellProfiler are computationally intensive and require frequent parameter adjustments. Inspired by recent advances in AI, we trained self-supervised learning (SSL) models DINO, MAE, and SimCLR on a subset of the JUMP Cell Painting dataset to obtain powerful representations for Cell Painting images. We assessed these SSL features for reproducibility, biological relevance, predictive power, and transferability to novel tasks and datasets. Our best model (DINO) surpassed CellProfiler in drug target and gene family classification, significantly reducing computational time and costs. DINO showed remarkable generalizability without fine-tuning, outperforming CellProfiler on an unseen dataset of genetic perturbations. In bioactivity prediction, DINO achieved comparable performance to models trained directly on Cell Painting images, with only a small gap between supervised and self-supervised approaches. Our study demonstrates the effectiveness of SSL methods for morphological profiling, suggesting promising research directions for improving the analysis of related image modalities.
Subject terms: Computational biology and bioinformatics, Drug discovery
Introduction
Morphological profiling uses image-based readouts to characterize the effect of chemical and genetic perturbations1–4 based on alterations in cell morphology. Offering high throughput and low cost, this technology has numerous applications in drug discovery such as mode of action identification5,6, off-target effect detection7,8, drug repurposing9–11, toxicity prediction12–14, and de novo molecular design15. One widely used assay for morphological profiling is Cell Painting, which utilizes 5 fluorescent dyes to stain 8 cellular compartments, generating thousands of morphological measurements per cell through automated image analysis. These high-dimensional readouts are used for hypothesis-free compound profiling, differentiating Cell Painting from target-based approaches. Despite significant progress in the visual representation learning field16–19, the analysis of Cell Painting images still largely relies on classical computer vision techniques20.
Conventional analysis of morphological profiling starts with single-cell segmentation, using CellProfiler20 or similar software tools21,22. The segmented cells are characterized using hand-crafted descriptors such as shape, size, intensity and texture23 among others. The descriptors are then aggregated to obtain a single vector of morphological features for each probed condition and feature selection methods are applied to reduce redundancy23. This multi-step workflow is computationally intensive and often requires adjustment of segmentation parameters when applied to new datasets. By contrast, deep learning models can offer a computationally efficient and segmentation-free alternative to morphological profiling.
The limited availability of biological labels has largely restricted the application of supervised learning in Cell Painting. Instead, morphological profiles are used for construction of biological maps24,25 to identify phenotypes and modes of action using the guilt-by-association principle. Specifically, clustering compounds or genes by morphological similarity provides mechanistic insights from annotated cluster members. Alternatively, classifiers trained on extracted features can predict downstream tasks, such as drug toxicity13 or cell health phenotypes26. However, label scarcity generally precludes end-to-end supervised learning from images, with only few exceptions27. The recently released JUMP Cell painting dataset28 (JUMP-CP) provides unprecedented opportunities for developing novel AI-based feature extraction methods. This large-scale image set (115 TB) contains approximately 117,000 chemical and 20,000 genetic perturbations. However, most compounds lack annotations, with only 4.5% having experimentally elucidated bioactivity. Thus, leveraging the full potential of this dataset requires techniques that do not rely on data curation or biological annotations.
Self-supervised learning (SSL) methods learn feature representations from unlabeled data through a pretext task. Early SSL pretext tasks focused on predicting image transformations29,30, but state-of-the-art performance has been achieved through methods that maximize the agreement between transformed views of the same image. For instance, PIRL31, MoCo32 and SimCLR17 use a contrastive loss to match paired views (“positives”) from the same image and repel unrelated views (“negatives”) from different images in the representation space. However, for optimal performance, contrastive methods require large minibatch sizes or memory banks, which can be computationally demanding. This limitation was overcome by recent non-contrastive approaches such as BYOL33 and DINO18. These approaches train a student network to predict the output of a teacher network while receiving different augmented views of an input image. Remarkably, DINO has been one of the best-performing SSL approaches across different domains18,34,35.
Recent advances in the SSL field have been accelerated by the adoption of the vision transformer36 (ViT) architecture. ViTs operate on image patches projected into tokens and use self-attention37 to capture global and local relationships between patches. The high computational cost of training ViT architectures inspired novel reconstruction-based pretext tasks, such as image masking, which provides a strong supervisory signal and improves training efficiency. This has been demonstrated by masked autoencoders19 and masked Siamese networks38, which achieved state-of-the-art results on natural images. Notably, ViT performance scales favorably with data volume and complexity39, making these models well-suited for the analysis of high-throughput imaging data.
Prior work has explored various approaches for single-cell feature extraction from high-content images, including transfer learning40, image inpainting41, variational autoencoders42, supervised43, self-supervised25,44,45 and weakly supervised learning46–48. However, existing single-cell methods require segmentation, leading to complex multistep workflows. Other approaches extract representations from whole images49–52, but rely on scarce labels or pretrained ImageNet weights, which restricts their application to 3-channel images or requires embedding concatenation for multichannel (> 3) assays such as Cell Painting. To date, only few approaches53–55 learn directly from microscopy images without segmentation or manually curated annotations. Moreover, a systematic study evaluating the benefits of SSL methods over classical analysis workflows for high-content imaging data is currently missing.
Here, we present the first comprehensive benchmark study of state-of-the-art SSL methods adapted for Cell Painting. We trained all SSL models directly in the applicability domain and assessed them on datasets with chemical and genetic perturbations. Our key contributions are:
Demonstrating that SSL approaches, particularly DINO, capture biologically meaningful image representations without segmentation and match or exceed the performance of cell-segmentation-derived features in drug target identification and gene clustering.
Showing in the context of morphological profiling that pretrained SSL models are transferable to new tasks and external datasets without fine-tuning.
Revealing a narrowing performance gap between supervised and SSL models for compound bioactivity prediction and identifying scenarios favoring each approach.
Shedding light on the advantages (reduction in data processing time) and limitations (higher susceptibility to cell count variations) of whole-image SSL approaches compared to segmentation-derived features.
Our results indicate that SSL methods provide a robust, efficient, and segmentation-free alternative to CellProfiler, and that SSL features demonstrate strong performance across a variety of clustering and predictive tasks.
Results
The central question of our study was whether segmentation-free SSL approaches could serve as a viable alternative to traditional cell-segmentation-based workflows, such as the standard CellProfiler pipeline56 for Cell Painting. To this end, we developed a framework for training SSL models on Cell Painting data and extracting features from images using these pretrained models. We established a rigorous evaluation strategy to assess SSL features across various computational tasks crucial to morphological profiling, including target identification5,6, similarity-based clustering24,25, and compound property prediction13,27. After a thorough comparison of several SSL methods, we focused on benchmarking the top-performing SSL approach (DINO) against CellProfiler, leveraging 2 public datasets28,57 with compound and gene perturbations. Additionally, we compared the performance of our best SSL model with supervised models27 trained directly on Cell Painting images to investigate whether transfer learning of SSL features could yield comparable results to resource-intensive, end-to-end training. Lastly, we examined potential limitations and practical advantages of segmentation-free SSL feature extraction methods.
SSL framework for segmentation-free morphological profiling
To establish a framework for training and evaluating self-supervised learning models on Cell Painting images, we adapted 3 state-of-the-art SSL approaches: SimCLR (simple framework for contrastive learning of visual representations)17, DINO (distillation with no labels)18 and MAE (masked autoencoder)19 (Fig. 1a). For benchmarking, we include two baselines (Fig. 1b): a standard CellProfiler20 pipeline for Cell Painting56 and transfer learning from a model pretrained on natural images36 (see “Methods”). Except for CellProfiler, all methods operate directly on image crops without requiring cell segmentation and use small (ViT-S) and base (ViT-B) vision transformer architectures as backbones.
Fig. 1.

Self-supervised learning for morphological profiling. (a) Schematic of the SSL models used in this study. All models are segmentation-free and use only image crops as input. SimCLR and DINO were trained on the pretext task of matching features from augmented views of the same image. MAE was trained on the image reconstruction task with partially masked input. (b) Schematic of the two baseline methods. CellProfiler pipeline for Cell Painting: conventional method based on single-cell segmentation and handcrafted features. Transfer learning: pretrained vision transformer that outputs per-channel features. (c) Training and evaluation workflow for SSL feature extraction models. The multisource compound dataset was used for training and validation. Two test datasets assessed model transferability: a genetic perturbation dataset from a JUMP-CP source not included in training/validation data and an external bioactivity prediction dataset. Evaluation metrics quantified reproducibility, biological relevance and predictive power of extracted features.
