

Summary
Purpose
Men with epilepsy often have sexual or reproductive abnormalities that are attributed to alterations in androgen levels, including subnormal free testosterone. Levels of the major metabolites of testosterone – androsterone (5α-androstan-3α-ol-17-one; 5α, 3α-A), a neurosteroid that acts as a positive allosteric modulator of GABAA receptors, and its 5β-epimer etiocholanolone (5β-androstan-3α-ol-17-one; 5β, 3α-A) – may also be reduced in epilepsy. 5α 3α-A has been found in adult brain and both metabolites, which can also be derived from androstenedione, are present in substantial quantities in serum along with their glucuronide and sulfate conjugates. This study sought to determine whether these endogenous steroid metabolites can protect against seizures.
Methods
The anticonvulsant activity of 5α 3α-A and 5β, 3α-A was investigated in electrical and chemoconvulsant seizure models in mice. The steroids were also examined for activity against extracellularly-recorded epileptiform discharges in the CA3 region of the rat hippocampal slice induced by perfusion with 55 μM 4-aminopyridine (4-AP).
Results
Intraperitoneal injection of 5α, 3α-A protected mice in a dose-dependent fashion from seizures in the following models (ED50, dose in mg/kg protecting 50% of animals): 6 Hz electrical stimulation (29.1), pentylenetetrazol (43.5), pilocarpine (105), 4-AP (215), and maximal electroshock (224). 5β, 3α-A was also active in the 6 Hz and pentylenetetrazol models, but was less potent (ED50 values, 76.9 and 139 mg/kg, respectively), whereas epiandrosterone (5α,3β-A) was inactive (ED50, ≤300 mg/kg). 5α, 3α-A (10–100 μM) also inhibited epileptiform discharges in a concentration-dependent fashion in the in vitro slice model, whereas 5β, 3α-A was active but of lower potency and 5α, 3β-A was inactive.
Conclusions
5α, 3α-A and 5β, 3α-A have anticonvulsant properties. Although of low potency, the steroids are present in high abundance and could represent endogenous modulators of seizure susceptibility.
Keywords: Androsterone, Etiocholanolone, Epiandrosterone, Pentylenetetrazol, Pilocarpine, 4-Aminopyridine, 6-Hz model, Seizure, Mouse
Male sex steroid hormones (“androgens”) were recognized in the early part of the last century (1). Androsterone (5α-androstan-3α-ol-17-one; 5α,3α-A), the first androgen to be identified, was isolated from urine in 1931 and chemically synthesized four years later at the same time as the first synthesis of testosterone (2). 5α,3α-A and its 5β-epimer etiocholanolone (5β,3α-A) are the major excreted metabolites of testosterone (3–5). The first step in testosterone metabolism is its irreversible reduction by 5α-reductase isoenzymes, which leads to the formation of the biologically potent androgen 5α-dihydrotestosterone (Fig. 1). Testosterone is also a good substrate for liver 5β-reductase (6), which synthesizes 5β-dihydrotestosterone. The reductions at the 5-position are followed by sequential 3α- and 17β-reductions resulting in the synthesis of 5α,3α-A and 5β,3α-A. The latter two reactions are catalyzed by 3α-hydroxysteroid dehydrogenase (3α-HSD) and 17β-hydroxysteroid dehydrogenase (17β-HSD), respectively (Fig. 1). 5α,3α-A and 5β,3α-A can also be derived from the metabolism of androstenedione through 5α- and 5β-reduction, followed by the action of 3α-HSD. Substantial quantities of both compounds (~3–4 mg/24 h in young men) are excreted in the urine as 3-glucuronide and 3-sulfate conjugates (7–10).
FIG. 1.

Metabolic pathways for conversion of testosterone and androstenedione to androsterone (5α-androstan-3α-ol-17-one; 5α,3α-A) and etiocholanolone (5β-androstan-3α-ol-17-one; 5β,3α-A). 5α-Reductase (5α-R) and 5β-reductase (5β-R) catalyze the rate-limiting irreversible initial steps, which are followed by sequential reductions by 3α-hydroxysteroid dehydrogenase (3α-HSD) and 17β-hydroxysteroid dehydrogenase (17β-HSD). 5α,3α-A and 5β,3α-A are conjugated by gucuronidation in the liver. 5α,3α-A has been identified as a substrate for human dehydroepiandrosterone (DHEA) sulfotransferase and some studies have indicated that 5α,3α-A sulfate may be more abundant in serum than the glucuronide (44,53).
5α,3α-A has an identical reduced A-ring as the neurosteroids allopregnanolone (5α,3α-P) and allotetrahydrodeoxycorticosterone (5α,3α-THDOC), which are potent positive allosteric modulators of GABAA receptors (11,12). 5β,3α-A is analogous to pregnanolone (5β,3α-P) and tetrahydrodeoxycorticosterone (5β,3α-THDOC), which also have activity at GABAA receptors, but are less potent (11,13). In fact, 5α,3α-A has been reported to interact with GABAA receptors in vitro in a fashion similar to other neurosteroid positive GABAA receptor modulators. Thus, 5α,3α-A enhances muscimol and flunitrazepam binding and inhibits t-butyl bicyclophosphorothionate binding in rat brain membranes, and also enhances muscimol-stimulated 36Cl− flux in intact neurons (14,15). Furthermore, in electrophysiological studies, 5α,3α-A potentiates GABAA receptor chloride currents (15–19). Neurosteroids, including 5α,3α-P and 5α,3α-THDOC and their 5β-epimers, that act as similar positive GABAA receptor modulators, have anticonvulsant properties in animal models (11,12,20–23) and recently we have demonstrated that they also protect against epileptiform activity in an in vitro brain slice system (24). Consequently, 5α,3α-A and 5β,3α-A could potentially have anticonvulsant properties although as far as we are aware this has only previously been demonstrated for 5α,3α-A in regard to ruthenium red convulsions in cats (25) and 3-mercaptoprionic acid seizures in Syrian hamsters (26).
There is accumulating evidence that neurosteroids with GABAA receptor modulating activity serve as endogenous regulators of seizure susceptibility (27). Men with temporal lobe epilepsy often have impaired sexual function, including diminished testicular efficiency and loss of libido in association with reduced androgen levels (28,29). The deficiency of androgens is attributed to the effects of antiepileptic medications and also to the suppression of the hypothalamic-pituitary-gonadal axis by recurrent seizures (30–32). If androgen metabolites have anti-seizure properties, alterations in androgen levels caused by antiepileptic drugs or inadequately controlled seizures could be relevant to seizure control. In fact, there is evidence that long-term antiepileptic drug therapy is associated with markedly reduced urinary excretion of both 5α,3α-A and 5β,3α-A (3,33). In the present study we characterize for the first time the anticonvulsant profile of these steroids in several animal seizure models and also in an in vitro slice model previously shown to be sensitive to neurosteroids (24). For comparison, we examined the activity 5α,3β-A (epiandrosterone) which is predicted to have very weak activity at GABAA receptors (11). Our results indicate that the 3α-epimers are effective anticonvulsants and the structure-activity comparisons indicate that this likely occurs through positive modulation of GABAA receptors.
