

Abstract
γδ T cells mediate demyelination in athymic (nude) mice infected with the neurotropic coronavirus mouse hepatitis virus strain JHM. Now, we show that these cells also mediate the same process in mice lacking αβ T cells (T-cell receptor β-deficient [TCRβ−/−] mice) and demyelination is gamma interferon (IFN-γ) dependent. Most strikingly, our results also show a major role for NKG2D, expressed on γδ T cells, in the demyelinating process with in vivo blockade of NKG2D interactions resulting in a 60% reduction in demyelination. NKG2D may serve as a primary recognition receptor or as a costimulatory molecule. We show that NKG2D+ γδ T cells in the JHM-infected central nervous system express the adaptor molecule DAP12 and an NKG2D isoform (NKG2D short), both required for NKG2D to serve as a primary receptor. These results are consistent with models in which γδ T cells mediate demyelination using the same effector cytokine, IFN-γ, as CD8 T cells and do so without a requirement for signaling through the TCR.
The human disease multiple sclerosis is an immune-mediated, multifocal demyelinating disease of unknown etiology (38). T cells, B cells, and macrophages/microglia can all be found in the lesions of multiple sclerosis (26). However, the specific role of the many immune system components in the human demyelinating process is only partially understood.
In rodents, the histopathology of multiple sclerosis is, in part, modeled by a number of virus-induced demyelinating diseases, including those induced by infection with mouse hepatitis virus strain JHM (55). As with multiple sclerosis, the disease induced by JHM is largely immune mediated (23). Mice with severe combined immunodeficiency (SCID mice) or mice lacking recombination activation gene 1 (RAG1−/− mice), both of which lack B and T cells, did not develop demyelination upon infection with JHM (6, 28, 60). T cells but not B cells were sufficient to mediate demyelination as reconstitution of RAG1−/− mice with JHM immune splenocytes depleted of either CD4 or CD8 T cells, but not both, resulted in demyelination (60). One apparent exception to this requirement for αβ T-cell receptor (TCR)-bearing cells was the presence of demyelination in athymic mice, which lack CD4 and CD8 T cells (4, 32). In a previous study, we demonstrated that demyelination in these mice was mediated by γδ T cells (14). These cells were critical for macrophage/microglia activation and infiltration into the white matter; depletion of γδ T cells abrogated demyelination.
Gamma interferon (IFN-γ) has a central role in the pathology of multiple sclerosis and several of its rodent models, including experimental autoimmune encephalomyelitis (EAE) and Theiler's murine encephalomyelitis virus-induced demyelination (49, 56, 59). In mice infected with JHM, IFN-γ was required for CD8 T-cell-mediated demyelination since myelin destruction was greatly reduced in JHM-infected RAG1−/− recipients of IFN-γ−/− CD8 T cells (44). In contrast, infected recipients of IFN-γ−/− CD4 T cells developed greater amounts of demyelination than their counterparts that received IFN-γ+/+ CD4 T cells, suggesting that, in the context of CD4 T cells, IFN-γ may be acting to down-regulate the immune response (43). These results are in agreement with those obtained from other experimental models, as IFN-γ−/− or IFN-γ receptor-deficient mice with CD4 T-cell-mediated EAE developed more severe disease than wild-type mice whereas neutralization of IFN-γ diminished EAE in models in which demyelination was CD8 T-cell dependent (13, 30, 56, 59).
γδ T cells have been identified in demyelinating lesions in the central nervous system (CNS) of patients with multiple sclerosis (50, 53). They have also been identified in rodents with EAE, although their function in these animals is controversial. Depletion of γδ T cells resulted in more severe disease or an inability to resolve EAE in one set of studies (34, 45). However, in other studies, the absence of γδ T cells resulted in milder disease (39, 47, 54). These different outcomes are explained in part by differences in experimental protocols. For example, two groups recently showed that inflammation was diminished during the early stages of EAE in the absence of γδ T cells (39, 45). While disease resolved if cells were transferred into normal mice, it did not if the recipient mice lacked γδ T cells (45). Thus, in the early stages of disease, the absence of γδ T cells resulted in milder disease, but their absence during the resolution stage resulted in more severe disease. However, in none of these models was the role of γδ T cells examined in the absence of αβ T cells.
The mechanisms by which γδ T cells are activated or function in the setting of demyelination, including that induced by JHM, remain unknown. No antigen-presenting molecule for γδ T cells, such as major histocompatibility complex class I and II molecules, has been identified. In common with CD8 T and NK cells, γδ T cells are able to lyse target cells and to produce IFN-γ in response to both infectious and tumor antigens (7, 17, 19, 58). Few ligands for murine γδ T cells have been identified and there is no identifiable molecular pattern among these putative ligands. Many of these ligands are proteins involved in the stress response, suggesting that γδ T cells, in some settings, do not require a foreign antigen for activation. In humans, a variety of small molecule ligands for γδ T cells have been identified, including prenoid derivatives and bisphosphonates (37).
A subset of γδ T cells have activity analogous to that of NK cells and may be activated through interactions with ligands for NK receptors. In particular, 25% of mouse splenic γδ T cells express NKG2D (31). Two isoforms of NKG2D have been identified in NK cells. The two proteins result from alternative splicing from a single gene and differ by 13 amino acids in their NH2 termini. The long form (NKG2D-L) is present in naïve cells, whereas the short form (NKG2D-S) is detected only in activated cells (19). In one study, cutaneous γδ T cells were shown to recognize two major histocompatibility complex class I-related molecules, Rae-1 and H60, via the activating NK receptor NKG2D (19). These NKG2D-ligand interactions resulted in activation of the cytolytic functions of γδ T cells and thereby effected a response to cutaneous malignancy (2, 19). In addition, γδ T cells have been demonstrated to express downregulatory molecules of the Ly49 family (22, 57), suggesting that these cells function more in the innate immune response than do traditional αβ T cells.
In NK cells, NKG2D may serve as a primary activating receptor, with cross-linking of the receptor sufficient for full cellular activation (48). This process involves signaling through DAP12 (DNAX-activating protein of 12 kDa), an adaptor molecule that contains a cytoplasmic immunoreceptor tyrosine-based activation motif. DAP12 is associated only with the short variant of NKG2D (NKG2D-S) (19). NK cells also express the adaptor protein DAP10 (DAP of 10 kDa), a protein that has motifs resembling those found in costimulatory molecules such as CD28 (16, 18). Activation via this adapter molecule is not believed to serve as a primary activating signal but rather to provide costimulatory activity. This differentiation between involvement of DAP10 in primary stimulatory and costimulatory pathways is not complete, however, since recent data suggest that DAP10 signaling is sufficient for some aspects of NK activation (64). In contrast to NK cells, CD8 T cells, which also express both isoforms of NKG2D, cannot be fully activated by cross-linking of the receptor. These cells express DAP10 but no DAP12, indicating that in the absence of DAP12, NKG2D serves only as a costimulatory receptor in this cell type. In support of this, expression of DAP12 by CD8 T cells from a transgene facilitates cellular activation by NKG2D ligation (16). Herein, we investigate the roles of IFN-γ and NKG2D in γδ T-cell-mediated demyelination.
MATERIALS AND METHODS
Mice.
Specific-pathogen-free TCRβ−/− (backcrossed 12 times onto a C57BL/6 background, as per the vendor) and C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME) and bred at the University of Iowa. Mice were assessed for clinical disease using the following scale: 0, asymptomatic; 1, limp tail; 2, wobbly gait or hunched or mild encephalitis; 3, hindlimb paresis or moderate encephalitis; 4, quadriparesis/paralysis or severe encephalitis; 5, moribund. In all experiments, mice were euthanized 14 days postinfection with JHM. All animal studies were approved by the University of Iowa Animal Care and Use Committee.
