

Abstract
It is a basic principle of genetics that each chromosome is transmitted from parent to offspring with a probability that is given by Mendel’s laws. However, several known biological processes lead to skewed transmission probabilities among surviving offspring and, therefore, to excess genetic sharing among relatives. Examples include in utero selection against deleterious mutations, meiotic drive, and maternal-fetal incompatibility. Although these processes affect our basic understanding of inheritance, little is known about their overall impact in humans or other mammals. In this study, we examined genome screen data from 148 nuclear families, collected without reference to phenotype, to look for departures from Mendelian transmission proportions. Using single-point and multipoint linkage analysis, we detected a modest but significant genomewide shift towards excess genetic sharing among siblings (average sharing of 50.43% for the autosomes; P=.009). Our calculations indicate that many loci with skewed transmission are required to produce a genomewide shift of this magnitude. Since transmission distortion loci are subject to strong selection, this raises interesting questions about the evolutionary forces that keep them polymorphic. Finally, our results also have implications for mapping disease genes and for the genetics of fertility.
Introduction
Various known biological processes can cause the probabilities of transmission of chromosomes to surviving offspring to be skewed away from Mendelian predictions. These processes include meiotic drive (biased segregation during meiosis), gametic selection (differential success of gametes in achieving fertilization), and postzygotic viability selection for or against particular genotypes (Pardo-Manuel de Villena et al. 2000; Pardo-Manuel de Villena and Sapienza 2001). All of these mechanisms produce offspring whose genotype proportions deviate from Mendelian predictions. Collectively, we will refer to these mechanisms as “transmission ratio distortion” (or “transmission distortion”) (LeMaire-Adkins and Hunt 2000; Pardo-Manuel de Villena and Sapienza 2001).
Some of the best-known examples of transmission distortion result from competition among male gametes, where sperm with a particular genotype manage to disrupt or otherwise outperform their competitors (as in the mouse t-haplotype system and the segregation distorter system in Drosophila). In females, the principal opportunity for prezygotic distortion occurs during meiosis, when each primary oocyte produces one functional gamete and three polar bodies. This asymmetry provides scope for “cheater” genotypes to subvert the segregation process in order to improve their chances of appearing in the functional gamete (Pardo-Manuel de Villena and Sapienza 2001). Finally, after fertilization, embryonic mortality can also lead to transmission distortion whenever the rate of loss depends on the genotype.
The existence of transmission distortion loci is intriguing from an evolutionary perspective, because selection on distortion alleles should be very strong (Crow 1991; Lyttle 1993; Westendorp et al. 2001). Variation at meiotic drive loci can be stable if the meiotic drive allele incurs some fitness cost at high frequencies (Lyttle 1993). For loci that cause transmission distortion through embryonic viability selection, variation could be maintained (at low frequencies) by mutation-selection balance (Polanski et al. 1998) or perhaps as the result of antagonistic pleiotropy, in which reductions in embryonic survival are balanced by improved survival later in life (Westendorp et al. 2001).
Although transmission distortion has now been documented in many species (Pardo-Manuel de Villena and Sapienza 2001), little is known about transmission distortion in humans. Occasional examples are known, though the mechanism is usually unclear, and in most cases the relevant variants are rare in the population (Evans et al. 1994; Chakraborty et al. 1996; Naumova et al. 1998; Eaves et al. 1999; Girardet et al. 2000; Naumova et al. 2001).
Although there are currently few data to document the overall frequency of events that lead to transmission distortion, it is biologically plausible that transmission distortion in humans might be widespread. It is known that a large fraction of conceptions—perhaps as many as 75%—end with early embryonic loss (Roberts and Lowe 1975; Edmonds et al. 1982; Regan and Rai 2000; Macklon et al. 2002). Some fraction of these losses are presumably due to genetic factors, including genetic incompatibility between the mother and fetus or inviability of the fetus’s genotype. These forms of selection would skew the observed transmission probabilities among those offspring that do survive to term, with fitter genotypes being transmitted more frequently.