Since all SSL methods were originally designed for RGB images, we implemented several adaptations to enable their application on 5-channel Cell Painting images (more details in “Methods”). We exclusively applied ‘Flip’ and ‘Color’ augmentations for methods that rely on paired augmented views, as recommended by the augmentation ablation study in Supplementary Fig. 1. We excluded crops without cells during both training and inference, and accelerated inference by dividing images into smaller patches followed by averaging the patch embeddings. Additionally, we post-processed image embeddings using the most effective strategy for each feature type, which was determined based on the normalization and feature selection comparisons presented in Supplementary Figs. 2 and 3.
We pretrained all SSL methods on a subset of the JUMP-CP28 dataset containing 10,000 compounds imaged across 4 experimental sources, which we refer to as the multisource dataset (see Fig. 1c and “Methods”). Using these pretrained models, we extracted features to construct morphological profiles of chemical and genetic perturbations in validation and test sets (Fig. 1c). We generated perturbation profiles by averaging normalized features across replicates of the same perturbation. As JUMP-CP datasets contain several data sources and experimental batches, this aggregation was conducted at the batch, source, and full dataset levels (see “Methods”), enabling assessment of reproducibility across batches and sources. Full dataset aggregation produced consensus profiles, which were used for drug target and gene family classification.
Evaluation of feature extraction methods across JUMP-CP and external datasets
We evaluated all feature extraction methods on 3 held-out datasets. The multisource validation set contained target-annotated compounds with 2 drugs per target class (see Fig. 1c and “Methods”). This allowed us to assess the suitability of pretrained features for few-shot learning, an important task in morphological profiling, where only few examples per class are available. The gene perturbation test set (Fig. 1c) consisted of gene overexpression perturbations (see Methods) from a JUMP-CP source not used for training or validation. Including genetic perturbations allowed us to test the models’ ability to generalize to previously unseen perturbations, since our SSL models were trained only on images of chemically perturbed cells (Fig. 1c). Additionally, we used a compound bioactivity test set (Fig. 1c) to compare the performance of pretrained SSL models against supervised models trained directly on Cell Painting images. This further enabled us to assess the transferability of pretrained SSL models to new tasks and external datasets.
We compared feature extractors based on two key criteria: reproducibility and biological relevance (see Fig. 1c and “Methods”). To assess reproducibility, we used perturbation mAP (mAP: mean average precision), which quantifies the agreement between replicates of the same perturbation across experimental batches (see Methods). Biological relevance was evaluated by the agreement between perturbations with the same biological annotation, using target mAP as the metric (see Methods). Additionally, we used nearest neighbor (NN) accuracy of matching across experimental batches (not-same-batch or NSB accuracy) and across experimental batches and distinct perturbations (not-same-batch-or-perturbation or NSBP accuracy). For genetic perturbations, we additionally evaluated the clustering quality with respect to gene family labels, using the adjusted mutual information (AMI) metric (see Methods). We quantified the accuracy of compound bioactivity predictions using AUCROC and F1-score values. As all JUMP-CP perturbations were screened across multiple experimental batches in different laboratories, we could additionally assess batch and data source effects (see “Methods”).
Comparison of feature extraction methods: DINO surpasses other SSL approaches
First, we evaluated the performance of DINO, MAE and SimCLR on the multisource validation set (Fig. 2). DINO achieved the best results among the SSL methods, demonstrating superior reproducibility (Fig. 2a) and biological relevance (Fig. 2b). Additionally, DINO surpassed CellProfiler by a margin of 29% in perturbation mAP, which was significantly higher than that of CellProfiler (Wilcoxon test:  ). Although the difference in target mAP was not statistically significant (Wilcoxon test:
). Although the difference in target mAP was not statistically significant (Wilcoxon test:  ), DINO achieved an improvement of 61% over CellProfiler in this metric.
), DINO achieved an improvement of 61% over CellProfiler in this metric.
Fig. 2.

Performance comparison of SSL models (DINO, MAE, and SimCLR) and two baselines (CellProfiler and transfer learning): (a) reproducibility metrics perturbation not-same-batch (NSB) accuracy and mean average precision (mAP), (b) biological relevance metrics target not-same-batch-or-perturbation (NSBP) accuracy and mean average precision (mAP). Colors indicate different models and two randomized baselines: Shuffled CellProfiler (CellProfiler features with shuffled labels) and Random (random normally distributed features). Dotted lines indicate CellProfiler performance. Shapes indicate the vision transformer (ViT) architecture.
The superiority of DINO was even more evident in F1-score curves (Supplementary Fig. 4) and further confirmed by comparing reproducibility across JUMP-CP data sources (Supplementary Fig. 5, see “Methods”). DINO yielded similar or better performance to CellProfiler in 3 out of 4 JUMP-CP data sources (Supplementary Fig. 5a-d) and achieved higher perturbation mAP (+ 73%) and NSB accuracy (+ 2%) on a cross-source matching task (Supplementary Fig. 5e, see “Methods”).
Interestingly, models based on the larger ViT-B architecture showed only a marginal improvement over the ViT-S models (Fig. 2). This was observed for both the SSL methods and the transfer learning baseline. Given DINO’s superior performance and the minimal gains from larger ViT architectures, we focused our subsequent evaluations on DINO with the ViT-S architecture.
DINO outperforms CellProfiler in gene clustering
To compare DINO with the standard CellProfiler pipeline for Cell Painting, we used an independent test set comprising gene overexpression perturbations from a new source that was not seen during training (see “Methods”). As for chemical perturbations, DINO features demonstrated superior reproducibility (Fig. 3a). We also assessed the ability to predict gene family labels (see “Methods”) and found that DINO outperformed CellProfiler on metrics of biological relevance (Fig. 3b,c). Notably, DINO’s gene family mAP was significantly higher than that of CellProfiler (Wilcoxon test:  ).
).
Fig. 3.

Evaluation of DINO and CellProfiler on a gene overexpression dataset. DINO was trained only on images of chemically perturbed cells. Colors indicate feature extraction methods and two randomized baselines: Shuffled CellProfiler (CellProfiler features with shuffled labels) and Random (random normally distributed features). Dotted lines in (a, b) indicate CellProfiler performance. (a) Reproducibility metrics: perturbation not-same-batch (NSB) accuracy and mean average precision (mAP). (b) Metrics of biological relevance: gene family not-same-batch-or-perturbation (NSBP) accuracy and mean average precision (mAP). (c) F1-scores for matching gene family labels based on gene consensus profiles for a range of nearest neighbors k. (d, e) Hierarchical clustering of the 20 gene families with the highest intragroup correlations in the DINO and CellProfiler representation spaces, respectively. Detailed versions of the heatmaps displaying gene and gene family annotations for each row are presented in Supplementary Figs. 5–6.
Using pairwise gene similarity analyses, we tested the ability of morphological features to recapitulate gene families. For a selection of 20 gene families (see “Methods”), we performed hierarchical clustering of gene profiles for DINO (Fig. 3d) and CellProfiler (Fig. 3e). The resulting similarity maps were annotated to highlight groups based on gene and gene family labels (see “Methods”). Our qualitative comparison (Fig. 3d and Supplementary Fig. 6) revealed that DINO features recovered a larger number of gene groups and produced more homogeneous clusters than CellProfiler. DINO recapitulated 11 gene groups, including MAP4K family, Src kinases, MAP3K family, coronins, voltage-gated potassium channels, mitochondrial proteins, MAP kinase phosphatases, DAN family, HOX transcription factors, glycosyltransferases, and interferons. By contrast, CellProfiler recovered only 3 gene groups (mitochondrial proteins, interferons, and MAP kinase phosphatases) and 2 large clusters of mixed gene families (Fig. 3e and Supplementary Fig. 7).
To provide a more objective assessment, we computed the adjusted mutual information (AMI) between cluster assignments and gene family labels (see “Methods”). We found that DINO (AMI = 0.51) outperformed CellProfiler (AMI = 0.19) on the gene clustering task, indicating that DINO features excel at capturing gene family information. Since DINO was trained on images with compound-treated cells, these results demonstrate the remarkable generalizability of DINO, enabling its application to unseen data sources and conditions without parameter adjustments.
DINO matches the performance of supervised models in bioactivity prediction
Next, we evaluated the performance of DINO features in bioactivity prediction, comparing it to CellProfiler features and additionally to supervised models trained directly on Cell Painting images. Using the bioactivity prediction dataset (see “Methods”), we assessed the ability of various models to predict compound activity across 209 ChEMBL assays from Cell Painting data as was done in27. To evaluate the performance gap between supervised and self-supervised learning, we compared models trained on DINO features versus images directly. Specifically, we assessed a neural network (NN) trained on DINO features (see “Methods”) against 6 convolutional neural networks (CNNs) trained on Cell Painting images from27. We used DINO pretrained on the JUMP-CP data, allowing us to probe its cross-dataset transferability. As an additional baseline, a NN trained on CellProfiler features was incorporated from27.