Virus.
We used the neuroattenuated variant of MHV JHM, strain 2.2-V-1, provided by J. Fleming (University of Wisconsin, Madison, WI), in all experiments. Mice were infected with 1,000 PFU of JHM intracerebrally. Viruses were titered by plaque reduction assay as previously described (42).
Flow cytometry.
Staining for γδ T cells and phenotypic markers was done as previously described (14). In brief, we made a single-cell suspension from the brains of JHM-infected mice and isolated leukocytes using a 30% Percoll (Pharmacia, Uppsala, Sweden) gradient. We blocked by treatment with 10% normal rat serum (Pel-Freez Biologicals, Rogers, AR) and 1:200 anti-FcγR (monoclonal antibody 2.4G2; BD Pharmingen, San Diego, CA). Cells were then stained with fluorescein isothiocyanate- or phycoerythrin-conjugated anti-γδ TCR (monoclonal antibody GL3) and anti-CD44 (monoclonal antibody IM-7), anti-CD62L (monoclonal antibody MEL-14), anti-CD69 (monoclonal antibody H1.2F3) (all from Pharmingen) or phycoerythrin-conjugated anti-NKG2D (monoclonal antibody CX5; eBioscience, San Diego, CA). Cells were washed and fixed with Cytofix (Pharmingen). A FACScan (Becton-Dickinson, Mountain View, CA) was used to analyze stained cells.
Depletion by antibody.
IFN-γ was depleted in vivo by administration of 750 μg of the anti-IFN-γ monoclonal antibody XMG1.2 (obtained from John Harty, University of Iowa) on days 0, 2, 5, 8, and 11 postinfection. The isotype-matched irrelevant antibody HOPC-1 (a gift from M. Dailey, University of Iowa) was used as a control. For blockade of CD154, we treated mice with 150 μg of monoclonal antibody MR-1 on the day of infection and every 3 days thereafter. For blockade of NKG2D, we treated mice with 250 μg of hamster anti-mouse NKG2D monoclonal antibody C7 (a gift from Wayne Yokoyama, Washington University, and Jon Heusel, University of Iowa) on days 0, 3, 6, 9, and 12 postinfection (27). γδ T cells were depleted with 250 μg of UC7-13D5 as previously described (14). As a control for treatment with either MR-1, C7, or UC7-13D5, we treated mice with equal quantities of hamster immunoglobulin (Ig) (Jackson ImmunoResearch, West Grove, PA).
Depletion of NK cells was performed using an anti-NK1.1 antibody (clone PK-136, obtained from Jon Heusel and Tim Ratliff, University of Iowa). The antibody was administered at an initial dose of 200 μg 1 day prior to infection and at maintenance doses of 100 μg every 3 days until harvest. An isotype-matched control antibody (Sigma, St. Louis, MO) was used at the same concentrations. Efficacy of depletion was monitored by flow cytometry using monoclonal antibodies to CD3 (clone 145-2C11, Pharmingen) and NKG2D (NK cells are CD3− NK1.1+ NKG2D+) and by cytotoxicity assays, using YAC-1 target cells in chromium release assays (9). Depletion was greater than 90% by flow cytometry. At an effector-to-target cell ratio of 25:1, residual NK cell cytotoxicity in depleted animals was less than 3% of that detected in controls.
Immunohistochemistry and immunofluorescence.
Spinal cords from JHM-infected mice were removed, fixed in zinc formalin (Labsco, Solon, OH), and embedded in paraffin. After blocking with CasBlock (Zymed, South San Francisco, CA), 8-μm sections were incubated with anti-JHM nucleocapsid monoclonal antibody 5B188.2 (a gift from Michael Buchmeier, Scripps Research Institute, La Jolla, CA), anti-F4/80 monoclonal antibody A3-1 (Serotec, Oxford, England), or a cocktail of anti-neurofilament monoclonal antibodies (SMI-312, Sternberger Monoclonal Antibodies, Lutherville, MD). For immunohistochemistry, samples were incubated with biotinylated anti-mouse IgG (for anti-JHM-N; Jackson Immunoresearch) or anti-rat IgG (for F4/80; Vector, Burlingame, CA) and developed with streptavidin-horseradish peroxidase (Jackson Immunoresearch) and diaminobenzidine (Sigma). For immunofluorescence, fluorescein isothiocyanate-conjugated anti-mouse IgG (Vector) was used.
Quantification of demyelination and macrophages.
Demyelination was quantified as previously described. In brief, 8-μm sections of zinc formalin-fixed, paraffin-embedded spinal cords were stained with Luxol fast blue and counterstained with hematoxylin and eosin. Sections were photographed using an Optronics (Goleta, CA) digital camera. The total area of intact myelin and demyelinated areas was quantified using NIH ImageJ. Macrophage infiltration into the white and gray matter was determined by counting F4/80+ cells in eight noncontiguous fields under 10X magnification as previously described (14).
RT-PCR for DAP10, DAP12, and NKG2D-S.
To determine whether mRNAs for DAP10, DAP12, or NKG2D-S were expressed in CNS-infiltrating γδ cells, we used nested reverse transcription (RT)-PCR. NKG2D+ γδ or CD8 T cells were isolated using flow cytometric sorting on a FACSDiva (Becton-Dickinson, Mountain View, CA). RNA was recovered from these cells using an RNeasy column kit (QIAGEN, Valencia, CA). cDNA was produced using Moloney murine leukemia virus reverse transcriptase (Invitrogen, Carlsbad, CA) and nested PCR was performed using Taq polymerase. Outer primers for DAP12 were TCTGGAGCCCTCCTGGTGCC (forward) and ATACTTCTGGTCTCTGACCC (reverse). Inner primers for DAP12 were CCTGTCCTCCTGACTGTGGG (forward) and TAAGGCGACTCAGTCTCAGC (reverse). Outer primers for DAP10 were AGGCTACCTCCTGTTCCTGC (forward) and ACTCTACCATCTTCTTGGGC (reverse). Inner primers were GGCTGCAAGTCAGACATC (forward) and GGCGCATACATACAAACACC (reverse). The outer primers for NKG2D-S were ACAAGAAACAGGATCTCCCTTCTCTGC (forward) and CAAACAGGAAGCTTGGCTCTGGTT (reverse). The inner primers were ACCTCAAGCCAGCAAAGTGGGATA (forward) and GAAACTGGGACTTCCTTGTTGCAC (reverse).
IFN-γ production by γδ T cells.
Brain-derived or splenic lymphocytes were treated with phorbol myristate acetate (10 ng/ml) and ionomycin (500 ng/ml) for 4 h at 37°C. Monensin was added for the final 2 h. Cells were immediately stained for intracellular IFN-γ production as described previously (60). In other experiments, tissue culture plates were coated with 1 mg/ml of N-[1-(2,3-dioleoloxy)propyl]-N,N,N-trimethylammonium chloride (DOTAP, Sigma) for 10 min at room temperature, washed with phosphate-buffered saline, and incubated with anti-NKG2D monoclonal antibody (clone A10. eBioscience, San Diego, CA). DOTAP was used to enhance antibody binding (14). CNS-derived or splenic cells were resuspended in RPMI supplemented with 10% fetal calf serum, 5% rat concanavalin A supernatant, and 50 mM α-methylmannoside and added to the coated plates. Samples were then incubated and analyzed as described above.
Statistics.
Statistical significance was determined by two-tailed unpaired t tests. All results are expressed as means ± standard error of the mean. Values of P < 0.05 were considered statistically significant.