We have used methods from linkage mapping to investigate the prevalence of transmission distortion effects at a genomewide scale. One effect of transmission distortion is that it increases the similarity of surviving siblings in affected regions. For example, suppose that both parents in a family are heterozygous for a recessive mutation that is lethal in utero. Then, one of the four possible inheritance combinations is not found among the surviving offspring and, on average, pairs of siblings share 1.11 chromosomes identical by descent at this locus (instead of the 1.0 predicted for Mendelian transmission). Thus, our approach to detecting transmission distortion is to use a set of markers across the genome to test whether the fraction of identical-by-descent sharing among siblings exceeds the Mendelian prediction.
Our prior expectation was that most alleles that cause transmission distortion would either be at low frequency in the population or have modest effects, and so the total fraction of sharing at each such locus would show only small excesses above Mendelian expectations. Thus, the linkage signal due to each transmission distortion locus would be small. However, if there are many such loci across the genome, we should find that, on average, sharing proportions across the genome are inflated above the null expectation.
We analyzed data from a genomewide linkage scan in 148 nuclear families, sampled from the Hutterite population of South Dakota (Ober et al. 1999, 2000; Newman et al. 2003). Families were ascertained without regard to phenotype. This founder population exhibits modest levels of inbreeding and, hence, may be slightly enriched for recessive factors that impact fertility or viability, relative to the general population (Ober et al. 1999). Our analysis and results below focused primarily on the autosomes, except where otherwise stated.
Methods
The Data
Our sample consisted of 148 Hutterite nuclear families (Ober et al. 1999, 2000). The average number of genotyped siblings per family was 4.7. All of these nuclear families were extracted from a single large Hutterite pedigree with 64 founders who lived in the early 1700s. Some individuals are included in more than one family (as offspring in one, as parents in another). This does not bias our results, since transmissions at independent meioses are independent under the null hypothesis.
Individuals had previously been genotyped at Marshfield at a total of 800 microsatellite markers (Marshfield marker sets 11 and 51 [Weber and Broman 2001]) and in C.O.'s lab at an additional 97 microsatellites and 145 biallelic markers. Except where otherwise stated, our results assume the Marshfield genetic map (Broman et al. 1998). Within each family, we deleted any marker locus where either parent had missing data, to avoid possible biases due to misspecified allele frequencies or linkage disequilibrium (LD) between markers. After we applied this criterion, 20% of the families had quite extensive missing data and contributed to Z scores on only a few of the chromosomes. These families were dropped from our analysis of the distribution of family-average Z scores, since the estimates for those families were much more noisy.
Prior to analysis, the data were preprocessed to remove genotype combinations that resulted in Mendelian incompatibilities (O’Connell and Weeks 1998) and to detect any pedigree errors. Since all of these families are part of a single large pedigree, our power to detect Mendelian errors is substantially higher than it would be with independent nuclear families. In addition, we used the multipoint method implemented in Merlin for detecting improbable genotypes. Genotypes that were considered unlikely by that method (according to the default setting) were deleted. Lastly, we removed all markers with extreme amounts of missing data to exclude loci where genotyping might have been problematic. Our final data set consisted of 888 markers.
Three pairs of identical twins were present in the sample and were used for an internal assessment of genotyping error rates: after Mendelian errors were removed, 2 differences were observed among 1,406 pairwise comparisons, indicating an error rate of ∼0.07%. This is consistent with a Marshfield lab estimate of a 0.5% error rate for these data prior to our extensive error checking, obtained by genotyping DNA samples with known genotypes in parallel with our own samples.
Analysis Methods
Multipoint nonparametric linkage analysis was performed using Merlin (Abecasis et al. 2002) and Genehunter (Kruglyak et al. 1996) for analyses with sex-averaged and sex-specific maps, respectively. For the purpose of the analysis, all offspring were treated as “affected.” The average Z score was calculated as the average across all markers, through use of the Spairs scoring function.
Merlin was also used to obtain an average single-point Z score. Sharing proportions for each pair of siblings were calculated from the estimated inheritance vectors; these were averaged over all loci and all 1,086 sib pairs. We estimated separate sharing proportions for maternal and paternal contributions through use of a single-point analysis implemented by ASPEX (Risch et al. 1999).
Significance testing of the average Z score and the average sharing was performed by simulation, as implemented in Merlin (this simulates data that have the observed family structures, allele frequencies, and pattern of missing data, under the null hypothesis of no genetic effects). The reported P values indicate the proportion of simulated data sets for which the average Z score or sharing was greater than or equal to the observed value in 2,500 simulations.