MATERIALS AND METHODS
In Vivo Studies
Animals
Male NIH Swiss mice (25–30 g) were housed five per cage and Sprague-Dawley rats (120–180 g) (Taconic Farms, Germantown, NY) were housed two per cage. Animals were kept in a vivarium under controlled laboratory conditions (temperature 22–26 °C, humidity 40–50%) with an artificial 12 h light/dark cycle and free access to food and water. Animals were allowed to acclimate to the vivarium for at least 5 days. For in vivo testing, the experimental groups consisted of 6–8 animals. The experiments were performed during the light phase of the light/dark cycle after at least a 30 min period of acclimation to the experimental room. Animals were maintained in facilities fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care and were performed under protocols approved by the Animal Care and Use Committee of the National Institute of Neurological Disorders and Stroke (NINDS) in strict compliance with the Guide for the Care and Use of Laboratory Animals of the National Research Council (National Academy Press, Washington, DC; http://www.nap.edu/readingroom/books/labrats/).
Test Substances and Drug Administration
Solutions of 5α,3α-A, 5β,3α-A, 5α,3β-A and 5α,3α-A d-glucuronide (Sigma-Aldrich, St. Louis, MO) were made fresh daily in 40% hydroxypropyl-β-cyclodextrin (Trappsol, Cyclodextrin Technologies Development, High Springs, FL) in sterile 0.9% saline. Further dilutions were made using sterile saline. The convulsant agents pentylenetetrazol (PTZ), 4-aminopyridine (4-AP) and pilocarpine were purchased from Sigma-Aldrich and were dissolved in saline immediately before use. All drug solutions were administered in a volume equaling 0.01 ml/kg. In the PTZ test and the 6 Hz model, the steroids or vehicle were administered intraperitoneally 15 min before PTZ or electrical stimulation. The pretreatment interval was based on the time of maximal effect in the time-course experiment in the 6 Hz model (Fig. 2). Pilot experiments indicated that 5α,3α-A was most effective against pilocarpine- and 4-AP-induced seizures with a 5 min pretreatment time. Vehicle alone did not affect seizures in any of the models.
FIG. 2.
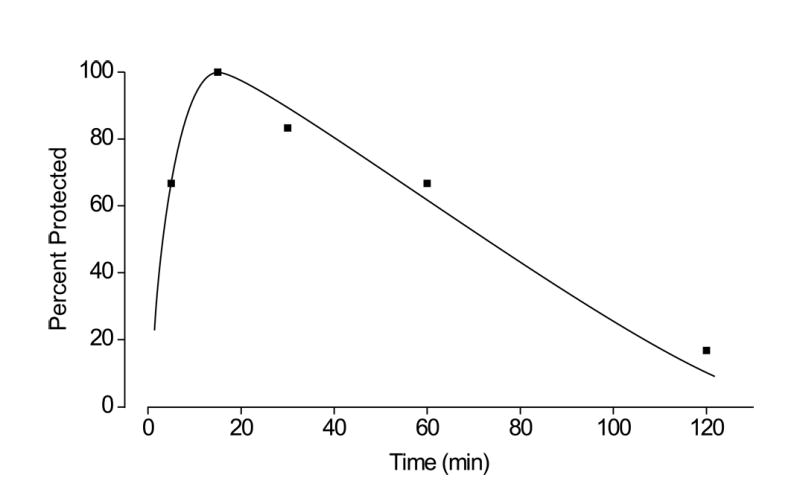
Time course for protection by androsterone (5α,3α-A) at a dose of 100 mg/kg in the 6 Hz electrical stimulation (32 mA, 3 s) model. The interval between the steroid injection and the electrical stimulus is plotted on the abscissa and the percentage of animals protected against seizures is plotted on the ordinate. Each point represents 6 mice.
6 Hz Seizure Test
Testing was carried out as previously described (23). In brief, 3-s corneal stimulation (200 μs-duration, 32-mA monopolar rectangular pulses at 6 Hz) was delivered by a constant current device (ECT Unit 5780, Ugo Basile, Comerio, Italy). Ocular anesthetic (0.5% tetracaine) was applied to the corneas 15 min before stimulation. Immediately before stimulation, the corneal electrodes were wetted with saline to provide good electrical contact. Following the stimulation the animals exhibited a “stunned” posture associated with rearing and automatic movements that lasted from 60 to 120 s in untreated animals. Animals resumed their normal exploratory behavior after the seizure. The experimental endpoint was protection against the seizure: an animal was considered to be protected if it resumed its normal exploratory behavior within 10 s of stimulation.
Pentylenetetrazol (PTZ) Seizure Test
Testing was carried out as previously described (11). In brief, mice were injected subcutaneously with PTZ (80 mg/kg) and were observed for a 30-min period. Mice failing to show clonic seizures lasting longer than 5 s were scored as protected.
Maximal Electroshock (MES) Seizure Test
Animals were subjected to a 0.2 s, 60-Hz electrical stimulus through corneal electrodes (as described above). The electroshock unit was adjusted to deliver a constant current of 50 mA. Animals failing to show tonic hindlimb extension were scored as protected (11).
4-Aminopyridine (4-AP) Seizure Test
Testing was carried out as previously described (34). Mice were injected subcutaneously with a CD97 dose (13.3 mg/kg) of 4-AP. The animals exhibited signs of behavioral activation progressing to clonic and tonic seizures. Animals failing to exhibit tonic hindlimb extension were scored as protected.
Pilocarpine Seizure Test
Testing was carried out as previously described (21). Mice were injected intraperitoneally with 1 mg/kg scopolamine methyl bromide to prevent the peripheral cholinergic activation. A few minutes later, seizures were induced by subcutaneous injection of a CD97 dose (dose producing seizures in 97% of animals) of pilocarpine (416 mg/kg). Animals were observed for 2 h after the pilocarpine injection. All control mice had severe generalized clonic seizures followed within 45 min by death. Mice failing to show generalized clonic seizures lasting longer than 10 s were scored as protected. The latencies to generalized clonic seizures and death were also recorded.
Motor Toxicity Test
Steroids were evaluated for motor toxicity using a modification of the horizontal screen test as previously described (11). Mice were placed on a horizontally oriented grid (consisting of parallel 1.5-mm diameter rods situated 1 cm apart) and the grid was inverted. Animals that fell from the grid within 10 s were scored as impaired.