RESULTS
In our previous study, we demonstrated that demyelination in nude mice was mediated by γδ T cells. Because the defect in these mice is a poorly characterized genetic defect that results in the lack of a thymus, conventional αβ T cells emerge in detectable numbers beginning at approximately 10 weeks of age (32). Although these cells did not contribute to demyelination at the age we studied, we were concerned that the presence of these cells could potentially confound further studies. To avoid this potential problem, we analyzed demyelination in JHM-infected mice lacking the TCRβ chain. These mice do not express αβ TCRs, so cells of the T-cell lineage are only capable of expressing the γδ TCR.
γδ T cells mediate demyelination in TCRβ−/− mice.
Initially, we characterized the disease induced by JHM infection in TCRβ−/− mice. These mice survived for up to 16 to 18 days postinfection, which was on average longer than JHM-infected nude mice (14). However, because mice began to die at day 14 postinfection, we analyzed mice at this time point in all experiments to ensure uniform comparisons.
TCRβ−/− mice infected with JHM exhibited clinical symptoms consistent with demyelination, including an inability to right themselves and limb paresis. These symptoms increased progressively with time (Fig. 1A). At day 14 postinfection, we observed robust amounts of demyelination in the spinal cords of these mice (26.8 ± 4.9%, Table 1), which was greater than what we observed previously in nude mice (14). Demyelination was initially detectable between days 7 and 10 postinfection (Fig. 1B) and was accompanied by a significant mononuclear cell infiltrate. The infiltrating cells did not effectively control the JHM infection, as the amount of virus in the brains of infected TCRβ−/− mice (Table 1) was similar to that observed in RAG1−/− mice (61). Similar to other models of JHM-induced disease, areas of demyelination were characterized by a robust macrophage/microglia infiltrate with JHM antigen present in areas adjacent to demyelination (Fig. 2A to C). Additionally, demyelination occurred with relative sparing of axons (Fig. 2D), indicating that demyelination was primary, and not secondary to neuronal loss.
FIG. 1.

Progression of clinical disease and demyelination in JHM-infected TCRβ−/− mice is dependent on γδ T cells and expression of IFN-γ and NKG2D. (A) TCRβ−/− mice were infected with JHM and treated with anti-γδ-TCR monoclonal antibody UC7-13D5 (n = 7, open squares), anti-IFN-γ monoclonal antibody XMG1.2 (n = 7, solid diamonds), anti-CD154 monoclonal antibody MR-1 (n = 4, hatched squares), or anti-NKG2D monoclonal antibody C7 (n = 8, hatched diamonds). Clinical signs were monitored as described in Materials and Methods. Mice that were not treated with antibody (n = 6, solid triangles) or were treated with control antibody (n = 20, open circles) developed the most severe disease. Each antibody was analyzed in at least two independent experiments. Samples were analyzed for statistical significance by comparing pairs of values at each day postinfection. Differences between mice receiving hamster Ig and those receiving anti-γδ TCR monoclonal antibody, anti-IFN-γ monoclonal antibody, or anti-NKG2D monoclonal antibody were statistically significant (day 5: P < 0.05; days 6 and 7: P < 0.01; days 8+: P < 0.001). The difference between treatment with anti-NKG2D monoclonal antibody and anti-IFN-γ monoclonal antibody or anti-γδ TCR monoclonal antibody was also statistically significant (day 7: P < 0.01; days 8+: P < 0.001). The clinical course after anti-IFN-γ monoclonal antibody versus anti-γδ TCR monoclonal antibody treatment was not significantly different (P > 0.05). (B) Demyelination was initially detectable between days 7 and 10 postinfection in JHM-infected TCRβ−/− mice.
TABLE 1.
Demyelination, viral titers, and lymphocyte infiltration in JHM-infected TCRβ−/− mice
| Group | Demyelination (%) (no. of samples analyzed) | Viral titer (log10 PFU/g) | γδ T cells in the CNS |
|---|---|---|---|
| TCRβ−/− untreated | 26.8 ± 4.9 (8) | 5.7 ± 0.1 | 5.2 ± 0.5 × 104 |
| Anti-γδ-TCR | 6.4 ± 3.0 (8)a | 6.0 ± 0.3 | (0) |
| Hamster Ig | 23.3 ± 5.2 (7) | 5.7 ± 0.1 | 4.8 ± 0.4 × 104 |
| Anti-IFN-γ | 4.2 ± 1.6 (7)a | 6.2 ± 0.2 | 5.7 ± 1.1 × 104 |
| HOPC-1 | 20.7 ± 5.3 (7) | 5.8 ± 0.3 | 5.2 ± 0.7 × 104 |
| Anti-CD154 | 19.1 ± 4.5 (4) | ndb | 5.0 ± 0.3 × 104 |
| Hamster Ig | 21.8 ± 3.9 (4) | nd | 4.7 ± 0.4 × 104 |
| Anti-NKG2D | 7.0 ± 3.1 (8)a | 5.6 ± 0.2 | 3.1 ± 0.4 × 104 |
| Hamster Ig | 19.7 ± 4.3 (8) | 5.8 ± 0.3 | 5.1 ± 0.6 × 104 |
| Wild-type C57BL/6 + anti-NKG2D | 16.1 ± 3.9 (5) | nd | nd |
| Wild-type C57BL/6 + hamster Ig | 15.7 ± 4.9 (5) | nd | nd |
| Anti-NK1.1 | 20.3 ± 6.5 (6) | nd | nd |
| Mouse Ig | 22.0 ± 9.9 (4) | nd | nd |
Significantly different from control (P < 0.05).
nd, not determined.
FIG. 2.

Demyelination in the spinal cord of JHM-infected TCRβ−/− mice. Spinal cords from infected mice at 14 days postinfection were removed, fixed with zinc formalin, and embedded in paraffin. Serial 8-μm sections were stained for (A) demyelination, (B) viral antigen, (C) macrophages or microglia, and (D) axons, as described in Materials and Methods. Areas of demyelination (A) are accompanied by robust macrophage/microglia infiltration (C). Little viral antigen is detectable in these areas, although some can be found in adjacent, normal-appearing white matter (B). The loss of myelin is primary, as axonal staining is preserved through the area of demyelination (D). Scale bar: 100 μm.
We anticipated that demyelination was induced by γδ T cells, based on the results obtained in JHM-infected nude mice. To address this formally, we depleted γδ T cells using the anti-γδ TCR monoclonal antibody UC7-13D5 in the same manner as described previously (14). Treatment with UC7-13D5 resulted in no detectable γδ T-cell infiltrate within the CNS (Fig. 3A and B). Depletion of γδ T cells resulted in a 73% reduction in demyelination (Table 1), indicating that these cells were involved in the demyelinating process. By contrast, mice treated with the same amount of hamster Ig developed demyelination at levels similar to that observed in untreated, JHM-infected TCRβ−/− mice. Mice depleted of γδ T cells additionally had a significantly improved clinical score compared to their control antibody-treated or untreated counterparts (Fig. 1A).
FIG. 3.

Phenotype and depletion of γδ T cells. (A and B) JHM-infected mice were treated with hamster Ig (A) or anti-γδ-TCR monoclonal antibody UC7-13D5 (B) on days 0, 3, 6, 9, and 12 postinfection. At 14 days postinfection, γδ T cells constituted 10 to 20% of the lymphocyte infiltrate into the CNS (A). No γδ T cells were detectable in UC7-13D5-treated mice (B). (C to F) Brains were removed from JHM-infected TCRβ−/− mice and CNS-derived lymphocytes were stained with anti-γδ-TCR and antibodies against CD69 (C), CD62L (D), CD44 (E), and NKG2D (F). Most γδ T cells within the CNS had marker levels consistent with an activated phenotype: CD44+, CD62L−, and CD69+. Approximately 40% of γδ T cells expressed the NK activating receptor NKG2D (F). Numbers indicate the percentage of γδ T cells in each group.