After ranking all methods by mean AUCROC across 209 assays (see “Methods”), we found (Table 1) that the model trained on DINO features (AUCROC = 0.72) achieved comparable performance to GapNet (AUCROC = 0.73), the third best method. Although the top 3 CNNs had slightly higher mean AUCROC values, DINO outperformed 3 additional CNNs and a model trained on CellProfiler features (Table 1). Among the 8 methods compared, DINO ranked 4th for the number of assays predicted with AUCROC above 0.7 and 0.8. This confirms that DINO can generalize to novel datasets and tasks without fine-tuning.
Table 1.
Performance comparison of bioactivity prediction models.
| Method | Input | Arch | AUCROC | F1-score | AUC > 0.9 | AUC > 0.8 | AUC > 0.7 |
|---|---|---|---|---|---|---|---|
| ResNet | Images | CNN | 0.731 ± 0.19 | 0.508 ± 0.30 | 68 | 94 | 119 |
| DenseNet | Images | CNN | 0.730 ± 0.19 | 0.530 ± 0.30 | 61 | 98 | 121 |
| GapNet | Images | CNN | 0.725 ± 0.19 | 0.510 ± 0.29 | 63 | 94 | 117 |
| DINO | SSL features | NN | 0.723 ± 0.18 | 0.507 ± 0.31 | 56 | 84 | 108 |
| MIL-Net | Images | CNN | 0.711 ± 0.18 | 0.445 ± 0.32 | 61 | 81 | 105 |
| M-CNN | Images | CNN | 0.705 ± 0.19 | 0.482 ± 0.31 | 57 | 78 | 105 |
| SC-CNN† | Images | CNN | 0.705 ± 0.20 | 0.362 ± 0.29 | 61 | 83 | 109 |
| CellProfiler† | Handcrafted features | NN | 0.675 ± 0.20 | 0.361 ± 0.31 | 55 | 71 | 90 |
The values of best-performing methods are highlighted in bold.
Bioactivity prediction evaluation results ranked by AUCROC, with the best values highlighted in bold. The evaluated methods included a neural network (NN) trained on DINO features, 6 CNNs trained directly on Cell Painting images, and a NN trained on CellProfiler features. Performance was assessed on held-out test data, reporting means and standard deviations of AUCROC and F1-score values across all assays. Additionally, the number of assays with AUCROC above 0.9, 0.8, and 0.7 is reported for each method. The DINO results were obtained by applying a DINO model pretrained on the JUMP-CP data to the same dataset used in27. The remaining results are from this table of Hofmarcher et al.27. Methods marked with †require cell-level segmentation, while the others use whole images as input. AUCROC area under the receiver operating characteristic curve.
Nevertheless, the top 3 CNNs surpassed the DINO-based model in the number of assays predicted with AUCROC > 0.9, indicating a performance gap. Further analysis revealed that the model trained on DINO features offered comparable performance on assays with limited data (Supplementary Fig. 8). For assays with less than 100 activity labels available, the DINO model produced a similar number of accurate predictions and median AUCROC as the top 3 CNNs. However, on assays with 100–500 labels, CNNs showed a higher number of accurate predictions than the DINO model, consistent with SSL features excelling in few-shot settings58. These results demonstrate that pretrained DINO features can serve as a computationally efficient and less data-greedy alternative to end-to-end supervised training.
DINO captures both biological and technical variation in the data
Beyond clustering and compound property prediction, morphological features are commonly used in exploratory analyses, such as dimensionality reduction, to provide a global dataset overview. To assess the suitability of DINO for such analyses, we used UMAP59 (see Methods) to embed DINO and CellProfiler features in 2 dimensions. To assess whether feature embeddings produced biologically meaningful clusters, we highlighted a selection of 20 drug targets in the UMAP space (see “Methods”). All embeddings grouped compounds with the same target to some extent (Fig. 4). DINO and CellProfiler embeddings yielded well-separated clusters in the UMAP, highlighting targets such as NAMPT, PAK1, and RET (Fig. 4). Additionally, DINO embeddings (Fig. 4a) demonstrated superior cluster separation for KRAS, AKT1, DNMT3A, TGFBR1, CDK2, and CDK7 compared with CellProfiler (Fig. 4b). These results further support that DINO features are biologically meaningful and at least as powerful as those from CellProfiler.
Fig. 4.

Two-dimensional UMAP embeddings of well-level features from DINO and the CellProfiler baseline on the multisource validation set. Colors highlight target labels for a selection of 20 targets (see “Methods”). Perturbations with other targets are depicted as grey hollow points.
We then examined feature robustness with respect to technical sources of variation by coloring UMAP embeddings by experimental batch and data source (Supplementary Fig. 9b,c). DINO embeddings showed higher sensitivity to technical variations in UMAP space compared to CellProfiler, displaying a stronger separation between experimental batches and sources. Upon closer inspection, we found that the most pronounced technical effects occurred within DMSO negative controls (Supplementary Fig. 9a,c) and that the differences strongly correlated with variations in cell count (Supplementary Fig. 9d). We hypothesize that CellProfiler is more robust towards cell count variations since the features are extracted from single cells. Recognizing that UMAPs provide only a two-dimensional view of the data, we further quantified the impact of technical variation on high-dimensional features (see “Methods”) and found that both DINO and CellProfiler features were similarly affected by experimental batch and source effects (Supplementary Fig. 10). These findings reveal potential limitations of whole-image SSL approaches, necessitating careful consideration of technical variation in morphological profiling data.
SSL pipeline is significantly faster than segmentation-based feature extraction
Efficient data processing is crucial in large-scale compound screens driving preclinical drug discovery. With robotic automation generating vast amounts of data, computational speed becomes a significant factor in accelerating research throughput. To this end, we additionally benchmarked the computation time and cloud costs of feature extraction using DINO versus CellProfiler. For this comparison, we used 12 GPU-accelerated cloud instances for DINO and 12 CPU-intensive instances for CellProfiler (Supplementary Table 1). We found that DINO was 50 times faster than CellProfiler, with an average processing time of 1.3 min per plate (Supplementary Table 1). Despite the need for GPU resources, the cloud costs per plate were over 50 times lower for DINO than for CellProfiler (Supplementary Table 1). Moreover, our approach offers a simpler workflow which processes images end-to-end, in contrast to the multi-step CellProfiler workflow which requires illumination correction, segmentation, feature extraction and selection.
Taken together, our findings show that image-level SSL methods are a viable alternative to traditional segmentation-based approaches, offering improved performance, generalizability to new datasets, speed, and lower workflow complexity and computational costs.
Discussion
To assess the applicability of SSL for Cell Painting, we trained and evaluated 3 state-of-the-art methods DINO, MAE, and SimCLR on complex datasets with chemical and genetic perturbations. Using reproducibility and biological relevance as our main evaluation criteria, we showed that our best model, DINO, outperformed the established feature extraction tool, CellProfiler, in drug target and gene family classification, with even greater improvements in gene clustering.
In particular, DINO captured informative cell-related features that generalized to unseen datasets without parameter fine-tuning. While trained only on compound perturbations, DINO achieved superior classification and clustering performance on an unseen gene overexpression set, facilitating the construction of biological maps24. For compound activity prediction, DINO features transferred well to a new dataset, with the model trained on DINO features achieving comparable performance to CNNs27 trained end-to-end on that dataset directly. The direct transferability of SSL models simplifies the analysis of new datasets in contrast to CellProfiler, which requires frequent parameter adjustments.
Previous studies46,48,52 fine-tuned ImageNet-pretrained networks to learn representations for Cell Painting, often curating the training set through compound preselection. These methods process each channel independently and output concatenated channel representations, increasing computational complexity and feature redundancy. By contrast, our SSL models are tailored for uncurated 5-channel images, resulting in compact representations with lower redundancy. More recently, DINO was applied for learning single-cell morphological representations44,45, with45 reporting superior performance over CellProfiler. However, unlike these approaches, our SSL framework operates without cell segmentation, streamlining feature extraction.
Our analysis revealed that models trained on SSL features excel in bioactivity prediction when ground-truth labels are scarce, while dedicated supervised methods achieve superior performance given ample labeled data. Since compound annotations are sparse, training supervised models directly from images remains infeasible in most but few27 cases, which makes the use of SSL features an attractive alternative to harness large-scale unlabeled data such as the JUMP-CP dataset.