We used two approaches to test whether the level of sharing of maternal and paternal contributions was correlated across loci. Testing this is not entirely straightforward, since sharing levels at neighboring loci are correlated because of linkage. First, we used a time series analysis (as implemented in the cross-correlation function in the statistical package R [R Project for Statistical Computing Web site]); loci with few informative meioses were dropped. However, the assumed model of dependence among neighboring markers is not entirely accurate, for example, because the marker spacing is uneven. Therefore, we also calculated the correlation in average paternal and maternal sharing on each chromosome. The latter approach is more robust but also less powerful, since there are only 22 observations.
Impact of Genotyping Error on Estimated Sharing
It is possible for certain types of genotyping errors to produce an upward bias in estimated sharing under the null hypothesis, although, as we show here, this effect is quite modest for the low genotyping error rate estimated for these data.
Let a, b, c, and d represent four different possible alleles in the parents. Genotyping errors tend to occur either as the result of “allelic dropout,” in which one allele in a heterozygote, (a,b), is dropped, leading to (a,a), for example; or as the result of “allelic shifting,” in which case one allele is misrecorded, either recording a homozygote as a heterozygote, (a,a)→(a,b), or changing one heterozygote to another, (a,b)→(a,c) (Weber and Broman 2001). Genotyping errors such as (a,a)→(b,b) will usually be detected as Mendelian errors.
Allelic dropout among the parents can often be detected as Mendelian errors among the offspring. If dropout is not detected, this implies that inferred sharing is actually 25% lower than it should be (negative bias). Allelic shifting among the parents is potentially more serious. If an (a,a) parent is recorded as (a,b), this increases sibling sharing estimates by 25%, on average. This type of error is not detected as a Mendelian incompatibility among the offspring, but, since our families are extracted from a single large pedigree, these errors may be detected as incompatibilities with parents or siblings. Allelic shifting from (a,b) parents to (a,c) is much less problematic, because it can potentially be detected as a Mendelian error by comparison with the offspring. When it is not detected, this implies that there really is increased sharing and therefore does not produce a bias. The exception is among (a,b)×(a,b) crosses. In that case, the error is detected among 1/4 of the offspring and, hence, is likely to be detected in large sibships. Conditional on not detecting the error, the estimated sharing at that marker is increased by 22.2%, on average.
Genotyping errors can also occur among the offspring. Many of these are detected as Mendelian errors. The bias caused by those that are not detected is negligible, being 0 for some parental configurations and of order ε2 for the rest (where ε is the genotyping error rate).
In summary, the major source of upward bias is the mistyping of homozygous parents as heterozygotes. Let ε1 represent the rate of allelic shift genotyping errors that are not detected by Mendelian checking, and let f be the proportion of homozygous parents. Then, the bias due to genotyping errors is roughly 2ε1f/4. In our data, the average homozygosity of parents is 33.2% (per locus), and the total genotyping error rate after Mendelian checking is in the range of 0.1%–0.5% (see above). The rate of allelic shift is probably considerably lower, since experience suggests that allelic dropout represents the most common type of genotyping error. Hence, the expected bias is (at most) in the range of 0.016%–0.08%. Heterozygous parent pairs, (a,b)×(a,b), constitute 9% of our data and contribute, at most, an extra .004%–0.02% to the bias (in practice, this will be substantially less, since we have good power to detect Mendelian errors in this case). These positive biases are balanced against the downward bias due to allelic dropout among the parents. Overall, the contribution of genotyping error is roughly an order of magnitude smaller than the effect that we find in the data (see the “Results” section).
Results
Our first analysis applied nonparametric multipoint linkage methods (Kruglyak et al. 1996; Abecasis et al. 2002) to examine average genomewide sharing among siblings (see the “Methods” section). A standard measure of the departure from Mendelian expectations at a given chromosomal position is the Z score. Under the null hypothesis, this statistic is drawn from a normal distribution with mean 0 and variance 1; positive values indicate excess sharing.
As shown in figures 1 and 2, we observed a substantial upward shift in the distribution of Z scores across the genome, with a mean of 0.332 (P=.017; see the “Methods” section). The average Z score is positive on 16 of the chromosomes—substantially so on some of them (fig. 3). We also estimated the average genomewide sharing among sibling pairs to be 50.43%, which is significantly higher than the expected sharing of 50% (P=.009).
Figure 1.