Previously we showed that diminished demyelination was accompanied by a decrease in macrophage/microglia infiltration into the white matter (14, 44). To determine whether this also occurred in JHM-infected TCRβ−/− mice after anti-γδ TCR antibody treatment, we counted the number of macrophages/microglia in tissue sections from three monoclonal antibody UC7-13D5-treated and three Ig-treated mice, as described in Materials and Methods. The number of F4/80+ cells in the white matter decreased by 61% after treatment with the anti-γδ TCR antibody.
Phenotype of CNS-infiltrating γδ T cells.
Unlike athymic mice, which had a relatively small lymphocyte infiltrate that made characterization of γδ T cells unfeasible, we were able to identify adequate numbers of lymphocytes within the CNS of TCRβ−/− mice to phenotype these cells. γδ T cells constitute 10 to 20% of the lymphocyte infiltrate in these mice (Fig. 3A), so there were 5 × 104 to 10 × 104 γδ T cells within the CNS. Consistent with an activated phenotype, these cells were CD69+, CD62Llo, and CD44hi (Fig. 3C to E). Additionally, approximately 40% of the γδ T cells within the CNS expressed the receptor NKG2D, an activating molecule found on NK cells, peritoneal macrophages, and some activated CD8 αβ T cells (Fig. 3F) (48).
Demyelination in TCRβ−/− mice requires IFN-γ.
We next sought to address the mechanisms by which γδ T cells induce demyelination in TCRβ−/− mice. Demyelination induced by CD8 T cells in JHM-infected mice is dependent on production of IFN-γ (44). Given similarities between NK cells, CD8 T cells, and γδ T cells in effector functions, in particular cytotoxicity and IFN-γ molecule expression (25), we reasoned that IFN-γ was likely to be a critical molecule in the demyelinating process. To test this hypothesis, TCRβ−/− mice were infected with JHM and treated with the depleting anti-IFN-γ monoclonal antibody XMG1.2 or a control antibody, as described in Materials and Methods.
TCRβ−/− mice treated with monoclonal antibody XMG1.2 exhibited a disease course different from that of mice treated with control antibody but nearly identical to that observed in mice treated with anti-γδ TCR antibody (Fig. 1A). These mice in general failed to develop clinical signs of demyelination but instead only had signs of mild encephalitis, such as hunching, irritability, lethargy, and ruffled fur. Consistent with the clinical signs, anti-IFN-γ antibody treatment resulted in an 80% reduction (4.2 ± 1.6% versus 20.7 ± 5.3%) in the amount of demyelination observed compared to control mice (Table 1). These results demonstrated that IFN-γ was necessary for demyelination in TCRβ−/− mice. We also observed minimal infiltration of macrophages/microglia into the white matter of JHM-infected, monoclonal antibody XMG1.2-treated TCRβ−/− mice (data not shown), consistent with a role for these cells in the demyelinating process.
Collectively, these results suggest that γδ T cells mediate demyelination via an IFN-γ-dependent mechanism. Consistent with this conclusion, we also demonstrated that CNS-derived and splenic γδ T cells from JHM-infected mice produced IFN-γ in response to phorbol myristate acetate and ionomycin (Fig. 4). IFN-γ is also produced by NK cells, but these cells are unlikely to be the source for the IFN-γ involved in demyelination. RAG1−/− mice contain normal numbers of NK cells, but demyelination does not occur in JHM-infected RAG1−/− mice in the absence of experimental interventions (44). To prove formally that NK cells were not critical for demyelination, we depleted these cells from JHM-infected TCRβ−/− mice. Depletion of NK cells had no effect on the amount of demyelination (Table 1).
FIG. 4.

IFN-γ production by CNS-derived and splenic γδ T cells. Lymphocytes were harvested from the spleens (A and B) and CNS (C and D) of infected TCRβ−/− mice at 14 days postinfection. Cells were either treated with phorbol myristate acetate and ionomycin directly ex vivo (B and D) or not stimulated (A and C) and then analyzed for intracellular IFN-γ production as described in Materials and Methods.
Failure to identify a target in JHM for γδ T cells.
We were unable to identify a JHM-specific target for γδ T cells in infected TCRβ−/− mice. In several experiments, we variously incubated CNS-derived γδ T cells from infected mice with either preparations of inactivated virus, structural proteins derived from JHM, or JHM-infected cell lysates. We used analogous methods to identify CD4 and CD8 αβ T-cell epitopes (10, 36, 62). However, we consistently observed no activation of γδ T cells above baseline as measured by either IFN-γ or tumor necrosis factor alpha production (data not shown). Although these data do not rule out direct recognition of JHM antigen by γδ T cells, they are consistent with previous data showing that γδ T cells often recognize host proteins that are upregulated during pathological conditions (24).
Based on these results, we pursued the hypothesis that the γδ T cells were activated, at least in part, through one of their NK cell-activating receptors. Although there are many such activating receptors, NKG2D is the most widely expressed among γδ T cells (48) and we showed that it was expressed on a fraction of γδ T cells in the JHM-infected CNS (Fig. 3F). We next asked whether demyelination was dependent on this molecule. As noted above, NK cells, which also express NKG2D, are not required for demyelination in TCRβ−/− mice (Table 1) and NK activity is not sufficient for demyelination, as SCID and RAG1−/− mice, in which NK cells develop normally, do not exhibit myelin destruction (28, 60).
NKG2D, but not CD40, is critical for the demyelinating process.
To investigate the role of NKG2D in γδ T-cell-mediated demyelination, we pursued an in vivo antibody blockade approach. TCRβ−/− mice were treated with 250 μg of the anti-NKG2D monoclonal antibody C7 (27) or hamster Ig on the day of infection and every 3 days thereafter. Although we expected monoclonal antibody C7 to block NKG2D, it resulted in a nearly complete loss of the NKG2D+ γδ T-cell population within the CNS with a concomitant diminution in total γδ T cells (Fig. 5A and B and data not shown). Apparent disappearance of NKG2D+ CD8 T cells after anti-NKG2D antibody treatment was also noted in a recent study (albeit with a different anti-NKG2D antibody) and was attributed to an effect on NKG2D+ cell proliferation or to an effect on cell surface expression of the receptor (40).
FIG. 5.