Data Analysis
To construct dose-response curves, steroids were tested at several doses spanning the dose producing 50% protection (ED50) or motor impairment (TD50). At least 6–8 mice were tested at each dose. ED50 and TD50 values and their corresponding 95% confidence limits were determined by log-probit analysis using the Litchfield and Wilcoxon method (PHARM/PCS Version 4.2, MicroComputer Specialists, Philadelphia, PA). The protective index (PI), a measure of relative toxicity, was taken as the ratio TD50/ED50. The mean latency to seizure values in the pilocarpine model was compared by one-way analysis of variance (one-way ANOVA). Comparisons of the mean time to seizure onset values with control were made with the Dunnett’s t-test. Comparisons of the percent lethality with control were made with the Fisher’s exact test.
In Vitro Studies
4-AP-Induced Epileptiform Activity in the Hippocampal Slice
Rats were decapitated under brief carbon dioxide narcosis. The brains were rapidly removed and immediately submerged in an ice-cold cutting solution. Transverse hippocampal slices (500 μm thick) were prepared with a Vibratome (Technical Products International, INC., St. Louis, MO) and equilibrated before recording for at least 1 h in artificial cerebrospinal fluid (ACSF) bubbled with carbogen gas (95% O2/5% CO2) at 32–33 °C. The composition of the ACSF (in mM) was: 130 NaCl; 26 NaHCO3; 3 KCl; 1.25 NaH2PO4; 2 CaCl2; 1 MgCl2; 10 d-glucose, pH 7.4. The cutting solution was similar to ACSF except that NaCl was replaced with isosmotic sucrose. After equilibration, the slices were transferred to an interface-type recording chamber that was continuously gravity-perfused during the 1 h of the experiment with warmed (33 °C) ACSF at a flow rate of 2.5–3 ml/min. The surface of the slice was exposed to a gentle stream of humidified carbogen gas. Extracellular field recordings were carried out in the CA3 pyramidal layer with glass micropipettes (2–10 M) filled with ACSF. The location of the recording electrode was optimized by maximizing the field potential amplitude evoked in response to a 100-ms test stimulus applied to the dentate gyrus mossy fibers. The amplified electrode signals (DAM-80, World Precision Instruments, Sarasota, FL) were digitized (Digidata 1322A) and acquired by pClamp 9.0 software (both from Axon Instruments, Union City, CA). Events were detected and counted off-line with Mini Analysis Program (Synaptosoft, Decatur, GA). The parameters were empirically set to detect a maximum number of population responses that were clearly discernible from the background noise. The amplitude threshold was always ±0.2 mV, which is 10-fold greater than the typical background noise level of 0.02 mV. The event rate during the course of each experiment is expressed by plotting the frequency of events in successive 10-min intervals against the time of the recording. To evoke spontaneous epileptiform discharges, the slice perfusion solution was changed from ACSF to ACSF supplemented with 55 μM 4-AP. The 4-AP solution also contained various concentrations of the test steroids (10–300 μM) or vehicle (0.2–0.3% 2-hydroxypropyl-β-cyclodextrin). The vehicle at these concentrations had no apparent effect on the epileptiform discharges.
Data Analysis
The effects of 5α,3α-A or its epimers on 4-AP-induced epileptiform activity in the hippocampal slice were compared by two-way analysis of variance with repeated measures (two-way ANOVA). Specific comparisons between the treatments were made by a post-hoc multiple comparisons procedure (Bonferroni t-test).
RESULTS
In Vivo Mouse Seizure Models
6 Hz Model
We first sought to determine whether 5α,3α-A is protective in the 6 Hz electrical stimulation-induced seizure model like other neurosteroids that act as positive modulators of GABAA receptors (23) and, if so, determine the time course of the effect. As shown in Fig. 2, 5α,3α-A at a dose of 100 mg/kg did protect mice against 6 Hz seizures and showed a maximal effect 15 min after injection; by 2 h, only minimal protective activity was evident. We next examined the dose-dependence for protection in this model in experiments with a 15 min interval between 5α,3α-A injection and testing. At doses of 10 to 100 mg/kg, 5α,3α-A exhibited dose-dependent protective activity (Fig. 3A); the ED50 value determined from the dose-effect relationship is presented in Table 1. 5β,3α-A was also protective in the 6 Hz model, but was less potent than 5α,3α-A, whereas 5α,3β-A was inactive at a dose of 300 mg/kg. 5α,3α-A glucuronide was also inactive in the 6 Hz model at a dose of 300 mg/kg when injected either 30 or 60 min before testing (3 mice each group).
FIG. 5.
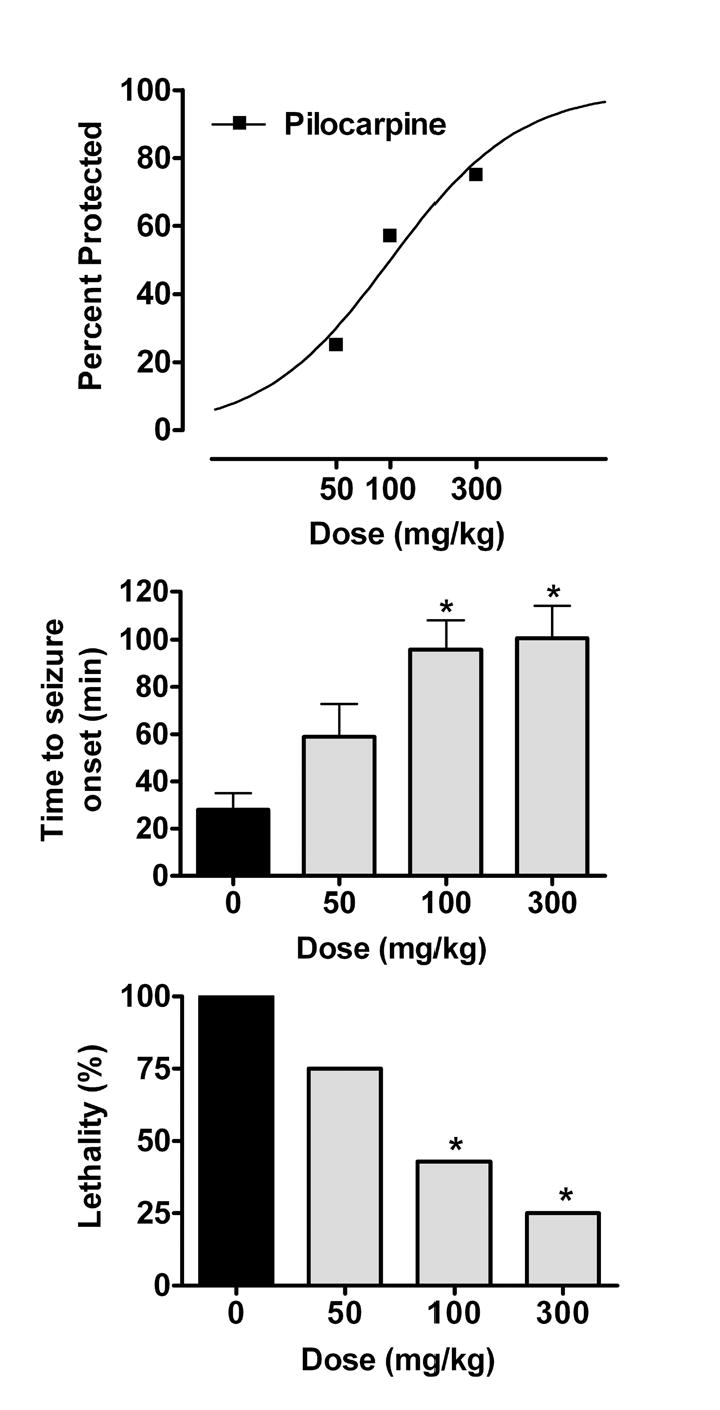
Dose-response relationships for androsterone (5α,3α-A) in the pilocarpine model. The steroid was administered 15 min before pilocarpine (416 mg/kg, s.c.). The percentage of animals protected against seizures at each dose is plotted at the top. The mean ± S.E.M. time to seizure onset is plotted in the middle bar chart. Steroid treatment significantly increased the latency (F3,31 = 8.0; p < 0.001). The percent lethality is shown in the lower bar chart. Six to 8 mice were tested at each dose. *Significantly different from control p < 0.05 (time to seizure onset by Dunnett’s t-test; percent lethality by Fisher’s exact test).