With a 50-fold reduction in compute time and costs compared to CellProfiler, SSL feature extraction methods can facilitate compound screening campaigns of unprecedented scale, revolutionizing the pace of early drug discovery. However, pretraining a DINO model demands substantial compute resources, requiring approximately 300 GPU hours. Given the intensive resource requirements, our study used only subsets of the JUMP-CP dataset, leaving room for exploration of the full dataset’s potential. Investigating the emerging properties of larger self-supervised ViTs trained on the complete JUMP-CP dataset offers promising research directions.
One limitation of our segmentation-free approach is that it operates at image-crop level and does not provide insights into cell heterogeneity, making CellProfiler a more suitable tool for single-cell analyses. Additionally, CellProfiler provides interpretability, by linking individual features to specific microscopy channels and mathematically defined morphological descriptors. However, even with self-supervised ViTs, we can gain some level of interpretability by examining self-attention maps (see Supplementary Fig. 11). Furthermore, in UMAPs, SSL methods showed higher susceptibility to experimental batch and laboratory effects compared to CellProfiler. Post-hoc approaches like Harmony60 can mitigate the effect of technical variation on SSL features. Alternatively, incorporating batch alignment as an additional objective during pretraining may produce more robust SSL representations.
Our SSL models showed generalizability across Cell Painting datasets but remain limited in transferability across other microscopy modalities, requiring fine-tuning for assays with different staining than Cell Painting. Drawing inspiration from natural language and image domains61–64, we encourage the development of assay-agnostic foundation models for microscopy images, which can standardize and expedite the analysis of high-content assays across various imaging modalities.
Methods
Technical terminology
Image features are high-dimensional readouts extracted from images using segmentation-based or deep learning approaches. We use the terms “representations” and “features” interchangeably. Feature vectors can be embedded into 2 dimensions for visualization; we refer to these projections as embeddings.
Cell Painting assay is conducted in 384-well plates, with each well imaged at several locations to produce multiple images or fields of view (FOVs). Well profiles or well features refer to features aggregated for each well across multiple FOVs. Cell Painting screening is performed in experimental batches containing groups of plates. Unless specified otherwise, the term “batch” refers to an experimental batch and not to a minibatch used for training deep learning models. In the JUMP Cell Painting consortium, several laboratories or data sources generated the data, adding a hierarchical level above the plate and batch levels.
Cells in each well are treated with a specific perturbation (e.g., compound or gene overexpression). This provides perturbation labels to assess reproducibility across repeated measurements or replicates. We used several subsets of the JUMP-CP data, which we refer to as datasets. Aggregating perturbation features across all replicates in a dataset produces a consensus profile. For a small subset of compounds, we have drug target labels, i.e., proteins targeted by these drugs. Gene overexpression perturbations correspond to individual genes which can be annotated and grouped in gene families. To evaluate biological relevance, we used target labels for compounds and gene family labels for genetic perturbations.
JUMP-CP training and validation sets
We used subsets of the JUMP Cell Painting dataset28 (cpg0016-jump) for training and evaluation of self-supervised learning (SSL) models. The complete JUMP-CP dataset (115 TB) includes 116,750 chemical perturbations, 12,602 gene overexpression and 7,975 CRISPR perturbations probed in a human cancer cell line (U2OS) in 5 replicates. Each chemical perturbation was screened by 5 out of the 10 consortium laboratories (“sources”) that used a standardized protocol but possibly different instrumentation. Genetic perturbations were screened solely by sources 4 and 7.
For model training, we used only images of cells treated with chemical perturbations. The multisource training set contains 564,272 images from 4 JUMP-CP sources: source 2, source 3, source 6, and source 8. The training set includes 5 replicates of 10,057 compounds from Selleckchem and MedChemExpress bioactive libraries, with two replicates originating from source 3. An overview of the JUMP-CP batches and plates used for model training is provided in Supplementary Data 1.
For model validation, we used JUMP Target2 plates28 that contain 306 compounds with drug target labels. These plates were imaged in every experimental batch, enabling us to not only assess model performance using biological labels, but also evaluate batch and laboratory effects. The multisource (16 batches, 75,545 images) validation set was constructed using JUMP-Target2 plates which were withheld from the training data. JUMP-CP batches and plates used for evaluation of models are listed in Supplementary Data 2.
JUMP-CP gene overexpression test set
The JUMP-CP28 gene overexpression data (source 4) with Open Reading Frames (ORFs) was used as a test set. A subset of the gene overexpression data was constructed by selecting ORF perturbations with high replicate correlations (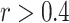 ) in the CellProfiler feature space, resulting in 5198 ORFs. Gene group memberships were assigned to each ORF perturbation using the HUGO Gene Nomenclature Committee (HGNC) gene annotation (hgnc_complete_set_2022-10-01.txt). To ensure robust evaluation based on gene annotations, we selected only those gene groups with at least 4 unique ORFs. 1970 ORF perturbations (Supplementary Data 3) satisfied this criterion. The complete list of batches and plates is provided in Supplementary Data 2.
) in the CellProfiler feature space, resulting in 5198 ORFs. Gene group memberships were assigned to each ORF perturbation using the HUGO Gene Nomenclature Committee (HGNC) gene annotation (hgnc_complete_set_2022-10-01.txt). To ensure robust evaluation based on gene annotations, we selected only those gene groups with at least 4 unique ORFs. 1970 ORF perturbations (Supplementary Data 3) satisfied this criterion. The complete list of batches and plates is provided in Supplementary Data 2.
Bioactivity prediction dataset
The Cell Painting dataset57 (cpg0012-wawer-bioactivecompoundprofiling) with bioactives profiled 30,000 small-molecule compounds in the U2OS cell line. For bioactivity prediction, we only used a subset of 10,000 compounds with activity labels from a study27, in which convolutional neural networks (CNNs) were trained to predict compound activity directly from Cell Painting images.
Image preprocessing
The JUMP-CP consortium28 generated Cell Painting images with 5 color channels (Mito, AGP, RNA, ER, DNA) that were stored as individual TIFF files. To optimize data loading, we combined the single-channel images into 5-channel TIFF files, resulting in a sixfold acceleration in training time. Prior to storage, we preprocessed the images: for each channel, intensities were clipped at 0.01st and 99.9th percentiles and scaled to the range [0,1]. Additionally, we calculated the Otsu threshold in the DNA staining channel and saved it as image metadata. During training, this threshold value enabled us to sample non-empty crops based on the minimum percentage of foreground area in the DNA channel.
During training, we sampled random crops (224 × 224 pixels) from the images and provided these as inputs to the models. We only used image crops with cells, which was ensured by imposing a lower bound of 1% on the Otsu-thresholded area in the DNA channel. For SimCLR and DINO, we additionally applied the augmentation pipeline, described in “Augmentations for multichannel images”, to generate multiple views from the sampled crops. All input crops were centered and scaled using channel intensity means and standard deviations estimated over the entire training set.
Augmentations for multichannel images
We systematically assessed augmentation strategies for contrastive learning approaches (Supplementary Fig. 1), exploring 6 augmentation transforms: ‘Flip’, ‘Resize’, ‘Color’, ‘Drop channel’, ‘Gaussian noise’ and ‘Gaussian blur’. ‘Flip’ rotates an image by 180 degrees along the horizontal or vertical axes. ‘Resize’ generates crops with dimensions varying between 12% and 47% of the whole microscopy image and rescales the output to 224 × 224 pixels. ‘Color’ introduces a random intensity shift:  and a random brightness change:
and a random brightness change:  with intensity values restricted to
with intensity values restricted to  . ‘Drop channel’ omits one channel from the image at random with probability
. ‘Drop channel’ omits one channel from the image at random with probability 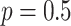 . The dropped channel is padded with zeros. ‘Gaussian noise’ adds random noise to the image:
. The dropped channel is padded with zeros. ‘Gaussian noise’ adds random noise to the image: 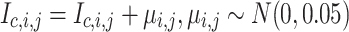 . ‘Gaussian blur’ applies a Gaussian filter with a kernel size of 23 pixels and a standard deviation uniformly sampled from
. ‘Gaussian blur’ applies a Gaussian filter with a kernel size of 23 pixels and a standard deviation uniformly sampled from  .
.
The ‘Color’ augmentation had the greatest positive impact both in terms of perturbation mAP and NSB accuracy, while the ‘Resize’ operation decreased performance. The remaining augmentations had a negligible effect relative to the ‘Color’ augmentation alone. Based on these findings, we selected the ‘Color’ and ‘Flip’ augmentations for training SimCLR and DINO. For training MAE, only the ‘Flip’ augmentation was used, as MAE learns representations through masked reconstruction rather than augmented view matching.