Genomewide plot of nonparametric Z scores across the 22 autosomes and the X chromosome. Under the null hypothesis, Z scores are symmetrically distributed around 0 (they are normal with mean 0 and variance 1). Instead, there is a general, genomewide shift towards positive Z scores, indicating an excess of genetic sharing among siblings. For example, much more of the genome has Z scores >1 (upper black lines) than <−1 (lower black lines); similarly, there are numerous peaks >2, but none <−2. The marker positions are indicated by the vertical black hash marks at the bottom of the plot, and the chromosomes by the numbers at the top. The highest peak is at 15q21.3.
Figure 2.

Quantile-quantile plot of nonparametric Z scores at each marker. The observed Z scores have been ordered from lowest to highest and plotted on the Y-axis. The X-axis is scaled in such a way that if the data were drawn from the null distribution they would sit on the diagonal line. Instead, the points are significantly above the line, indicating a strong upward skew of Z scores, corresponding to excess sharing. The slope of the diagonal has been modified to account for the autocorrelation of linked markers.
Figure 3.

Average Z scores, by chromosome, indicate a general shift towards excess sharing across many of the chromosomes, with peaks on chromosomes 5, 6, and 15.
The signal of increased sharing is spread broadly across the genome, and no single chromosomal location reaches genomewide significance, indicating that the observed effect is due to many different loci. There is no indication that the effect is restricted to a subset of the families (fig. 4), since the distribution of average Z scores within families shows a general shift towards excess sharing with no clear outliers. Thus, in summary, we find strong evidence for genomewide transmission distortion, spread across most or all of the chromosomes and present in most or all families.
Figure 4.

Quantile-quantile plot of Z scores of each family, averaged over all markers. The observed Z scores have been ordered from lowest to highest and plotted on the Y-axis. The X-axis is scaled in such a way that if the data were drawn from the null distribution they would sit on the diagonal line. Instead, the points are significantly above the line, indicating a strong upward skew of Z scores, corresponding to excess sharing. The slope of the diagonal has been modified to account for the reduced variance in genomewide averages.
Controlling for Bias
In linkage mapping, genotyping errors and other problems in data quality tend to lead to reduced power and not to upward biases in inferred sharing. However, two factors that might possibly lead to upward bias are (1) certain types of genetic map errors in multipoint analysis (Daw et al. 2000) and (2) certain types of genotyping errors. In studies in which parental genotypes must be inferred, misspecified allele frequencies or intermarker LD can also lead to upward bias. Therefore, we analyzed only markers with full genotype information for both parents (see the “Methods” section).
To control for the possible effect of map error, we repeated the analysis through use of the single-point approach implemented in ASPEX (which does not use a genetic map). The average single-point sharing estimate (50.32%) was essentially the same as that obtained from the multipoint analysis. In addition, we were able to integrate most of our markers (some by interpolation) into the more accurate, sex-specific genetic map from deCode (Kong et al. 2002). This led to a slightly higher average Z score (increased from 0.332 to 0.373), as might be expected for a true positive signal.
Certain types of undetected genotyping errors could also, in principle, bias the results towards higher inferred sharing, though other genotyping errors create downward biases. However, our calculations (see the “Methods” section) indicate that this effect is about an order of magnitude smaller than the excess sharing that we observed.
As an additional control for genotyping error, we performed the following quite conservative test of sharing in sibships with four or more sibs. As described in the “Methods” section, the major bias from genotyping error arises when a homozygous parent is called as a heterozygote. We reasoned that if a parent is genotyped as heterozygous at a locus and if both alleles are transmitted to at least one offspring each, then we have high confidence that the parent really is a heterozygote (apart from the unlikely possibility of two identical genotyping errors at the same locus in the same family). Therefore, for each family we restricted the analysis to loci where the transmissions could be scored unambiguously and where both alleles appeared among the offspring. This test is clearly extremely wasteful of power, since much of the data set is discarded, and loci where there is excess sharing are discarded preferentially. Nonetheless, we found that there was still an excess of single-point sharing compared with what would be expected under these conditions (P=.03 [P value obtained by simulation]).