FIG. 3.

Dose-response relationships for protective activity of androsterone (5α,3α-A) and its 5β- and 3β-epimers in the 6 Hz electrical stimulation model (A) and the pentylenetetrazol (PTZ) model (B). Steroids were administered 15 min before electrical stimulation or injection of PTZ (see Methods). Data points indicate percentage of animals protected. Each point represents 6 to 8 mice.
TABLE 1.
ED50 values of androsterone (5α,3α-A), etiocholanolone (5β,3α-A) and epiandrosterone (5α,3β-A) for protection in the 6 Hz electrical stimulation, pentylenetetrazol (PTZ), pilocarpine, 4-aminopyridine (4-AP) and maximal electroshock (MES) seizure models in mice
| Androsterone epimer | Seizure model | ED50 (mg/kg)a | PIb |
|---|---|---|---|
| 5α,3α-A | 6 Hz | 29.1(16.2–52.1) | 5.23 |
| 5β,3α-A | 6 Hz | 76.9 (53.7–109.9) | 1.98 |
| 5α,3β-A | 6 Hz | > 300 | N.D. |
| 5α,3α-A | PTZ | 43.5 (31.6–60.0) | 3.50 |
| 5β,3α-A | PTZ | 138.7 (110.5–169.8) | 1.53 |
| 5α,3β-A | PTZ | > 300 | N.D.c |
| 5α,3α-A | Pilocarpine | 105.4 (47.7–232.7) | 1.44 |
| 5α,3α-A | 4-AP | 215.3 (173.8–266.6) | 0.70 |
| 5α,3α-A | MES | 223.7 (182.5–274.1) | 0.68 |
ED50 values (with 95% confidence intervals) represent the dose in mg/kg that is estimated to protect 50% of animals.
PI (protective index) is the ratio between TD50 (the dose estimated to produce motor impairment in 50% of animals) and the ED50. The TD50 value for 5α,3α-A was 152.3 (133.9–173.1) mg/kg, while the TD50 for its 5β-epimer was 212.3 (150.8–298.7). 5α,3β-A did not cause motor impairment at doses as high as 300 mg/kg.
N.D., not possible to determine.
PTZ-Induced Seizures
5α,3α-A (10–100 mg/kg) conferred dose-dependent protection in the PTZ model (Fig. 3B; Table 1). The 5β,3α-A epimer had approximately 3-fold lower potency and 5α,3β-A was inactive at a dose of 300 mg/kg.
MES Test
5α,3α-A at higher doses (150–300 mg/kg) exhibited dose-dependent activity in the mouse MES test with nearly complete protection conferred at 300 mg/kg (Fig. 4; Table 1). The 5α,3β-A epimer was inactive at 300 mg/kg (data not shown).
FIG. 4.
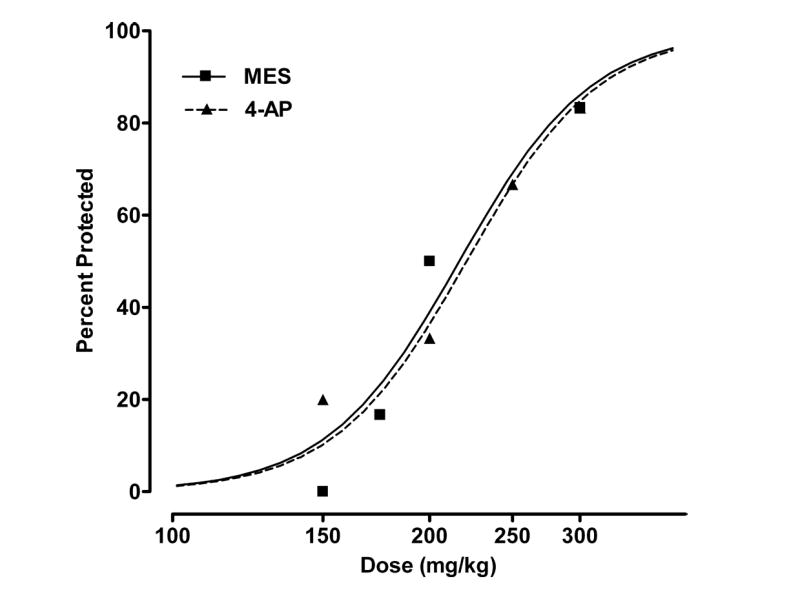
Dose-response relationships for protection by androsterone (5α,3α-A) in the maximal electroshock (MES; 50 mA, 0.2 s, 60 Hz) and 4-aminopyridine (4-AP; 13.3 mg/kg, s.c.) models. 5α,3α-A was administered 15 min before electrical stimulation in the MES test and 5 min before 4-AP (see Methods). Data points indicate percentage of animals protected against seizures. Each point represents 6 to 8 mice.
4-AP-Induced Seizures
5α,3α-A conferred protection against tonic hindlimb extension elicited by s.c. injection of 4-AP at doses comparable to those that were effective in the MES test (150–300 mg/kg) (Fig. 4; Table 1).
Pilocarpine-induced Status Epilepticus
As illustrated in Fig. 5, pretreatment with the 5α,3α-A (50–300 mg/kg) conferred dose-dependent protection against pilocarpine-induced limbic motor seizures (Table 1). 5α,3α-A pretreatment significantly increased the latency to seizure onset as well as the survival rate in a dose-dependent fashion.
Motor Toxicity
5α,3α-A at doses substantially higher than those that were protective in the 6 Hz and PTZ models caused motor impairment as assessed by the horizontal screen test. The TD50 value was 152 mg/kg (Table 1). 5β,3α-A also caused motor impairment at higher doses (150–300 mg/kg) but only 4 of 5 animals were affected even at the 300 mg/kg dose. 5α,3β-A did not affect motor performance at a dose of 300 mg/kg. The ratios between the protective doses in the seizure models (ED50 values) and the TD50 values for motor toxicity are given in Table 1.