Model training details
We implemented distributed training with 2 NVIDIA Tesla V100 GPUs (32 GB VRAM) per SSL model. We used small (ViT-S/16) and base (ViT-B/16) variants of the vision transformer, with a patch size of 16 pixels. The models with ViT-S/16 were trained for 200 epochs, while those with ViT-B/16 were trained for 400 epochs. We used the AdamW optimizer and saved checkpoints every 20 epochs. A brief exposition of model-specific hyperparameters is provided below. For a comprehensive overview of training hyperparameters refer to Supplementary Table 2.
DINO was trained with a total minibatch size of 128 (192 for ViT-B), a learning rate of 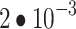 (
( for ViT-B), and a weight decay linearly increasing from 0.04 to 0.4. The learning rate followed a 20-epoch linear warmup followed by a cosine decay. For each image, 8 local crops (96 × 96) and 2 global crops (224 × 224) were sampled. DINO is a joint-embedding model with a student–teacher architecture18. DINO projects representations into a high-dimensional (here 20,000-dimensional) space where the temperature-scaled cross-entropy loss is optimized using a temperature of 0.1 for the student and 0.04 for the teacher network. The teacher temperature followed a linear warmup starting from 0.01 for 30 epochs.
for ViT-B), and a weight decay linearly increasing from 0.04 to 0.4. The learning rate followed a 20-epoch linear warmup followed by a cosine decay. For each image, 8 local crops (96 × 96) and 2 global crops (224 × 224) were sampled. DINO is a joint-embedding model with a student–teacher architecture18. DINO projects representations into a high-dimensional (here 20,000-dimensional) space where the temperature-scaled cross-entropy loss is optimized using a temperature of 0.1 for the student and 0.04 for the teacher network. The teacher temperature followed a linear warmup starting from 0.01 for 30 epochs.
Masked autoencoder (MAE) was trained with a total minibatch size of 1024 (1536 for ViT-B), a learning rate of 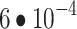 (
(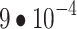 for ViT-B), and a weight decay of 0.05. The learning rate followed a 30-epoch linear warmup followed by a cosine decay. Given a partially masked input image, MAE reconstructs the missing regions using an asymmetric encoder-decoder architecture19, with a significantly smaller decoder. To accelerate data loading, 4 random crops were sampled from each image during training. The masking ratio was set to 50%, and image augmentation was performed using only horizontal and vertical flips.
for ViT-B), and a weight decay of 0.05. The learning rate followed a 30-epoch linear warmup followed by a cosine decay. Given a partially masked input image, MAE reconstructs the missing regions using an asymmetric encoder-decoder architecture19, with a significantly smaller decoder. To accelerate data loading, 4 random crops were sampled from each image during training. The masking ratio was set to 50%, and image augmentation was performed using only horizontal and vertical flips.
SimCLR was trained with a total minibatch size of 256, a learning rate of  , and a weight decay of 0.1. The learning rate followed a 30-epoch linear warmup followed by a cosine decay. SimCLR is a contrastive approach17 that aims to match augmented views from the same image in the representation space (“positives”), while pushing away representations from different images (“negatives”). The temperature-scaled cross-entropy loss was used as the objective function with a constant temperature value of 0.2.
, and a weight decay of 0.1. The learning rate followed a 30-epoch linear warmup followed by a cosine decay. SimCLR is a contrastive approach17 that aims to match augmented views from the same image in the representation space (“positives”), while pushing away representations from different images (“negatives”). The temperature-scaled cross-entropy loss was used as the objective function with a constant temperature value of 0.2.
SSL inference and postprocessing
A feature extraction model maps an input image 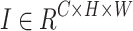 to a d-dimensional feature space through a mapping function
to a d-dimensional feature space through a mapping function 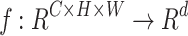 . In DINO, MAE, and SimCLR, the mapping
. In DINO, MAE, and SimCLR, the mapping  is performed by a vision transformer (ViT) backbone. The dimensionality
is performed by a vision transformer (ViT) backbone. The dimensionality 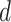 of learned features depends on the network architecture, with
of learned features depends on the network architecture, with 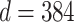 for ViT-S and
for ViT-S and  for ViT-B.
for ViT-B.
At inference, each microscopy image 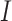 , corresponding to a single field of view (FOV), was split into 224 × 224 image crops
, corresponding to a single field of view (FOV), was split into 224 × 224 image crops  . All image crops were passed through a pretrained ViT backbone to generate crop features
. All image crops were passed through a pretrained ViT backbone to generate crop features 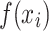 . Crops with no cells were excluded following the same criteria used during training. The resulting image crop features were aggregated using the arithmetic mean:
. Crops with no cells were excluded following the same criteria used during training. The resulting image crop features were aggregated using the arithmetic mean: 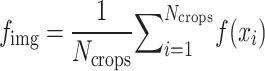 . Well features were obtained by taking the mean across all FOV images:
. Well features were obtained by taking the mean across all FOV images: 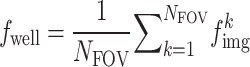 .
.
We tested several feature postprocessing methods (Supplementary Figs. 2–3) and chose “sphering + MAD robustize” for SSL features. First, we removed well features with variance less than  . We then applied a sphering transformation40
. We then applied a sphering transformation40
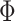 computed on dataset-wide negative controls. After sphering, well profiles were normalized using whole-plate median (
computed on dataset-wide negative controls. After sphering, well profiles were normalized using whole-plate median ( ) and median absolute deviation (
) and median absolute deviation ( ):
):
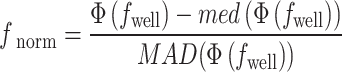 |
To generate perturbation profiles for downstream analyses, we averaged normalized well features  across multiple replicates. Aggregation was performed at several levels: batch-aggregated profiles average all replicates within an experimental batch, source-aggregated profiles average all replicates within a JUMP-CP data source, and consensus profiles average all replicates within an entire dataset (multisource/gene overexpression/bioactivity prediction).
across multiple replicates. Aggregation was performed at several levels: batch-aggregated profiles average all replicates within an experimental batch, source-aggregated profiles average all replicates within a JUMP-CP data source, and consensus profiles average all replicates within an entire dataset (multisource/gene overexpression/bioactivity prediction).
CellProfiler features
We used CellProfiler features provided by the JUMP-CP consortium (https://registry.opendata.aws/cellpainting-gallery/, for details see28). CellProfiler features were normalized using whole-plate median and MAD (“MAD robustize” normalization), which was the best postprocessing method for this feature type (Supplementary Fig. 2e). We tested several feature selection approaches (Supplementary Fig. 3) and selected the set of 560 features from the CPJUMP1 study65, in which low-variance and redundant features were removed based on a dataset with chemical and genetic perturbations.
Transfer learning
A vision transformer36 (ViT-S/16 or ViT-B/16) pretrained on the image classification task on ImageNet-1 K was used to extract ‘transfer learning’ features. Each of the 5 channels was duplicated 3 times to generate pseudo-RGB images, which were individually passed through a pretrained ViT. The transfer learning features were obtained by concatenating individual channel features, resulting in 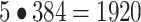 -dimensional feature vectors. As for SSL methods, low variance features
-dimensional feature vectors. As for SSL methods, low variance features 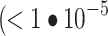 ) were removed before normalization. The transfer learning features were normalized using whole-plate median and MAD (“MAD robustize” normalization), which was the best postprocessing method for transfer learning (Supplementary Fig. 2d).
) were removed before normalization. The transfer learning features were normalized using whole-plate median and MAD (“MAD robustize” normalization), which was the best postprocessing method for transfer learning (Supplementary Fig. 2d).
Evaluation of reproducibility and biological relevance
We evaluated all features based on two key criteria: reproducibility using perturbation labels and biological relevance using drug target or gene family labels (see “Technical terminology”). All evaluations excluded negative controls and considered only conditions with compound treatments and non-control genetic perturbations. To assess the sensitivity and precision of inferring ground-truth labels based on pairwise feature distances 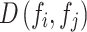 , we followed an approach similar to65. For each perturbation
, we followed an approach similar to65. For each perturbation 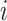 , we define a neighborhood
, we define a neighborhood 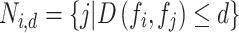 consisting of all other perturbations
consisting of all other perturbations 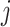 within a cosine distance threshold
within a cosine distance threshold  of perturbation
of perturbation 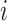 . We then compared the label
. We then compared the label 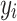 of perturbation
of perturbation 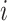 with the labels
with the labels 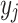 of its nearest neighbors {
of its nearest neighbors { . The precision
. The precision  and recall
and recall  of matching labels for perturbation
of matching labels for perturbation 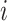 at distance threshold
at distance threshold 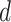 were calculated as:
were calculated as:
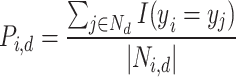 |
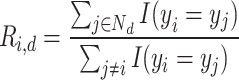 |
where 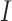 is an indicator function, and
is an indicator function, and 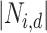 is the size of the neighborhood of perturbation
is the size of the neighborhood of perturbation 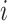 .