Investigating the Mechanism
We have conducted further analyses to gain some insight into the likely biological mechanisms. Possible mechanisms can be divided into processes that depend on the sex of the parent (including meiotic drive and gametic selection, maternal-fetal incompatibility, and transmission distortion connected with genetic imprinting [Pardo-Manuel de Villena et al. 2000; Naumova et al. 2001]) and processes that do not depend on the sex of the parent (including viability selection against particular fetal genotypes). If most of the effect is due to viability selection, then (1) we should see that average genomewide sharing of maternal and paternal alleles is similar, and (2) maternal and paternal sharing patterns should be correlated, because transmission distortion loci should produce increased sharing for both parental contributions. Conversely, if most of the bias is due to processes that depend on the sex of the parent, then regions of high maternal sharing will occur independently of regions of high paternal sharing, and the total maternal and paternal sharing proportions may be different.
Overall, our results provide some support for the viability selection hypothesis as an important mechanism of transmission distortion. Using a single-point analysis method implemented in ASPEX (Risch et al. 1999), we found that sharing of maternal and paternal alleles was similar: 50.23% and 50.42%, respectively (P>.1). Furthermore, we evaluated the correlation between maternal and paternal sharing across loci; this was positive but not significant.
However, when we looked at the correlation of maternal and paternal sharing across markers within each chromosome, we found that these correlation scores are themselves significantly correlated with chromosome-average sharing from ASPEX (P=10-4) (fig. 5). Thus, chromosomes with increased overall sharing (and therefore higher estimated transmission distortion) show greater correspondence between regions of increased maternal and paternal sharing. In chromosomes with little evidence for transmission distortion, maternal and paternal sharing patterns are essentially independent, as expected under the null hypothesis.
Figure 5.

Correlation of the average single-point sharing from ASPEX with the correlation coefficient of maternal and paternal sharing of markers on the same chromosome. Regions of high average sharing also show a high correlation between maternal and paternal patterns of sharing (see text), indicating that a substantial part of the effect is not due to parent-specific processes such as meiotic drive.
As an additional test of the maternal-fetal incompatibility mechanism, we hypothesized that, when incompatibility is due to a maternal immune response (as with Rh factor), the probability of incompatibility would often increase with birth number in a sibship. Under this hypothesis, later-born siblings would have greater average sharing with each other than they would with first-born siblings. Analysis of the data indicates no significant difference in sharing among sib pairs as a function of birth order.
We also examined X chromosome sharing, for which transmission distortion due to viability selection would likely be reduced because of the more immediate selection against recessive mutations but where there is still scope for other types of transmission distortion. In fact, we see no evidence for excess sharing on the X chromosome. The average Z score on the X chromosome was slightly lower than on any other chromosome (fig. 3).
There is a previous report of an X-linked locus that shows transmission distortion in male offspring only (Naumova et al. 1998). Loci such as this, where the transmission distortion occurs in only one sex, would produce increased sharing between brothers or between sisters, but not between brothers and sisters.
It is curious that, in our data, the average genomewide sharing between sisters is substantially higher than between brothers (50.78% and 50.12%, respectively), whereas the sharing between brothers and sisters is intermediate (50.36%). There is considerable uncertainty in these estimates (the two-sided P value comparing 50.78% and 50.12% is 0.12), but results from the CEPH show the same qualitative result and order of sharing proportions (see below). If these sex-specific results are general, then they suggest a model in which many of the same loci are involved in the viability of both sexes but in which the effects are stronger, on average, in females.
Number of Transmission Distortion Loci
To gain some insight into how many loci would be required to produce a skew of the observed magnitude, we performed the following calculation. Suppose that all of the transmission distortion is due to loci at which both parents are heterozygous for recessive lethal mutations (many of these mutations might be active in the earliest stages of embryonic development, before pregnancy is detected [Roberts and Lowe 1975]). We have calculated (see the Appendix) that each recessive lethal locus where both parents are heterozygous increases the genomewide sharing in that family by 0.068%, on average. Thus, to account for the observed sharing of 50.43% by this mechanism alone, we would have to posit that, in each family, there is an average of 6.3 recessive lethal mutations that are heterozygous in both parents. In practice, it is likely that the total distortion effect is due to numerous mechanisms, of which recessive lethals are just one (and, indeed, the estimate of 6.3 such loci seems implausibly high for recessive mutations to account for the entire effect). However, since recessive lethals produce a relatively strong distortion effect compared with other mechanisms, our estimate of approximately six transmission distortion loci per family is probably an underestimate. Furthermore, since most distortion alleles may be at low frequency, the total number of distortion loci in the genome is likely to be large.