In Vitro Hippocampal Slice Model
Perfusion of hippocampal slices with 55 μM 4-AP elicited positive going discharges in the CA3 region that increased in frequency from about 30 min−1 to nearly 60 min−1 at the end of the 60 min experimental period (F5,179 = 11.0; p < 0.001) (Fig. 6A). Coperfusion of 5α,3α-A with 4-AP (10–100 μM) resulted in a concentration-dependent reduction in the number of epileptiform discharges at each time point (F3,179 = 18.5; p < 0.001) (Fig. 6A). In addition, there was a significant interaction between treatment and time in slices treated with 5α,3α-A (F15,179 = 3.4; p < 0.001). Thus, for 50 and 100 μM 5α,3α-A there was a progressive reduction in discharge frequency after the 20 min epoch whereas in control recordings the frequency showed a monotonic increase over the 60 min recording. At 100 μM, 5α,3α-A almost completely eliminated the discharges.
FIG. 6.

Effects of androsterone (5α,3α-A) and its 5β and 3β epimers on the frequency of 4-aminopyridine (55 μM)-induced spontaneous epileptiform discharges in the CA3 region of the in vitro hippocampal slice. Co-application of various concentrations of 5α,3α-A along with 4-AP significantly reduced bursting in a concentration dependent fashion (F3,179 = 18.5; p < 0.001; panel A). 5β,3α-A (100 μM) also significantly reduced bursting activity (F1,107 = 5.4; p < 0.05; panel B), while 5α,3β-A did not have a significant effect (F5,107 = 0.75; p > 0.05; panel C). Each point represents the mean ± S.E.M. of event frequency values during the preceding 10 min epoch (6 slices). Frequency values from a common control group collected throughout the experiments are depicted on all three graphs (12 slices). Concentrations higher than 100 μM could note be tested due to the limited solubility of the steroids. *p < 0.05 vs. control group (post-hoc Bonferroni t-test).
Coperfusion with 100 μM 5β,3α-A also reduced the discharge frequency at all time points (Fig. 6B) but the magnitude of the effect was substantially less than for 5α,3α-A. Two-way ANOVA revealed a significant time effect for 100 μM 5β,3α-A (F5,107 = 21.3; p < 0.001) and also a significant treatment effect (F1,107 = 5.4; p < 0.05), but no treatment vs. time effect (F5,107 = 0.23; p > 0.05) (Fig. 6B). Individual time points for 5β,3α-A were only significantly different from vehicle at 30 and 60 min (Fig. 6B). Higher concentrations of 5β,3α-A were not tested due to limitations of solubility. Perfusion with 5α,3β-A did not significantly affect the discharge frequency (F5,107 = 0.75; p > 0.05) (Fig. 6C).
DISCUSSION
Men with epilepsy often have altered androgen levels in comparison with control subjects who do not have epilepsy (28). In particular, serum levels of free testosterone and adrostenedione are reduced in men with uncontrolled temporal lobe seizures (3,32,35,36). Remarkably, the androgen levels normalize following epilepsy surgery that results in successful seizure control (36), suggesting that uncontrolled seizures are responsible for the endocrine abnormalities, although enzyme-inducing antiepileptic drugs are also likely to play an important role through effects on sex hormone binding globulin (30,32). Whatever the cause, subnormal serum free testosterone and androstenedione would be expected to result in reduced production of the metabolites 5α,3α-A and 5β,3α-A. In fact, men with epilepsy receiving treatment with antiepileptic medications excrete diminished quantities of 5α,3α-A and 5β,3α-A in comparison with age-matched control subjects (3,33).
In the present study, we have found that 5α,3α-A is an effective anticonvulsant in several seizure models. Similarly, 5β,3α-A has anticonvulsant properties but is less potent, as is consistent with the usual structure-activity relationship for GABAA receptor modulatory activity of A-ring reduced neurosteroids (27). The 5α,3β-epimer, which is expected to have minimal GABAA receptor modulatory activity, was inactive both in vivo and in vitro. Overall, the structure-activity relationship suggests that the protective activity in the 6 Hz and PTZ models is due predominantly to effects on GABAA receptors. In fact, various studies have confirmed that 5α,3α-A is a positive modulator of GABAA receptors (14–19); we would predict that 5β,3α-A has similar, but weaker activity, although this remains to be demonstrated experimentally. It is unlikely that the weak androgenic activity of 5α,3α-A contributes to its anticonvulsant properties since testosterone, which has approximately 10-times greater androgenic activity (37), is inactive in the seizure models (unpublished observations). Since 5α,3α-A and 5β,3α-A are present in substantial quantities in serum (39–44), cerebrospinal fluid (45) and brain (46), it seems plausible that they could play a role as endogenous regulators of seizure susceptibility. Neither steroid was exceptionally potent, although in the 6 Hz and PTZ models, the potency of 5α,3α-A was only 2- and 3-fold less than that of 5α,3α-P and 5α,3α-THDOC (11,23), which have been proposed as endogenous regulators of seizure susceptibility in catamenial epilepsy and stress (27). Nevertheless, the extent to which the potencies are predictive of activity in human epilepsy is unclear, since in human epilepsy even small reductions in the excitability of critical brain regions may reduce seizure frequency. Serum levels of unconjugated 5α,3α-A in men and women are 3–5 nM (40,41,44), which is in the same range as the concentrations of 5α,3α-P in women in the luteal phase of the menstrual cycle (47–49), and one study indicated that the amounts of 5α,3α-A may be far higher (45). A considerable portion of the androsterone epimers may be as the glucouronide (7,44,50–52) and sulfate (42,44,53) conjugates. Serum androsterone glucuronide levels are ~15–60-fold greater than the unconjugated amounts (40,42) and the levels of the sulfate may be >300-fold the unconjugated form (42,44). Whether the conjugates can serve as sources of the free steroids for delivery to the brain remains to be determined.
Androgens have previously been implicated in the regulation of seizure activity in men with epilepsy (54). In particular, the reduced metabolite of testosterone 5α-androstane-3α,17β-diol (3α-diol; 3α-androstanediol; Fig. 1) has been shown to have anticonvulsant activity and has been proposed as an endogenous anticonvulsant substance (55,56). However, the body burden of 5α,3α-A and 5β,3α-A is likely to be substantially greater than that of the 17-OH intermediates. In men, while the serum free levels of 5α,3α-A modestly exceed those of free 3α-diol (44), the ratio for glucuronide conjugates in serum is about 5-fold (57) and the ratio of conjugated 5α,3α-A to conjugated 3α-diol in urine is 20 or more (10). Therefore, 5α,3α-A and possibly 5β,3α-A are even more likely to be relevant in the regulation of seizure susceptibility in persons with epilepsy. It is noteworthy that women also produce substantial amounts of 5α,3α-A and 5β,3α-A (9,10,42,57), suggesting that the steroids could also play a role in the regulation of seizure susceptibility in women.