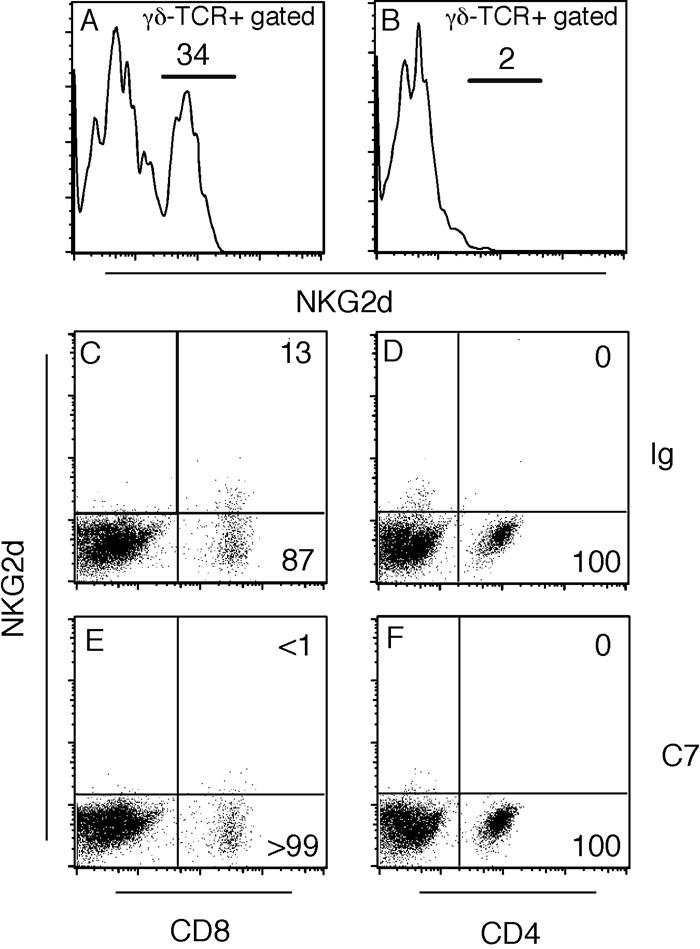
Depletion of NKG2D+ cells. (A and B) JHM-infected TCRβ−/− mice were treated with hamster Ig (A) or the anti-NKG2D monoclonal antibody C7 (B) on days 0, 3, 6, 9, and 12 postinfection. There were no residual γδ T cells expressing NKG2D protein in C7-treated mice (B). (C to F) JHM-infected C57BL/6 mice were treated with anti-NKG2D monoclonal antibody C7 (E and F) or Ig (C and D). CD8 (C and E) and CD4 (D and F) cells were analyzed for expression of NKG2D. Only CD8 T cells expressed NKG2D (C) and treatment with monoclonal antibody C7 depleted this population (E).
Anti-NKG2D monoclonal antibody-treated mice developed a clinical picture that was different from that of either control mice or those treated with anti-γδ TCR or anti-IFN-γ antibody (Fig. 1A). Anti-NKG2D monoclonal antibody-treated mice all exhibited gait difficulties and righting inability. However, none of these mice developed limb paralysis as did untreated or Ig-treated mice. Additionally, unlike the anti-IFN-γ-treated mice, these mice did not have clinical signs of encephalitis, consistent with no change in virus titer compared to Ig-treated controls (Table 1). Treatment with anti-NKG2D antibody resulted in a significant reduction in the amount of demyelination (Table 1) compared to the control limb (7.0 ± 3.1% versus 19.7 ± 4.3%). Notably, demyelination was not completely abrogated, indicating that NKG2D− γδ T cells contributed to demyelination in the JHM-infected mouse.
NKG2D is expressed on several other cell types, including macrophages. Because macrophages and microglia are critical for demyelination (33, 55), we next addressed the possibility that macrophage/microglia and not γδ T-cell expression of NKG2D was important in myelin destruction. To do this, we infected C57BL/6 mice with JHM and treated them with C7 or hamster Ig as described above. Demyelination in these mice is mediated largely by CD4 and CD8 αβ T cells (23). Only a subset of CNS-derived CD8 T cells in these mice express NKG2D at 14 days postinfection (Fig. 5C), a time when most anti-JHM CD8 T cells in the infected CNS have lost cytolytic activity (5). NKG2D expression was not detected on these cells after anti-NKG2D monoclonal antibody treatment (Fig. 5E), showing the efficacy of the antibody treatment. There was no difference in the amount of demyelination observed between C7- and Ig-treated wild-type mice (16.1% versus 15.7%, respectively, Table 1). These results indicate that depletion of NKG2D+ macrophages did not contribute to the reduction of demyelination in anti-NKG2D monoclonal antibody-treated C57BL/6 mice. They suggest that the key effect of anti-NKG2D antibody treatment in TCRβ−/− mice is to diminish γδ T-cell and not macrophage function and thereby to prevent demyelination.
CD40-CD154 interactions have been implicated in the pathogenesis of CNS inflammation in the experimental autoimmune encephalitis model (3) and CD154 is expressed by γδ T cells (7). To investigate the potential role of these interactions in demyelination induced by γδ T cells in JHM-infected mice, we treated mice with the anti-CD154 monoclonal antibody MR-1 as described in Materials and Methods. Treatment with this antibody did not result in a change in the amount of demyelination (Table 1) or clinical disease (Fig. 1A), suggesting that CD40/CD40L interactions were not critical for γδ T-cell-mediated demyelination.
DAP12 and NKG2D-S mRNAs are expressed in NKG2D+ γδ T cells in the CNS.
The adaptor signaling proteins DAP10 and DAP12 associate with NKG2D in NK cells (48) and DAP12 is expressed by at least some splenic γδ T cells (41). Cross-linking NKG2D is sufficient by itself to activate NK cells with signaling occurring via DAP12-dependent pathways (16).
To probe the expression of DAP10 and DAP12 in NKG2D+ γδ T cells within the CNS, we used nested RT-PCR to identify mRNA expression. We sorted NKG2D+ γδ T cells from the JHM-infected TCRβ−/− CNS. As a control, we also sorted NKG2D+ CD8 T cells from the spleens of uninfected C57BL/6 mice. RNA was harvested from these cells and subjected to RT-PCR as described in Materials and Methods. NKG2D+ γδ T cells from three independent samples all showed evidence of both DAP12 and DAP10 mRNAs, while CD8 T cells derived from C57BL/6 mice expressed DAP10 but not DAP12 (Fig. 6A). DAP12 is associated with NKG2D-S but not NKG2D-L. As shown in Fig. 6B, NKG2D-S mRNA is expressed in CNS-derived NKG2D+ γδ T cells. These results suggest that the pathway for complete activation is present in these CNS-derived γδ T cells.
FIG. 6.

Identification of DAP10, DAP12, and NKG2D-S mRNA in CNS-derived γδ T cells. NKG2D+ γδ T cells from the CNS of JHM-infected TCRβ−/− mice or NKG2D+ CD8 T cells from an uninfected C57BL/6 spleen were sorted as described in Materials and Methods. (A) mRNA was extracted from the sorted cells and subjected to RT-PCR for DAP10, DAP12, and hypoxanthine phosphoribosyltransferase (HPRT). Lanes 1 to 3, CNS-derived γδ T cells from three individual JHM-infected TCRβ−/− mice. Lane 4, splenic NKG2D+ CD8 T cells from an uninfected C57BL/6 mouse. Lane 5, negative control. (B) mRNA was subjected to RT-PCR for NKG2D-S. Lanes 1 and 2, CNS-derived γδ T cells from two individual JHM-infected mice. Lane 3, negative control.
DISCUSSION
In this study, we confirmed and extended our previous finding that γδ T cells mediate demyelination. In both nude mice and TCRβ−/− mice, infection with JHM results in a robust γδ T-cell infiltrate, with subsequent activation of macrophages and microglia, resulting in demyelination. Furthermore, we have begun to elucidate the mechanisms by which these cells induce demyelination. γδ T cells, like CD8 T cells, induce demyelination by an IFN-γ-dependent mechanism. Most importantly, NKG2D+ cells are critical for virus-induced demyelination and are consistent with previous results indicating a role for this receptor molecule in autoimmune diabetes (40).
The function of murine γδ T cells remains largely elusive. In humans, γδ T cells appear to increase in number after infection with a number of bacterial agents, including mycobacteria and rickettsiae, and various γδ T-cell nonpeptide target antigens have been identified (37). No such antigenic patterns have been found in mice, although, in one case, murine γδ T cells have been shown to interact with herpes simplex virus glycoprotein I (51, 52). This interaction did not require antigen presentation by major histocompatibility complex class I or II molecules. Likewise, few descriptions of a pathogenic role for γδ T cells exist. In mice with EAE, depletion of γδ T cells resulted in decreased IFN-γ expression by αβ T cells in the CNS, with effects on both the initiation and resolution of disease (39, 45). Disease outcome was worse when γδ T cells were not present during the resolution phase (45). The absence of these cells during the initial stages resulted in less severe disease with a concomitant reduction in the levels of several proinflammatory cytokines and chemokines (39, 46, 47).