.
The distance threshold 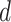 was uniformly sampled between 0 and 1 at 100 increments to compute aggregate metrics. For each perturbation
was uniformly sampled between 0 and 1 at 100 increments to compute aggregate metrics. For each perturbation  , average precision (
, average precision ( ) was computed as the sum of precision values weighted by changes in recall across these threshold values:
) was computed as the sum of precision values weighted by changes in recall across these threshold values:
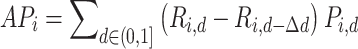 |
Mean average precision (mAP) was then calculated by averaging the AP values across all perturbations:
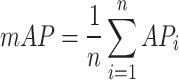 |
Perturbation mAP, measuring reproducibility, was estimated on batch-aggregated profiles (see “SSL inference and postprocessing”), thus quantifying the ability to match perturbations across batches. Target mAP was estimated on consensus profiles (see “SSL inference and postprocessing”) using drug target labels, focusing on biological content after technical variations were averaged out. For genetic perturbations, biological relevance was estimated using gene family mAP, calculated on consensus profiles with gene family labels. To evaluate the cross-source matching ability of features (Supplementary Fig. 5e), perturbation mAP was calculated on source-aggregated profiles. Along with AP values, F1-scores at k nearest neighbors were computed and visualized (Supplementary Fig. 4) to investigate whether some features worked better in specific k ranges.
The second class of metrics, widely used in morphological profiling40,54,66, reports the nearest neighbor (NN) accuracy estimated on well profiles with restrictions on the possible match. To evaluate reproducibility, the not-same-batch (NSB) accuracy restricts true positive matches to well profiles from different experimental batches. The not-same-batch-or-perturbation (NSBP) accuracy restricts true positive matches to profiles from both different batches and distinct perturbations. We used perturbation NSB accuracy to evaluate feature reproducibility across batches and target NSBP accuracy to evaluate biological relevance.
UMAP embeddings
To visualize features in 2 dimensions, we generated UMAP (Uniform Manifold Approximation and Projection)59 embeddings using the first 200 principal components as input, with the correlation distance as the metric. For optimal visualization, we set the number of nearest neighbors to 50 and the minimum distance between points to 0.7 in the UMAP algorithm.
We selected 20 drug targets with the highest mean F1-scores from the JUMP-Target2 plate28 annotation set. The F1-scores were determined by performing target classification using CellProfiler and DINO features. Supplementary Table 3 provides the list of these 20 targets and their F1-scores stratified by feature type.
Quantification of technical biases
The impact of technical variation was assessed by examining well profiles of the multisource validation dataset, which contained 24 replicates of each perturbation (see “Technical terminology”). To quantify batch and source effects for each feature type, we compared within- and between-cluster similarity and connectivity, using batch and source information as cluster labels. We used 3 metrics: Silhouette scores67, Graph Connectivity (GC)68 and Local Inverse Simpson’s Index (LISI)68.
The silhouette score measures the similarity of an observation 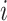 to its own cluster (batch/source) relative to the nearest cluster67. It calculates the relative difference between the mean intra-cluster distance
to its own cluster (batch/source) relative to the nearest cluster67. It calculates the relative difference between the mean intra-cluster distance 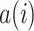 and the mean nearest-cluster distance
and the mean nearest-cluster distance 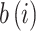 :
:
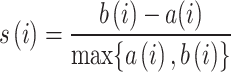 |
The silhouette score ranges from -1 to 1, with higher values indicating the observation is well matched to its own cluster and poorly matched to neighboring clusters. We compared distributions of silhouette scores for all well profiles clustered by batch or source (Supplementary Fig. 10c,f).
The GC and LISI metrics are based on a k-nearest neighbor (kNN) graph G(V, E). This graph consists of vertices V corresponding to well profiles. Each vertex is connected to its k nearest neighbors based on pairwise cosine distances defining the edge set E. Let  be a set of clusters, such as batches or sources. Taking only the vertices of a specific cluster
be a set of clusters, such as batches or sources. Taking only the vertices of a specific cluster 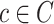 induces a subgraph
induces a subgraph  . GC measures the ratio between the number of vertices in the largest connected component (LCC) of
. GC measures the ratio between the number of vertices in the largest connected component (LCC) of  and the total number of vertices in
and the total number of vertices in  , averaged across all clusters:
, averaged across all clusters:
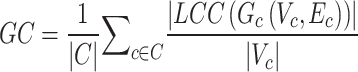 |
If the LCC of 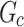 is almost as large as
is almost as large as  itself, this indicates that vertices from the same cluster are close together—a sign of batch/source effects. We reported GC for k = 1, 2, 3, 5, 10, 15 (Supplementary Fig. 10a,d).
itself, this indicates that vertices from the same cluster are close together—a sign of batch/source effects. We reported GC for k = 1, 2, 3, 5, 10, 15 (Supplementary Fig. 10a,d).
LISI quantifies neighborhood diversity in the kNN-graph G using the inverse Simpson’s index:
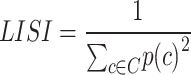 |
where 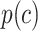 is the relative abundance of cluster
is the relative abundance of cluster  . LISI can be interpreted as the expected number of profiles to be sampled before two are drawn from the same cluster60. Higher LISI implies more diverse neighborhoods and lower batch/source effects. LISI was calculated for k = 15, 30, 60, 90 (Supplementary Fig. 10b,e). For both GC and LISI, the values for k are based on68.
. LISI can be interpreted as the expected number of profiles to be sampled before two are drawn from the same cluster60. Higher LISI implies more diverse neighborhoods and lower batch/source effects. LISI was calculated for k = 15, 30, 60, 90 (Supplementary Fig. 10b,e). For both GC and LISI, the values for k are based on68.
For the sake of interpretability, we incorporated two baselines: (1) a Gaussian baseline, which simulates non-overlapping Gaussian ( ) clusters with the number of clusters equal to the number of batches/sources and with feature dimensionality identical to that of CellProfiler features; (2) a random baseline, which corresponds to the Gaussian baseline but with randomized cluster assignments.
) clusters with the number of clusters equal to the number of batches/sources and with feature dimensionality identical to that of CellProfiler features; (2) a random baseline, which corresponds to the Gaussian baseline but with randomized cluster assignments.
Hierarchical clustering of genetic perturbations
For the clustering analysis, we only considered HGNC gene families (see “Gene overexpression data”) that contained between 4 and 10 unique ORF perturbations. Since many of the gene families were heterogeneous and uncorrelated, we only selected the top 20 gene families with the highest within-family correlations in the respective feature space, resulting in 2 gene sets for DINO and CellProfiler (Supplementary Data 4).
To cluster these gene sets, we calculated the gene–gene correlation matrix within the respective representation space, which was then provided as input for hierarchical clustering using the complete linkage method and the Euclidean distance metric. To highlight biologically meaningful clusters in box frames, adjacent gene groups with at least 3 perturbations were identified visually and labeled with the majority gene family label.
To evaluate the quality of hierarchical clustering of genetic perturbations, we used adjusted mutual information (AMI). For a given number of clusters, mutual information (MI) quantifies the dependence between cluster assignment labels 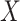 and gene family labels
and gene family labels  :
:
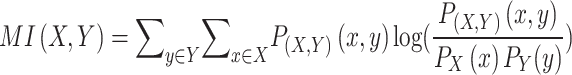 |
Adjusting mutual information (MI) for random chance results in AMI:
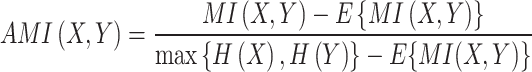 |
Assessing the performance gap between supervised and self-supervised learning
We used DINO pretrained on the multisource JUMP-CP dataset to extract morphological features from Cell Painting images of the bioactivity prediction dataset. We trained a 3-layer fully connected neural network (FNN) on the extracted DINO features to predict compound activity. To ensure comparability with the CNNs trained on Cell Painting images directly, we used the same code base (https://github.com/ml-jku/hti-cnn), activity labels and train/validation/test splits as27.