5α,3α-A had the greatest potency in the 6 Hz and PTZ models, and was of lower potency against pilocarpine-, 4-AP- and MES-induced seizures. The relative potency ranking in the various in vivo models is similar to that of other GABAA receptor modulating neurosteroids (11,21,23). Indeed, we recently reported that such neurosteroids are particularly effective in the 6 Hz model (23), and this model was most sensitive to 5α,3α-A. In the 6 Hz model, 5α,3α-A had a rapid time course of action, comparable to that of other similar neurosteroids (11,12).
Although 5α,3α-A has an anticonvulsant profile similar to that of other GABAA receptor modulating neurosteroids, some differences were noted. At high doses, 5α,3α-A was protective in the MES and 4-AP seizure models, unlike 5α,3α-P which is inactive at comparable doses (11). Whether high doses of 5α,3α-A exert pharmacological actions on targets other than GABAA receptors that account for the activity in these models remains to be determined. GABAergic agents are not typically active in the 4-AP model in vivo, where seizures occur in the face of GABA synaptic tone that is strongly enhanced by the release-potentiating action of 4-AP (34). In contrast, GABA potentiating neurosteroids are effective in the in vitro 4-AP model (24). We have postulated that the in vitro activity may be due to tonic activation of GABAA receptors, which directly suppresses the firing of principal cells and interneurons (24). The in vitro activity of 5α,3α-A and 5β,3α-A in the present study presumably occurs through a similar tonic action.
The protective index (PI) values reported in Table 1 provide a measure of therapeutic potency in relation to the potency for induction of motor impairment. In the 6 Hz and PTZ models, 5α,3α-A exhibited PI values comparable to or higher than those reported for progesterone and deoxycorticosterone derivatives (11). This suggests that exogenously administered 5α,3α-A or related compounds could be of potential utility as therapeutic agents. The PI of the 5β epimer was substantially less than that of 5α,3α-A, indicating that the 5α-stereoisomer would be preferred therapeutically. A similar preference for the 5α epimers has been previously reported for other neurosteroids (11,21).
In conclusion, our results demonstrate that 5α,3α-A and 5β,3α-A are protective in various seizure models in vivo and in vitro. Both steroids are present endogenously in serum and brain at concentrations that may exceed those of other neurosteroids. Moreover, there is evidence that the levels are reduced in some men with epilepsy and could therefore be a cause of increased seizure susceptibility. Therapeutic strategies seeking to replace the diminished “neurosteroid tone” in men with epilepsy are worthy of investigation.
References
- 1.Tausk M. Androgens and anabolic steroids. In: Parnham MJ, Bruinvels J, eds. Discoveries in pharmacology; haemodynamics, hormons and inflammation; Vol. 2 Amsterdam: Elsevier, 1984:307–320.
- 2.Kochakian CD. History, chemistry and pharmacodynamics of anabolic-androgenic steroids. Wien Med Wochenschr. 1993;143:359–363. [PubMed] [Google Scholar]
- 3.Brunet M, Rodamilans M, Martinez-Osaba MJ, Santamaria J, To-Figueras J, Torra M, Corbella J, Rivera F. Effects of long-term antiepileptic therapy on the catabolism of testosterone. Pharmacol Toxicol. 1995;76:371–375. doi: 10.1111/j.1600-0773.1995.tb00164.x. [DOI] [PubMed] [Google Scholar]
- 4.Schänzer W. Metabolism of anabolic androgenic steroids. Clin Chem. 1996;42:1001–1020. [PubMed] [Google Scholar]
- 5.Vierhapper H, Nowotny P. The stress of being a doctor: steroid excretion rates in internal medicine residents on and off duty. Am J Med. 2000;109:492–494. doi: 10.1016/s0002-9343(00)00578-7. [DOI] [PubMed] [Google Scholar]
- 6.Charbonneau A, The VL. Genomic organization of a human 5β-reductase and its pseudogene and substrate selectivity of the expressed enzyme. Biochim Biophys Acta. 2001;1517:228–235. doi: 10.1016/s0167-4781(00)00278-5. [DOI] [PubMed] [Google Scholar]
- 7.Slaunwhite WR, Jr, Sandberg AA. Metabolism of 4-C14-testosterone in human subjects. III. Fate of androsterone and etiocholanolone. J Clin Endocrinol Metab. 1958;18:1056–1066. doi: 10.1210/jcem-18-10-1056. [DOI] [PubMed] [Google Scholar]
- 8.Rittmaster RS, Leopold CA, Thompson DL. Androgen glucuronyl transferase activity in rat liver, evidence for the importance of hepatic tissue in 5α-reduced androgen metabolism. J Steroid Biochem. 1989;33:1207–1212. doi: 10.1016/0022-4731(89)90431-7. [DOI] [PubMed] [Google Scholar]
- 9.Callies F, Arlt W, Siekmann L, Hubler D, Bidlingmaier F, Allolio B. Influence of oral dehydroepiandrosterone (DHEA) on urinary steroid metabolites in males and females. Steroids. 2000;65:98–102. doi: 10.1016/s0039-128x(99)00090-2. [DOI] [PubMed] [Google Scholar]
- 10.Poór V, Juricskay S, Gáti A, Osváth P, Tényi T. Urinary steroid metabolites and 11β-hydroxysteroid dehydrogenase activity in patients with unipolar recurrent major depression. J Affect Disord. 2004;81:55–59. doi: 10.1016/S0165-0327(03)00199-X. [DOI] [PubMed] [Google Scholar]
- 11.Kokate TG, Svensson BE, Rogawski MA. Anticonvulsant activity of neurosteroids: correlation with γ-aminobutyric acid-evoked chloride current potentiation. J Pharmacol Exp Ther. 1994;270:1223–1229. [PubMed] [Google Scholar]