Transmission Distortion in Other Populations
Since the Hutterites are a founder population, it is natural to wonder whether similar effects occur in other populations. First, levels of inbreeding in the Hutterites are somewhat elevated (individual inbreeding coefficients average ∼0.03 [Ober et al. 1999]). Second, the founding bottleneck could have allowed some deleterious variants to increase in frequency (Risch et al. 1995).
Two additional data sets support the hypothesis that our results are general. First, a previous study of autism reported a genome average of 50.8% sharing among discordant sib pairs in a nationwide U.S. sample, albeit in a much smaller data set (Risch et al. 1999). Those authors dismissed their observation as possibly resulting from genotyping error, but it now seems likely that they, too, observed a transmission distortion effect. Second, we have repeated our analysis using nuclear families and publicly available genotype data extracted from the CEPH resource (Fondation Jean Dausset–CEPH Web site). There were 65 nuclear families available with multiple offspring and both parents. As before, the average Z score was positive (0.221). Average sharing was estimated at 50.2%. Sharing was 50.4% between sisters, 50.2% between brothers and sisters, and 50.0% between brothers. In contrast, analysis of the Icelandic data used by deCode to build their recent genetic map (Kong et al. 2002) provided no evidence of excess sharing (mean sharing 49.9%).
Discussion
In this study, we have identified a modest but highly significant genomewide signal of transmission distortion in humans, by looking at excess sharing among siblings in our sample of Hutterite families. Similar effects are also reported for two other samples from outbred populations. The effect appears to be spread across most chromosomes and is due to the combined participation of many loci across the genome. Although some types of genotyping error can produce an upward bias in estimated sharing, the estimated error rates for these data are low, and, in addition, we have performed extensive error checking to remove dubious genotypes. In summary, the effect of genotyping error seems to be small for these data, and most of the signal is likely due to biological causes.
A full elucidation of the underlying mechanism will require further study, but our findings are consistent with the hypothesis that viability selection plays an important role. First, overall sharing of maternal and paternal contributions is similar. Second, we find that, on the chromosomes with highest average sharing (i.e., where transmission distortion appears to be highest), the regions of maternal and paternal sharing are strongly correlated. In this context, the human leukocyte antigen (HLA) cluster on chromosome 6 is of obvious interest, since it has previously been shown that there is an increased rate of fetal loss among Hutterite couples that share HLA alleles (Ober 1998). Indeed, our data show higher than expected sharing in this region (Z score=1.13). Apart from the possibility of recessive lethals, this selection could also include loci of weaker effects or selection against certain multilocus combinations of alleles. However, these results supporting the role of viability selection do not exclude the possibility that other mechanisms, notably including meiotic drive, also make some smaller contribution to the overall transmission distortion.
Alleles that modify postzygotic viability would be subject to strong natural selection (Crow 1991; Westendorp et al. 2001). In light of this, it may seem counterintuitive to find evidence of many such loci across the genome. There are at least two plausible mechanisms that could maintain such variation. First, mutation-selection balance can allow for low equilibrium frequencies of deleterious variants at many loci (Polanski et al. 1998). Second, it is possible that some genetic variants could be subject to pleiotropic effects, where occasional embryonic loss is balanced by fitness gains at later stages in development (Westendorp et al. 2001).
Our observation that transmission distortion may be a general feature of the human genome has implications for gene mapping methods, since some of the most commonly used methods are not robust to the presence of transmission distortion. For example, we have shown that, with standard linkage mapping techniques, the distribution of the test statistic lies substantially above the null distribution. No single region reached genomewide significance in our study, but it is clear that the effect observed here could lead to increased rates of false positives (Greenwood and Morgan 2000; Edwards 2003). For the level of distortion that we have observed, this is likely to be quite a modest effect in most studies, but it may become substantial in very large studies. In such studies it may be advisable to first assess the average genomewide sharing and then use that value as the baseline for detecting excess transmission.
The transmission disequilibrium test (TDT), used to detect association, is also sensitive to transmission distortion (Spielman et al. 1993). If the marker is in linkage disequilibrium with variation at the distortion locus, then the TDT is prone to reproducible false positives. It has been argued that, for this reason, it is important to test unaffected siblings for transmission distortion at loci where significant TDT results have been obtained, and we concur (Spielman et al. 1993; Eaves et al. 1999).