In our experiments, treatment with anti-IFN-γ antibody resulted in an 80% decrease in γδ T-cell-mediated demyelination (Table 1). Although γδ T cells act, in part, to regulate IFN-γ production by αβ T cells in mice with EAE (39, 45), we think that it is more likely that IFN-γ produced by γδ T cells is directly involved in the demyelinating process in JHM-infected RAG1−/− mice since these mice lack αβ T cells and γδ T cells from the infected CNS are able to express IFN-γ (Fig. 4). Also, it is unlikely that IFN-γ production by NK cells is required for demyelination, since depletion of NK cells did not diminish the amount of demyelination that we detected (Table 1).
IFN-γ expressed by γδ T cells may directly activate macrophages/microglia, cells which are important for demyelination. Alternatively, these cells may interact with infected target cells in the CNS to release proinflammatory cytokines or chemokines. The latter would then serve as chemoattractants for macrophages/microglia and thereby propagate the demyelinating process. Expression of chemokines such as CCL2/monocyte chemotactic protein 1 and CCL3/macrophage inflammatory protein 1α has been identified as crucial for macrophage/microglia infiltration in EAE (29, 35, 63). CCL5 (RANTES)/CCR5 and CCL2/CCR2 interactions were identified as important in T-cell and macrophage infiltration into the JHM-infected CNS (12, 20). Consistent with these reports, we recently showed that infection of RAG1−/− mice with a recombinant JHM that expressed CCL2 resulted in demyelination in the absence of any other experimental interventions (33).
In addition to the critical role for IFN-γ in demyelination in TCRβ−/− mice, our results suggest that NKG2D+ γδ T cells play a central role in effecting demyelination. Mice treated with the anti-NKG2D monoclonal antibody exhibited diminished infiltration of macrophages and microglia into the white matter (data not shown). In mice, NKG2D binds to members of the retinoic acid-inducible early transcript 1 (Rae-1) family, to H-60, a minor histocompatibility antigen, and to murine UL16 binding-protein-like transcript 1 (MULT-1), a homologue of a human cytomegalovirus protein (8, 11). Of note, H-60 is not expressed in C57BL/6 mice (15, 48). Rae-1 is detected in the CNS on some neurons, particularly dorsal ganglia (1); our results show that demyelination is primary so that Rae-1, if it is the relevant ligand, would also need to be expressed on another cell type, such as oligodendrocytes or macrophages/microglia.
Notably, a recent study demonstrated that macrophages upregulate Rae-1 upon stimulation through Toll-like receptors, but not through treatment with IFN-γ (21). This raises the intriguing possibility that macrophages and γδ T cells in the JHM-infected TCRβ−/− CNS act in concert to activate each other, leading to demyelination. Our results also show incomplete abrogation of demyelination after NKG2D blockade (Table 1). This result is not unexpected since only a fraction of γδ T cells in the JHM-infected CNS express NKG2D (Fig. 3) and suggests that γδ T cells are also activated via NKG2D-independent mechanisms. These results are in agreement with others showing that NK cell lysis of tumor cells expressing NKG2D ligands is only partially blocked by anti-NKG2D antibody treatment (31).
These results, in conjunction with the detection of DAP12 in γδ T cells, raise the possibility that these cells are activated directly through NKG2D in JHM-infected TCRβ−/− mice. In the case of NK cells, cross-linking NKG2D results in cell activation as measured by cytokine expression and cytotoxicity (31). This process requires DAP12 and the short isomer of NKG2D (16). The presence of the mRNA encoding these 2 molecules within CNS-localized NKG2D+ γδ T cells (Fig. 6) suggests a mechanism by which the requirement for TCR recognition of an antigen might be bypassed in these cells.
Collectively, our results provide additional evidence that IFN-γ is critical for demyelination to occur and also are consistent with the notion that γδ T-cell activation in the JHM-infected mouse occurs via a mechanism, ligation of the NKG2D receptor, which does not involve recognition of a viral antigen. γδ T cells appear to have primarily a regulatory role in rodents with EAE (39, 45). Our results suggest that γδ T cells also function as effector cells in the demyelinating process. These results may be relevant for understanding the role of γδ T cells in multiple sclerosis and, together with previous reports in the EAE model, raise the possibility that γδ T cells have both regulatory and effector roles in the CNS of affected humans.
Acknowledgments
We thank Jon Heusel and Julianne Grant for helpful discussions and Taeg Kim and Andrew Brooks for review of the manuscript.
This research was supported in part by grants from the N.I.H. (NS40438) and National Multiple Sclerosis Society (RG2864). A.A.D. was supported by an individual National Research Service Award from the N.I.H. (NS42981).
REFERENCES
- 1.Backstrom, E., B. J. Chambers, E. L. Ho, O. V. Naidenko, R. Mariotti, D. H. Fremont, W. M. Yokoyama, K. Kristensson, and H. G. Ljunggren. 2003. Natural killer cell-mediated lysis of dorsal root ganglia neurons via RAE1/NKG2D interactions. Eur. J. Immunol. 33:92-100. [DOI] [PubMed] [Google Scholar]
- 2.Bauer, S., V. Groh, J. Wu, A. Steinle, J. H. Phillips, L. L. Lanier, and T. Spies. 1999. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 285:727-729. [DOI] [PubMed] [Google Scholar]
- 3.Becher, B., B. G. Durell, A. V. Miga, W. F. Hickey, and R. J. Noelle. 2001. The clinical course of experimental autoimmune encephalomyelitis and inflammation is controlled by the expression of CD40 within the central nervous system. J. Exp. Med. 193:967-974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benveniste, P., B. S. Chadwick, R. G. Miller, and J. Reimann. 1990. Characterization of cells with T-cell markers in athymic nude bone marrow and of their in vitro-derived clonal progeny. Comparison with euthymic bone marrow. J. Immunol. 144:411-419. [PubMed] [Google Scholar]
- 5.Bergmann, C. C., J. D. Altman, D. Hinton, and S. A. Stohlman. 1999. Inverted immunodominance and impaired cytolytic function of CD8+ T cells during viral persistence in the central nervous system. J. Immunol. 163:3379-3387. [PubMed] [Google Scholar]
- 6.Bergmann, C. C., B. Parra, D. R. Hinton, C. Ramakrishna, K. C. Dowdell, and S. A. Stohlman. 2004. Perforin and gamma interferon-mediated control of coronavirus central nervous system infection by CD8 T cells in the absence of CD4 T cells. J. Virol. 78:1739-1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bluestone, J. A., R. Khattri, R. Sciammas, and A. I. Sperling. 1995. TCR gamma delta cells: a specialized T-cell subset in the immune system. Annu. Rev. Cell Dev. Biol. 11:307-353. [DOI] [PubMed] [Google Scholar]