- 12.Reddy DS, Rogawski MA. Stress-induced deoxycorticosterone-derived neurosteroids modulate GABAA receptor function and seizure susceptibility. J Neurosci. 2002;22:3795–3805. doi: 10.1523/JNEUROSCI.22-09-03795.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morrow AL, Pace JR, Purdy RH, Paul SM. Characterization of steroid interactions with γ-aminobutyric acid receptor-gated chloride ion channels: evidence for multiple steroid recognition sites. Mol Pharmacol. 1990;37:263–270. [PubMed] [Google Scholar]
- 14.Turner DM, Ransom RW, Yang JS, Olsen RW. Steroid anesthetics and naturally occurring analogs modulate the γ-aminobutyric acid receptor complex at a site distinct from barbiturates. J Pharmacol Exp Ther. 1989;248:960–966. [PubMed] [Google Scholar]
- 15.Hawkinson JE, Kimbrough CL, Belelli D, Lambert JJ, Purdy RH, Lan NC. Correlation of neuroactive steroid modulation of [35S]t-butylbicyclophosphorothionate and [3H]flunitrazepam binding and γ-aminobutyric acidA receptor function. Mol Pharmacol. 1994;46:977–985. [PubMed] [Google Scholar]
- 16.Turner JP, Simmonds MA. Modulation of the GABAA receptor complex by steroids in slices of rat cuneate nucleus. Br J Pharmacol. 1989;96:409–417. doi: 10.1111/j.1476-5381.1989.tb11832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Twyman RE, Macdonald RL. Neurosteroid regulation of GABAA receptor single-channel kinetic properties of mouse spinal cord neurons in culture. J Physiol. 1992;456:215–245. doi: 10.1113/jphysiol.1992.sp019334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peters JA, Kirkness EF, Callachan H, Lambert JJ, Turner AJ. Modulation of the GABAA receptor by depressant barbiturates and pregnane steroids. Br J Pharmacol. 1988;94:1257–1269. doi: 10.1111/j.1476-5381.1988.tb11646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park-Chung M, Malayev A, Purdy RH, Gibbs TT, Farb DH. Sulfated and unsulfated steroids modulate γ-aminobutyric acid receptor A function through distinct sites. Brain Res. 1999;830:72–87. doi: 10.1016/s0006-8993(99)01381-5. [DOI] [PubMed] [Google Scholar]
- 20.Belelli D, Bolger MB, Gee KW. Anticonvulsant profile of the progesterone metabolite 5α-pregnan-3α-ol-20-one. Eur J Pharmacol. 1989;166:325–329. doi: 10.1016/0014-2999(89)90077-0. [DOI] [PubMed] [Google Scholar]
- 21.Kokate TG, Cohen AL, Karp E, Rogawski MA. Neuroactive steroids protect against pilocarpine- and kainic acid–induced limbic seizures and status epilepticus in mice. Neuropharmacology. 1996;35:1049–1056. doi: 10.1016/s0028-3908(96)00021-4. [DOI] [PubMed] [Google Scholar]
- 22.Frye CA, Scalise TJ. Anti-seizure effects of progesterone and 3α,5α-THP in kainic acid and perforant pathway models of epilepsy. Psychoneuroendocrinology. 2000;25:407–420. doi: 10.1016/s0306-4530(99)00068-2. [DOI] [PubMed] [Google Scholar]
- 23.Kaminski RM, Livingood MR, Rogawski MA. Allopregnanolone analogs that positively modulate GABAA receptors protect against partial seizures induced by 6 Hz electrical stimulation in mice. Epilepsia. 2004;45:1–4. doi: 10.1111/j.0013-9580.2004.04504.x. [DOI] [PubMed] [Google Scholar]
- 24.Salazar P, Tapia R, Rogawski MA. Effects of neurosteroids on epileptiform activity induced by picrotoxin and 4-aminopyridine in the rat hippocampal slice. Epilepsy Res. 2003;55:71–82. doi: 10.1016/s0920-1211(03)00112-8. [DOI] [PubMed] [Google Scholar]
- 25.Kubli-Garfias C, Canchola E, Arauz-Contreras J, Feria-Velasco A. Depressant effect of androgens on the cat brain electrical activity and its antagonism by ruthenium red. Neuroscience. 1982;7:2777–2782. doi: 10.1016/0306-4522(82)90100-2. [DOI] [PubMed] [Google Scholar]
- 26.Naum G, Cardozo J, Golombek DA. Diurnal variation in the proconvulsant effect of 3-mercaptopropionic acid and the anticonvulsant effect of androsterone in the Syrian hamster. Life Sci. 2002;71:91–98. doi: 10.1016/s0024-3205(02)01577-1. [DOI] [PubMed] [Google Scholar]
- 27.Rogawski MA, Reddy DS. Neurosteroids: endogenous modulators of seizure susceptibility. In: Rho JM, Sankar R, Cavazos J, eds. Epilepsy: scientific foundations of clinical practice. New York: Marcel Dekker, 2004; 319–355.
- 28.Herzog AG, Seibel MM, Schomer DL, Vaitukaitis JL, Geschwind N. Reproductive endocrine disorders in men with partial seizures of temporal lobe origin. Arch Neurol. 1986;43:347–350. doi: 10.1001/archneur.1986.00520040035015. [DOI] [PubMed] [Google Scholar]
- 29.Bauer J, Blumenthal S, Reuber M, Stoffel-Wagner B. Epilepsy syndrome, focus location, and treatment choice affect testicular function in men with epilepsy. Neurology. 2004;62:243–246. doi: 10.1212/01.wnl.0000091866.48962.79. [DOI] [PubMed] [Google Scholar]
- 30.Connell JM, Rapeport WG, Beastall GH, Brodie MJ. Changes in circulating androgens during short term carbamazepine therapy. Br J Clin Pharmacol. 1984;17:347–351. doi: 10.1111/j.1365-2125.1984.tb02352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Edwards HE, MacLusky NJ, Burnham WM. The effect of seizures and kindling on reproductive hormones in the rat. Neurosci Biobehav Rev. 2000;24:753–762. doi: 10.1016/s0149-7634(00)00034-8. [DOI] [PubMed] [Google Scholar]
- 32.Herzog AG, Drislane FW, Schomer DL, Pennell PB, Bromfield EB, Kelly KM, Farina EL, Frye CA. Differential effects of antiepileptic drugs on sexual function and reproductive hormones in men with epilepsy: Interim analysis of a comparison between lamotrigine and enzyme-inducing antiepileptic drugs. Epilepsia. 2004;45:764–768. doi: 10.1111/j.0013-9580.2004.60703.x. [DOI] [PubMed] [Google Scholar]
- 33.Christiansen P, Lund M. Sexual potency, testicular function and excretion of sexual hormones in male epileptics. In: Dieter J, ed, with contributions by P. Ahlgren et al. Epileptology: proceedings of the seventh International Symposium on Epilepsy, Berlin (West) 1975;190–191.