We included 6 CNNs from27 as fully supervised baselines for bioactivity prediction: GapNet, ResNet, DenseNet, MIL-Net, M-CNN and SC-CNN. All CNNs, except Single-Cell CNN (SC-CNN), were trained end-to-end on Cell Painting images without segmentation. A 3-layer FNN trained on CellProfiler features was incorporated as an additional baseline from27. The results for the CNNs and CellProfiler FNN were taken from the original publication. For details on the CNN and FNN architectures and training methodology, refer to27.
Following27, we evaluated the performance of the FNN trained on DINO features using area under the receiver operating characteristic curve (AUCROC) as the primary metric. The AUCROC values for each assay were obtained by averaging across 3 test splits. The mean AUCROC across 209 assays and the standard deviation are reported in Table 1. Similarly, F1-scores were computed and the mean and standard deviation across all assays are also provided. To illustrate the performance gap across different data availability regimes, we grouped the assays into 5 bins based on the number of activity labels: [< 50, 50–100, 100–500, 500–1000, 1000–3000]. AUCROC values were visualized for these 5 assay groups in Supplementary Fig. 8.
Implementation details
All self-supervised learning (SSL) models were implemented in Python 3.9.7 using PyTorch69 v1.10.2 and PyTorch Lightning v1.6.3. Feature postprocessing (sphering, MAD robustize, standardize) was carried out using pycytominer v0.2.0. Mean average precision (mAP), NSB and NSBP accuracies were computed using custom Python functions, with average precision (AP) and accuracy scores computed using the scikit-learn70 v1.0.2 implementation. PCA and UMAP were performed using scikit-learn v1.0.2 and umap-learn v0.5.3. The silhouette scores, kNN graphs, and the simulated Gaussian baseline for batch and source effect quantification were computed using scikit-learn v1.0.2. The LISI scores were calculated using HarmonyPy60 v0.0.9. Adjusted mutual information (AMI) of gene family clustering was calculated using scikit-learn v1.0.2. The visualization of UMAP was performed using matplotlib v3.5.0 and seaborn v0.11.2. The visualization of evaluation metrics and hierarchical clustering of ORF perturbations was performed in R 4.1.2.
Supplementary Information
Acknowledgements
We thank Bayer AG, which enabled the successful completion of this work through the Life Science Collaboration (LSC) project ‘Picasso’. We thank all members of the JUMP-CP consortium for producing the Cell Painting dataset that was used for training SSL models in this study. Finally, we thank all members of the Machine Learning Research group at Bayer AG for their kind advice and support.
Author contributions
VK and PAMZ designed the study. PAMZ supervised the study. VK prepared training and evaluation data. VK and NA implemented the self-supervised learning (SSL) framework including data loading, augmentation, and training pipelines. VK and NA trained SSL models. MO and VK designed and implemented a cloud-based inference pipeline. VK and MO produced representations for the gene overexpression data using the inference pipeline. VK conducted an augmentation study. VK and NA performed evaluations of SSL representations. FM conducted batch and laboratory effect analysis on the SSL representations. MH trained a bioactivity prediction model on DINO features and compared it with supervised CNNs. VK designed the figures, with contributions from PAMZ, MO, FM and NA. VK, NA and PAMZ wrote the manuscript with inputs from MO, FM, TK and DG. All authors reviewed and approved the final version.
Data availability
The image data (cpg0016-jump and cpg0012-wawer-bioactivecompoundprofiling) used in the study are available from the Cell Painting Gallery on the Registry of Open Data on AWS (https://registry.opendata.aws/cellpainting-gallery/).
Code availability
The code for training, inference, and evaluation of the self-supervised learning (SSL) models used in this study is publicly available in a GitHub repository (https://github.com/Bayer-Group/CellPaintSSL). The code is distributed under the BSD 3-Clause License. The model weights are provided and intended for non-commercial use only.
Declarations
Competing interests
VK, MO, FM, TK, DG, and PAMZ are employees of Bayer AG. NA and MH are former employees of Bayer AG.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Vladislav Kim and Nikolaos Adaloglou.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-025-88825-4.
References
- 1.Boutros, M., Heigwer, F. & Laufer, C. Microscopy-based high-content screening. Cell163, 1314–1325 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Loo, L.-H., Wu, L. F. & Altschuler, S. J. Image-based multivariate profiling of drug responses from single cells. Nat. Methods4, 445–453 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Carpenter, A. E. Image-based chemical screening. Nat. Chem. Biol.3, 461–465 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Mattiazzi Usaj, M. et al. High-content screening for quantitative cell biology. Trends Cell Biol.26, 598–611 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Reisen, F. et al. Linking phenotypes and modes of action through high-content screen fingerprints. Assay Drug Dev. Technol.13, 415–427 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Ziegler, S., Sievers, S. & Waldmann, H. Morphological profiling of small molecules. Cell Chem. Biol.28, 300–319 (2021). [DOI] [PubMed] [Google Scholar]
- 7.MacDonald, M. L. et al. Identifying off-target effects and hidden phenotypes of drugs in human cells. Nat. Chem. Biol.2, 329–337 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Chow, Y. L., Singh, S., Carpenter, A. E. & Way, G. P. Predicting drug polypharmacology from cell morphology readouts using variational autoencoder latent space arithmetic. PLoS Comput. Biol.18, e1009888 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jin, G. & Wong, S. T. C. Toward better drug repositioning: prioritizing and integrating existing methods into efficient pipelines. Drug Discov. Today19, 637–644 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karaman, B. & Sippl, W. Computational drug repurposing: Current trends. Curr. Med. Chem.26, 5389–5409 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Mirabelli, C. et al. Morphological cell profiling of SARS-CoV-2 infection identifies drug repurposing candidates for COVID-19. Proc. Natl. Acad. Sci.118, e2105815118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nyffeler, J. et al. Bioactivity screening of environmental chemicals using imaging-based high-throughput phenotypic profiling. Toxicol. Appl. Pharmacol.389, 114876 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Lomana, M. G., Marin Zapata, P. A. & Montanari, F. Predicting the mitochondrial toxicity of small molecules: Insights from mechanistic assays and cell painting data. Chem. Res. Toxicol.36, 1107–1120 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Camilleri, F., Wenda, J. M., Pecoraro-Mercier, C., Comet, J.-P. & Rouquié, D. Cell painting and chemical structure read-across can complement each other for rat acute oral toxicity prediction in chemical early derisking. Chem. Res. Toxicol.37, 1851–1866 (2024). [DOI] [PubMed] [Google Scholar]
- 15.Zapata, P. A. M. et al. Cell morphology-guided de novo hit design by conditioning GANs on phenotypic image features. Digit. Discov.2, 91–102 (2023). [Google Scholar]
- 16.Jing, L. & Tian, Y. Self-Supervised visual feature learning with deep neural networks: A survey. IEEE Trans. Pattern Anal. Mach. Intell.43, 4037–4058 (2021). [DOI] [PubMed] [Google Scholar]
- 17.Chen, T., Kornblith, S., Norouzi, M. & Hinton, G. A Simple Framework for Contrastive Learning of Visual Representations. In Proceedings of the 37th International Conference on Machine Learning, 1597–1607 (PMLR, 2020).
- 18.Caron, M. et al.Emerging Properties in Self-Supervised Vision Transformers, 9650–9660 (2021).
- 19.He, K. et al.Masked Autoencoders Are Scalable Vision Learners, 16000–16009 (2022).
- 20.McQuin, C. et al. Cell Profiler 3.0: Next-generation image processing for biology. PLOS Biol.16, e2005970 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berg, S. et al. ilastik: interactive machine learning for (bio)image analysis. Nat. Methods16, 1226–1232 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Schindelin, J. et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Caicedo, J. C. et al. Data-analysis strategies for image-based cell profiling. Nat. Methods14, 849–863 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Celik, S. et al.Biological Cartography: Building and Benchmarking Representations of Life. 10.1101/2022.12.09.519400 (2022).
- 25.Sivanandan, S. et al.A Pooled Cell Painting CRISPR Screening Platform Enables de novo Inference of Gene Function by Self-supervised Deep Learning. 10.1101/2023.08.13.553051 (2023).
- 26.Way, G. P. et al. Predicting cell health phenotypes using image-based morphology profiling. Mol. Biol. Cell32, 995–1005 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hofmarcher, M., Rumetshofer, E., Clevert, D.-A., Hochreiter, S. & Klambauer, G. Accurate prediction of biological assays with high-throughput microscopy images and convolutional networks. J. Chem. Inf. Model.59, 1163–1171 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Chandrasekaran, S. N. et al.JUMP Cell Painting Dataset: Morphological Impact of 136,000 Chemical and Genetic Perturbations. 10.1101/2023.03.23.534023 (2023).