- 8.Carayannopoulos, L. N., O. V. Naidenko, D. H. Fremont, and W. M. Yokoyama. 2002. Cutting edge: murine UL16-binding protein-like transcript 1: a newly described transcript encoding a high-affinity ligand for murine NKG2D. J. Immunol. 169:4079-4083. [DOI] [PubMed] [Google Scholar]
- 9.Castro, R. F., G. D. Evans, A. Jaszewski, and S. Perlman. 1994. Coronavirus-induced demyelination occurs in the presence of virus-specific cytotoxic T cells. Virology 200:733-743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Castro, R. F., and S. Perlman. 1995. CD8+ T-cell epitopes within the surface glycoprotein of a neurotropic coronavirus and correlation with pathogenicity. J. Virol. 69:8127-8131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cerwenka, A., A. B. Bakker, T. McClanahan, J. Wagner, J. Wu, J. H. Phillips, and L. L. Lanier. 2000. Retinoic acid early inducible genes define a ligand family for the activating NKG2D receptor in mice. Immunity 12:721-727. [DOI] [PubMed] [Google Scholar]
- 12.Chen, B. P., W. A. Kuziel, and T. E. Lane. 2001. Lack of CCR2 results in increased mortality and impaired leukocyte activation and trafficking following infection of the central nervous system with a neurotropic coronavirus. J. Immunol. 167:4585-4592. [DOI] [PubMed] [Google Scholar]
- 13.Chu, C. Q., S. Wittmer, and D. K. Dalton. 2000. Failure to suppress the expansion of the activated CD4 T-cell population in interferon γ-deficient mice leads to exacerbation of experimental autoimmune encephalomyelitis. J. Exp. Med. 192:123-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dandekar, A. A., and S. Perlman. 2002. Virus-induced demyelination in nude mice is mediated by gamma delta T cells. Am. J. Pathol. 161:1255-1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Diefenbach, A., A. M. Jamieson, S. D. Liu, N. Shastri, and D. H. Raulet. 2000. Ligands for the murine NKG2D receptor: expression by tumor cells and activation of NK cells and macrophages. Nat. Immunol. 1:119-126. [DOI] [PubMed] [Google Scholar]
- 16.Diefenbach, A., E. Tomasello, M. Lucas, A. M. Jamieson, J. K. Hsia, E. Vivier, and D. H. Raulet. 2002. Selective associations with signaling proteins determine stimulatory versus costimulatory activity of NKG2D. Nat. Immunol. 3:1142-1149. [DOI] [PubMed] [Google Scholar]
- 17.Gao, Y., W. Yang, M. Pan, E. Scully, M. Girardi, L. H. Augenlicht, J. Craft, and Z. Yin. 2003. Gamma delta T cells provide an early source of interferon gamma in tumor immunity. J. Exp. Med. 198:433-442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilfillan, S., E. L. Ho, M. Cella, W. M. Yokoyama, and M. Colonna. 2002. NKG2D recruits two distinct adapters to trigger NK cell activation and costimulation. Nat. Immunol. 3:1150-1155. [DOI] [PubMed] [Google Scholar]
- 19.Girardi, M., D. E. Oppenheim, C. R. Steele, J. M. Lewis, E. Glusac, R. Filler, P. Hobby, B. Sutton, R. E. Tigelaar, and A. C. Hayday. 2001. Regulation of cutaneous malignancy by γδ T cells. Science 294:605-609. [DOI] [PubMed] [Google Scholar]
- 20.Glass, W. G., and T. E. Lane. 2003. Functional expression of chemokine receptor CCR5 on CD4+ T cells during virus-induced central nervous system disease. J. Virol. 77:191-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamerman, J. A., K. Ogasawara, and L. L. Lanier. 2004. Cutting edge: toll-like receptor signaling in macrophages induces ligands for the NKG2D receptor. J. Immunol. 172:2001-2005. [DOI] [PubMed] [Google Scholar]
- 22.Hara, T., H. Nishimura, Y. Hasegawa, and Y. Yoshikai. 2001. Thymus-dependent modulation of Ly49 inhibitory receptor expression on NK1.1+gamma/delta T cells. Immunology 102:24-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haring, J., and S. Perlman. 2001. Mouse hepatitis virus. Curr. Opin. Microbiol. 4:462-466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hayday, A., and R. Tigelaar. 2003. Immunoregulation in the tissues by gammadelta T cells. Nat. Rev. Immunol. 3:233-242. [DOI] [PubMed] [Google Scholar]
- 25.Hayday, A. C. 2000. γδ cells: a right time and a right place for a conserved third way of protection. Annu. Rev. Immunol. 18:975-1026. [DOI] [PubMed] [Google Scholar]
- 26.Hemmer, B., J. J. Archelos, and H. P. Hartung. 2002. New concepts in the immunopathogenesis of multiple sclerosis. Nat. Rev. Neurosci. 3:291-301. [DOI] [PubMed] [Google Scholar]
- 27.Ho, E. L., L. N. Carayannopoulos, J. Poursine-Laurent, J. Kinder, B. Plougastel, H. R. Smith, and W. M. Yokoyama. 2002. Costimulation of multiple NK cell activation receptors by NKG2D. J. Immunol. 169:3667-3675. [DOI] [PubMed] [Google Scholar]
- 28.Houtman, J. J., and J. O. Fleming. 1996. Dissociation of demyelination and viral clearance in congenitally immunodeficient mice infected with murine coronavirus JHM. J. Neurovirol. 2:101-110. [DOI] [PubMed] [Google Scholar]
- 29.Huang, D. R., J. Wang, P. Kivisakk, B. J. Rollins, and R. M. Ransohoff. 2001. Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific T helper cell type 1 immune response in experimental autoimmune encephalomyelitis. J. Exp. Med. 193:713-726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huseby, E. S., D. Liggitt, T. Brabb, B. Schnabel, C. Ohlen, and J. Goverman. 2001. A pathogenic role for myelin-specific CD8(+) T cells in a model for multiple sclerosis. J. Exp. Med. 194:669-676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jamieson, A. M., A. Diefenbach, C. W. McMahon, N. Xiong, J. R. Carlyle, and D. H. Raulet. 2002. The role of the NKG2D immunoreceptor in immune cell activation and natural killing. Immunity 17:19-29. [DOI] [PubMed] [Google Scholar]
- 32.Kennedy, J. D., C. W. Pierce, and J. P. Lake. 1992. Extrathymic T-cell maturation. Phenotypic analysis of T-cell subsets in nude mice as a function of age. J. Immunol. 148:1620-1629. [PubMed] [Google Scholar]
- 33.Kim, T. S., and S. Perlman. 2005. Viral expression of CCL2 is sufficient to induce demyelination in RAG1−/− mice infected with a neurotropic coronavirus. J. Virol. 79:7113-7120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kobayashi, Y., K. Kawai, K. Ito, H. Honda, G. Sobue, and Y. Yoshikai. 1997. Aggravation of murine experimental allergic encephalomyelitis by administration of T-cell receptor gammadelta-specific antibody. J. Neuroimmunol. 73:169-174. [DOI] [PubMed] [Google Scholar]
- 35.Mahad, D. J., and R. M. Ransohoff. 2003. The role of MCP-1 (CCL2) and CCR2 in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). Semin. Immunol. 15:23-32. [DOI] [PubMed] [Google Scholar]
- 36.Mobley, J., G. Evans, M. O. Dailey, and S. Perlman. 1992. Immune response to a murine coronavirus: Identification of a homing receptor-negative CD4+ T-cell subset that responds to viral glycoproteins. Virology 187:443-452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morita, C. T., R. A. Mariuzza, and M. B. Brenner. 2000. Antigen recognition by human γδ T cells: pattern recognition by the adaptive immune system. Springer Semin. Immunopathol. 22:191-217. [DOI] [PubMed] [Google Scholar]