- 34.Yamaguchi S, Rogawski MA. Effects of anticonvulsant drugs on 4-aminopyridine-induced seizures in mice. Epilepsy Res. 1992;11:9–16. doi: 10.1016/0920-1211(92)90016-m. [DOI] [PubMed] [Google Scholar]
- 35.Stoffel-Wagner B, Bauer J, Flügel D, Brennemann W, Klingmüller D, Elger CE. Serum sex hormones are altered in patients with chronic temporal lobe epilepsy receiving anticonvulsant medication. Epilepsia. 1998;39:1164–1173. doi: 10.1111/j.1528-1157.1998.tb01307.x. [DOI] [PubMed] [Google Scholar]
- 36.Bauer J, Stoffel-Wagner B, Flugel D, Kluge M, Schramm J, Bidlingmaier F, Elger CE. Serum androgens return to normal after temporal lobe epilepsy surgery in men. Neurology. 2000;55:820–824. doi: 10.1212/wnl.55.6.820. [DOI] [PubMed] [Google Scholar]
- 37.Nussey SS, Whitehead SA. Endocrinology : an integrated approach Oxford, U.K.: Bios Scientific Publishers, 2001. [PubMed]
- 38.Hammond GL, Ruokonen A, Kontturi M, Koskela E, Vihko R. The simultaneous radioimmunoassay of seven steroids in human spermatic and peripheral venous blood. J Clin Endocrinol Metab. 1977;45:16–24. doi: 10.1210/jcem-45-1-16. [DOI] [PubMed] [Google Scholar]
- 39.Sato K, Ohsawa N. Radioimmunoassay for etiocholanolone. Steroids. 1977;29:295–308. doi: 10.1016/0039-128x(77)90001-0. [DOI] [PubMed] [Google Scholar]
- 40.Rittmaster RS, Stoner E, Thompson DL, Nance D, Lasseter KC. Effect of MK-906, a specific 5α-reductase inhibitor, on serum androgens and androgen conjugates in normal men. J Androl. 1989;10:259–262. doi: 10.1002/j.1939-4640.1989.tb00097.x. [DOI] [PubMed] [Google Scholar]
- 41.Gormley GJ, Stoner E, Rittmaster RS, Gregg H, Thompson DL, Lasseter KC, Vlasses PH, Stein EA. Effects of finasteride (MK-906), a 5α-reductase inhibitor, on circulating androgens in male volunteers. J Clin Endocrinol Metab. 1990;70:1136–1141. doi: 10.1210/jcem-70-4-1136. [DOI] [PubMed] [Google Scholar]
- 42.Carmina E, Lobo RA. Evidence for increased androsterone metabolism in some normoandrogenic women with acne. J Clin Endocrinol Metab. 1993;76:1111–1114. doi: 10.1210/jcem.76.5.8496299. [DOI] [PubMed] [Google Scholar]
- 43.Ueshiba H, Takeda S, Segawa M, Ueshiba H, Miyachi Y. Serum androsterone levels in children. Horm Metab Res. 1996;28:190–192. doi: 10.1055/s-2007-979158. [DOI] [PubMed] [Google Scholar]
- 44.Labrie F, Belanger A, Cusan L, Gomez JL, Candas B. Marked decline in serum concentrations of adrenal C19 sex steroid precursors and conjugated androgen metabolites during aging. J Clin Endocrinol Metab. 1997;82:2396–2402. doi: 10.1210/jcem.82.8.4160. [DOI] [PubMed] [Google Scholar]
- 45.Kim YS, Zhang H, Kim HY. Profiling neurosteroids in cerebrospinal fluids and plasma by gas chromatography/electron capture negative chemical ionization mass spectrometry. Anal Biochem. 2000;277:187–195. doi: 10.1006/abio.1999.4384. [DOI] [PubMed] [Google Scholar]
- 46.Hammond GL, Hirvonen J, Vihko R. Progesterone, androstenedione, testosterone, 5α-dihydrotestosterone and androsterone concentrations in specific regions of the human brain. J Steroid Biochem. 1983;18:185–189. doi: 10.1016/0022-4731(83)90086-9. [DOI] [PubMed] [Google Scholar]
- 47.Wang M, Seippel L, Purdy RH, Bäckström T. Relationship between symptom severity and steroid variation in women with premenstrual syndrome: study on serum pregnenolone, pregnenolone sulfate, 5α-pregnane-3,20-dione and 3α-hydroxy-5α-pregnan-20-one. J Clin Endocrinol Metab. 1996;81:1076–1082. doi: 10.1210/jcem.81.3.8772579. [DOI] [PubMed] [Google Scholar]
- 48.Schmidt PJ, Purdy RH, Moore PH, Jr, Paul SM, Rubinow DR. Circulating levels of anxiolytic steroids in the luteal phase in women with premenstrual syndrome and in control subjects. J Clin Endocrinol Metab. 1994;79:1256–1260. doi: 10.1210/jcem.79.5.7962316. [DOI] [PubMed] [Google Scholar]
- 49.Galli R, Luisi M, Pizzanelli C, Monteleone P, Casarosa E, Iudice A, Murri L. Circulating levels of allopregnanolone, an anticonvulsant metabolite of progesterone, in women with partial epilepsy in the postcritical phase. Epilepsia. 2001;42:216–219. doi: 10.1046/j.1528-1157.2001.07600.x. [DOI] [PubMed] [Google Scholar]
- 50.Horton R, Rosner JM, Forsham PH. Urinary excretion pattern of injection H3-testosterone. Proc Soc Exp Biol Med. 1963;114:400–402. doi: 10.3181/00379727-114-28690. [DOI] [PubMed] [Google Scholar]
- 51.Brochu M, Belanger A. Increase in plasma steroid glucuronide levels in men from infancy to adulthood. J Clin Endocrinol Metab. 1987;64:1283–1287. doi: 10.1210/jcem-64-6-1283. [DOI] [PubMed] [Google Scholar]
- 52.Belanger A, Brochu M, Lacoste D, Noel C, Labrie F, Dupont A, Cusan L, Caron S, Couture J. Steroid glucuronides: human circulatory levels and formation by LNCaP cells. J Steroid Biochem Mol Biol. 1991;40:593–598. doi: 10.1016/0960-0760(91)90281-9. [DOI] [PubMed] [Google Scholar]
- 53.Zwicker H, Rittmaster RS. Androsterone sulfate: physiology and clinical significance in hirsute women. J Clin Endocrinol Metab. 1993;76:112–116. doi: 10.1210/jcem.76.1.8380602. [DOI] [PubMed] [Google Scholar]
- 54.Rhodes ME, Harney JP, Frye CA. Gonadal, adrenal, and neuroactive steroids’ role in ictal activity. Brain Res. 2004;1000:8–18. doi: 10.1016/j.brainres.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 55.Frye CA, Reed TA. Androgenic neurosteroids: anti-seizure effects in an animal model of epilepsy. Psychoneuroendocrinology. 1988;23:385–399. doi: 10.1016/s0306-4530(98)00009-2. [DOI] [PubMed] [Google Scholar]
- 56.Reddy DS. Anticonvulsant activity of the testosterone-derived neurosteroid 3α-androstanediol. Neuroreport. 2004;15:515–518. doi: 10.1097/00001756-200403010-00026. [DOI] [PubMed] [Google Scholar]
- 57.Lookingbill DP, Demers LM, Wang C, Leung A, Rittmaster RS, Santen RJ. Clinical and biochemical parameters of androgen action in normal healthy Caucasian versus Chinese subjects. J Clin Endocrinol Metab. 1991;72:1242–1248. doi: 10.1210/jcem-72-6-1242. [DOI] [PubMed] [Google Scholar]