- 29.Gidaris, S., Singh, P. & Komodakis, N. Unsupervised Representation Learning by Predicting Image Rotations. 10.48550/arXiv.1803.07728 (2018).
- 30.Noroozi, M. & Favaro, P. Unsupervised learning of visual representations by solving jigsaw puzzles. In Computer Vision—ECCV 2016 (eds. Leibe, B., Matas, J., Sebe, N. & Welling, M.) 69–84 (Springer, 2016). 10.1007/978-3-319-46466-4_5.
- 31.Misra, I. & van der Maaten, L. Self-Supervised Learning of Pretext-Invariant Representations, 6707–6717 (2020).
- 32.He, K., Fan, H., Wu, Y., Xie, S. & Girshick, R. Momentum Contrast for Unsupervised Visual Representation Learning. 10.48550/arXiv.1911.05722 (2020).
- 33.Grill, J.-B. et al. Bootstrap your own latent: A new approach to self-supervised learning. in Advances in Neural Information Processing Systems vol. 33, 21271–21284 (Curran Associates, Inc., 2020).
- 34.Chen, R. J. & Krishnan, R. G. Self-supervised vision transformers learn visual concepts in histopathology. 10.48550/arXiv.2203.00585 (2022).
- 35.Xie, Y., Zhang, J., Xia, Y. & Wu, Q. UniMiSS: Universal medical self-supervised learning via breaking dimensionality barrier. In Computer Vision – ECCV 2022 (eds. Avidan, S., Brostow, G., Cissé, M., Farinella, G. M. & Hassner, T.) 558–575 (Springer, 2022). 10.1007/978-3-031-19803-8_33.
- 36.Dosovitskiy, A. et al.An Image is Worth 16x16 Words: Transformers for Image Recognition at Scale. 10.48550/arXiv.2010.11929 (2021).
- 37.Vaswani, A. et al. Attention is all you need. in Advances in Neural Information Processing Systems, vol. 30 (Curran Associates, Inc., 2017).
- 38.Assran, M. et al. Masked siamese networks for label-efficient learning. In Computer Vision—ECCV 2022 (eds. Avidan, S., Brostow, G., Cissé, M., Farinella, G. M. & Hassner, T.) 456–473 (Springer, 2022). 10.1007/978-3-031-19821-2_26.
- 39.Dehghani, M. et al.Scaling Vision Transformers to 22 Billion Parameters. http://arxiv.org/abs/2302.05442 (2023).
- 40.Ando, D. M., McLean, C. Y. & Berndl, M. Improving Phenotypic Measurements in High-Content Imaging Screens. 10.1101/161422 (2017).
- 41.Lu, A. X., Kraus, O. Z., Cooper, S. & Moses, A. M. Learning unsupervised feature representations for single cell microscopy images with paired cell inpainting. PLOS Comput. Biol.15, e1007348 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lafarge, M. W. et al. Capturing single-cell phenotypic variation via unsupervised representation learning. in Proceedings of The 2nd International Conference on Medical Imaging with Deep Learning, 315–325 (PMLR, 2019). [PMC free article] [PubMed]
- 43.Dürr, O. & Sick, B. Single-cell phenotype classification using deep convolutional neural networks. J. Biomol. Screen.21, 998–1003 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Pfaendler, R., Hanimann, J., Lee, S. & Snijder, B. Self-supervised Vision Transformers Accurately Decode Cellular State Heterogeneity. 10.1101/2023.01.16.524226 (2023).
- 45.Doron, M. et al. Unbiased Single-Cell Morphology With Self-Supervised Vision Transformers. 10.1101/2023.06.16.545359 (2023).
- 46.Caicedo, J. C., McQuin, C., Goodman, A., Singh, S. & Carpenter, A. E. Weakly Supervised Learning of Single-Cell Feature Embeddings. In Proceedings of the IEEE conference on computer vision and pattern Recognition. 2018, 9309–9318 (2018). [DOI] [PMC free article] [PubMed]
- 47.Le, T. et al. Analysis of the human protein atlas weakly supervised single-cell classification competition. Nat. Methods19, 1221–1229 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moshkov, N. et al.Learning Representations for Image-Based Profiling of Perturbations.10.1101/2022.08.12.503783 (2022). [DOI] [PMC free article] [PubMed]
- 49.Kraus, O. Z., Ba, J. L. & Frey, B. J. Classifying and segmenting microscopy images with deep multiple instance learning. Bioinform. Oxf. Engl.32, i52–i59 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Godinez, W. J., Hossain, I., Lazic, S. E., Davies, J. W. & Zhang, X. A multi-scale convolutional neural network for phenotyping high-content cellular images. Bioinform. Oxf. Engl.33, 2010–2019 (2017). [DOI] [PubMed] [Google Scholar]
- 51.Pawlowski, N., Caicedo, J. C., Singh, S., Carpenter, A. E. & Storkey, A. Automating Morphological Profiling with Generic Deep Convolutional Networks. 10.1101/085118 (2016).
- 52.Cross-Zamirski, J. O. et al.Self-Supervised Learning of Phenotypic Representations from Cell Images with Weak Labels. 10.48550/arXiv.2209.07819 (2022).
- 53.Kobayashi, H., Cheveralls, K. C., Leonetti, M. D. & Royer, L. A. Self-supervised deep learning encodes high-resolution features of protein subcellular localization. Nat. Methods19, 995–1003 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Janssens, R., Zhang, X., Kauffmann, A., de Weck, A. & Durand, E. Y. Fully unsupervised deep mode of action learning for phenotyping high-content cellular images. Bioinform. Oxf. Engl.10.1093/bioinformatics/btab497 (2021). [DOI] [PubMed] [Google Scholar]
- 55.Kraus, O. et al.Masked Autoencoders for Microscopy are Scalable Learners of Cellular Biology. 10.48550/arXiv.2404.10242 (2024).
- 56.Cimini, B. A. et al. Optimizing the cell painting assay for image-based profiling. Nat. Protoc.18, 1981–2013 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bray, M.-A. et al. A dataset of images and morphological profiles of 30 000 small-molecule treatments using the cell painting assay. GigaScience6, 1–5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen, T., Kornblith, S., Swersky, K., Norouzi, M. & Hinton, G. Big Self-Supervised Models are Strong Semi-Supervised Learners. 10.48550/arXiv.2006.10029 (2020).
- 59.McInnes, L., Healy, J. & Melville, J. UMAP: Uniform manifold approximation and projection for dimension reduction. Preprint at 10.48550/arXiv.1802.03426 (2020).
- 60.Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods16, 1289–1296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brown, T. B. et al.Language Models are Few-Shot Learners. 10.48550/arXiv.2005.14165 (2020).
- 62.Radford, A. et al.Learning Transferable Visual Models From Natural Language Supervision. 10.48550/arXiv.2103.00020 (2021).
- 63.Yuan, L. et al.Florence: A New Foundation Model for Computer Vision. 10.48550/arXiv.2111.11432 (2021).
- 64.Wu, C. et al.Visual ChatGPT: Talking, Drawing and Editing with Visual Foundation Models. 10.48550/arXiv.2303.04671 (2023).
- 65.Chandrasekaran, S. N. et al.Three Million Images and Morphological Profiles of Cells Treated with Matched Chemical and Genetic Perturbations. 10.1101/2022.01.05.475090 (2022). [DOI] [PMC free article] [PubMed]
- 66.Ljosa, V. et al. Comparison of methods for image-based profiling of cellular morphological responses to small-molecule treatment. J. Biomol. Screen.18, 1321–1329 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rousseeuw, P. J. Silhouettes: A graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math.20, 53–65 (1987). [Google Scholar]
- 68.Luecken, M. D. et al. Benchmarking atlas-level data integration in single-cell genomics. Nat. Methods19, 41–50 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Paszke, A. et al.PyTorch: An Imperative Style, High-Performance Deep Learning Library. 10.48550/arXiv.1912.01703 (2019).
- 70.Pedregosa, F. et al. Scikit-learn: Machine learning in python. J. Mach. Learn. Res.12, 2825–2830 (2011). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The image data (cpg0016-jump and cpg0012-wawer-bioactivecompoundprofiling) used in the study are available from the Cell Painting Gallery on the Registry of Open Data on AWS (https://registry.opendata.aws/cellpainting-gallery/).
The code for training, inference, and evaluation of the self-supervised learning (SSL) models used in this study is publicly available in a GitHub repository (https://github.com/Bayer-Group/CellPaintSSL). The code is distributed under the BSD 3-Clause License. The model weights are provided and intended for non-commercial use only.