- 38.Noseworthy, J. H., C. Lucchinetti, M. Rodriguez, and B. G. Weinshenker. 2000. Multiple sclerosis. N. Engl. J. Med. 343:938-952. [DOI] [PubMed] [Google Scholar]
- 39.Odyniec, A., M. Szczepanik, M. P. Mycko, M. Stasiolek, C. S. Raine, and K. W. Selmaj. 2004. Gammadelta T cells enhance the expression of experimental autoimmune encephalomyelitis by promoting antigen presentation and IL-12 production. J. Immunol. 173:682-694. [DOI] [PubMed] [Google Scholar]
- 40.Ogasawara, K., J. A. Hamerman, L. R. Ehrlich, H. Bour-Jordan, P. Santamaria, J. A. Bluestone, and L. L. Lanier. 2004. NKG2D blockade prevents autoimmune diabetes in NOD mice. Immunity 20:757-767. [DOI] [PubMed] [Google Scholar]
- 41.Pennington, D. J., B. Silva-Santos, J. Shires, E. Theodoridis, C. Pollitt, E. L. Wise, R. E. Tigelaar, M. J. Owen, and A. C. Hayday. 2003. The inter-relatedness and interdependence of mouse T-cell receptor gammadelta+ and alphabeta+ cells. Nat. Immunol. 4:991-998. [DOI] [PubMed] [Google Scholar]
- 42.Perlman, S., R. Schelper, E. Bolger, and D. Ries. 1987. Late onset, symptomatic, demyelinating encephalomyelitis in mice infected with MHV-JHM in the presence of maternal antibody. Microb. Pathog. 2:185-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pewe, L., J. Haring, and S. Perlman. 2002. CD4 T-cell-mediated demyelination is increased in the absence of gamma interferon in mice infected with mouse hepatitis virus. J. Virol. 76:7329-7333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pewe, L. L., and S. Perlman. 2002. CD8 T-cell-mediated demyelination is IFN-γ dependent in mice infected with a neurotropic coronavirus. J. Immunol. 168:1547-1551. [DOI] [PubMed] [Google Scholar]
- 45.Ponomarev, E. D., M. Novikova, M. Yassai, M. Szczepanik, J. Gorski, and B. N. Dittel. 2004. Gamma delta T-cell regulation of IFN-gamma production by central nervous system-infiltrating encephalitogenic T cells: correlation with recovery from experimental autoimmune encephalomyelitis. J. Immunol. 173:1587-1595. [DOI] [PubMed] [Google Scholar]
- 46.Rajan, A. J., V. C. Asensio, I. L. Campbell, and C. F. Brosnan. 2000. Experimental autoimmune encephalomyelitis on the SJL mouse: effect of γδ T-cell depletion on chemokine and chemokine receptor expression in the central nervous system. J. Immunol. 164:2120-2130. [DOI] [PubMed] [Google Scholar]
- 47.Rajan, A. J., Y. L. Gao, C. S. Raine, and C. F. Brosnan. 1996. A pathogenic role for γδ T cells in relapsing-remitting experimental allergic encephalomyelitis in the SJL mouse. J. Immunol. 157:941-949. [PubMed] [Google Scholar]
- 48.Raulet, D. H. 2003. Roles of the NKG2D immunoreceptor and its ligands. Nat. Rev. Immunol. 3:781-790. [DOI] [PubMed] [Google Scholar]
- 49.Rodriguez, M., K. Pavelko, and R. L. Coffman. 1995. Gamma interferon is critical for resistance to Theiler's virus-induced demyelination. J. Virol. 69:7286-7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Salerno, A., and F. Dieli. 1998. Role of γδ T lymphocytes in immune response in humans and mice. Crit. Rev. Immunol. 18:327-357. [DOI] [PubMed] [Google Scholar]
- 51.Sciammas, R., and J. A. Bluestone. 1998. HSV-1 glycoprotein I-reactive TCR γδ cells directly recognize the peptide backbone in a conformationally dependent manner. J. Immunol. 161:5187-5192. [PubMed] [Google Scholar]
- 52.Sciammas, R., P. Kodukula, Q. Tang, R. L. Hendricks, and J. A. Bluestone. 1997. T-cell receptor-γ/δ cells protect mice from herpes simplex virus type 1-induced lethal encephalitis. J. Exp. Med. 185:1969-1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Selmaj, K., C. F. Brosnan, and C. S. Raine. 1991. Colocalization of lymphocytes bearing gamma delta T-cell receptor and heat shock protein hsp65+ oligodendrocytes in multiple sclerosis. Proc. Natl. Acad. Sci. USA 88:6452-6456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spahn, T. W., S. Issazadah, A. J. Salvin, and H. L. Weiner. 1999. Decreased severity of myelin oligodendrocyte glycoprotein peptide 33-35-induced experimental autoimmune encephalomyelitis in mice with a disrupted TCR δ chain gene. Eur. J. Immunol. 29:4060-4071. [DOI] [PubMed] [Google Scholar]
- 55.Stohlman, S. A., C. C. Bergmann, and S. Perlman. 1998. Mouse hepatitis virus, p. 537-557. In R. Ahmed and I. Chen (ed.), Persistent viral infections. John Wiley & Sons, New York, NY.
- 56.Tran, E. H., E. N. Prince, and T. Owens. 2000. IFN-gamma shapes immune invasion of the central nervous system via regulation of chemokines. J. Immunol. 164:2759-2768. [DOI] [PubMed] [Google Scholar]
- 57.Van Beneden, K., A. De Creus, F. Stevenaert, V. Debacker, J. Plum, and G. Leclercq. 2002. Expression of inhibitory receptors Ly49E and CD94/NKG2 on fetal thymic and adult epidermal TCR V gamma 3 lymphocytes. J. Immunol. 168:3295-3302. [DOI] [PubMed] [Google Scholar]
- 58.Wang, T., E. Scully, Z. Yin, J. H. Kim, S. Wang, J. Yan, M. Mamula, J. F. Anderson, J. Craft, and E. Fikrig. 2003. IFN-gamma-producing gamma delta T cells help control murine West Nile virus infection. J. Immunol. 171:2524-2531. [DOI] [PubMed] [Google Scholar]
- 59.Willenborg, D. O., S. Fordham, C. C. Bernard, W. B. Cowden, and I. A. Ramshaw. 1996. IFN-gamma plays a critical down-regulatory role in the induction and effector phase of myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. J. Immunol. 157:3223-3227. [PubMed] [Google Scholar]
- 60.Wu, G. F., A. A. Dandekar, L. Pewe, and S. Perlman. 2000. CD4 and CD8 T cells have redundant but not identical roles in virus-induced demyelination. J. Immunol. 165:2278-2286. [DOI] [PubMed] [Google Scholar]
- 61.Wu, G. F., and S. Perlman. 1999. Macrophage infiltration, but not apoptosis, is correlated with immune-mediated demyelination following murine infection with a neurotropic coronavirus. J. Virol. 73:8771-8780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xue, S., and S. Perlman. 1997. Antigen specificity of CD4 T-cell response in the central nervous system of mice infected with mouse hepatitis virus. Virology 238:68-78. [DOI] [PubMed] [Google Scholar]
- 63.Youssef, S., G. Wildbaum, G. Maor, N. Lanir, A. Gour-Lavie, N. Grabie, and N. Karin. 1998. Long-lasting protective immunity to experimental autoimmune encephalomyelitis following vaccination with naked DNA encoding C-C chemokines. J. Immunol. 161:3870-3879. [PubMed] [Google Scholar]
- 64.Zompi, S., J. A. Hamerman, K. Ogasawara, E. Schweighoffer, V. L. Tybulewicz, J. P. Di Santo, L. L. Lanier, and F. Colucci. 2003. NKG2D triggers cytotoxicity in mouse NK cells lacking DAP12 or Syk family kinases. Nat. Immunol. 4:565-572. [DOI] [PubMed] [Google Scholar]