Our results imply that it will be extremely interesting to identify the loci underlying the observed transmission distortion. These loci are potentially involved in human infertility and therefore the study of these loci may lead to useful insight into the science of human reproduction.
Acknowledgments
We thank N. Cox, D. Falush, I. Hellman, C. Langley, G. Montana, N. Rosenberg, D. Schneider, J. Weber, and two anonymous reviewers, for comments on the manuscript and/or helpful discussions; and J. Weber and the National Heart, Lung, and Blood Institute Mammalian Genotyping Service for genotyping. S.Z. was supported by a Katz Fellowship to the Department of Human Genetics, University of Chicago. This work was supported, in part, by National Institutes of Health grants HL56399 and HL66533 (to C.O.) and GR 2772 (to J.K.P.).
Appendix: Average Increase in Sharing Due to Each Recessive Lethal
Suppose that both parents are heterozygous for a recessive lethal mutation. Then, the expected increase in sharing can be calculated as follows. Denote the genotypes of the mother and father by Wm,Lm and Wp,Lp respectively, where W is the wild-type allele and L is the recessive lethal allele. There are three possible labeled genotypes for the surviving offspring, each occurring with a probability of 1/3. Therefore, the probability that two children share one allele identical by descent is 4(1/3)2=4/9, and the probability of sharing two alleles is 3(1/3)2=3/9. Thus, the expected sharing among surviving offspring is 10/9 at that locus.
Let y denote the map position of the recessive lethal and s(x) denote the observed sharing at position x on the same chromosome. Let R be the probability of an odd number of recombination events between x and y; it can be shown under the Haldane mapping function that this is 1/2(1-e-2|x-y|), where |x-y| denotes the distance between x and y in morgans (for simplicity, we assume equal recombination rates in males and females ). We use the notation Wmx to indicate that the maternal contribution at position x is from the same original chromosome as the W allele. Then, the probabilities for the four possible genotypes at x are as follows: Pr(Lmx,Lpx)=(2R-R2)/3, Pr(Lmx,Wpx)=Pr(Wmx,Lpx)=(1-R+R2)/3, and Pr(Wmx,Wpx)=(1-R2)/3. The probability that two children share one chromosome identical by descent at x is 2×[Pr(LmxWpx)Pr(WmxWpx)+Pr(WmxWpx)Pr(WmxLpx)+Pr(WmxLpx)Pr(LmxLpx)+Pr(LmxLpx)Pr(LmxWpx)]. The probability that they share two chromosomes identical by descent is Pr(WmxWpx)2+Pr(LmxLpx)2+Pr(LmxWpx)2+Pr(WmxLpx)2. The expected sharing at x is the probability of sharing one chromosome plus twice the probability of sharing two:
 |
Suppose that chromosome i is of length li (in morgans) and that both parents carry a recessive lethal at a random location. Then, the expected excess of sharing on this chromosome, 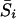 , is
, is
 |
if we assume that the mutation is equally likely to occur anywhere on the chromosome. Let L represent the total length of the genome (in morgans). Then, a mutation on chromosome i increases average genomewide sharing by 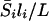 . Then, since the probability that a recessive mutation is on chromosome i is li/L, the expected genomewide excess in sharing is
. Then, since the probability that a recessive mutation is on chromosome i is li/L, the expected genomewide excess in sharing is 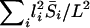 . On the basis of the average chromosome lengths reported by deCode (Kong et al. 2002), each recessive mutation increases the genomewide sharing by 0.068%. Under the approximation that loci increase sharing independently of each other, it would take 6.3 such loci to account for the observed excess sharing of 0.43%.
. On the basis of the average chromosome lengths reported by deCode (Kong et al. 2002), each recessive mutation increases the genomewide sharing by 0.068%. Under the approximation that loci increase sharing independently of each other, it would take 6.3 such loci to account for the observed excess sharing of 0.43%.
Electronic-Database Information
The URLs for data presented herein are as follows:
- Fondation Jean Dausset–CEPH, http://www.cephb.fr/ (for genotype data)
- R Project for Statistical Computing, http://www.r-project.org/ (for the statistical package R)
References
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR (2002) Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 30:97–101 10.1038/ng786 [DOI] [PubMed] [Google Scholar]
- Broman KW, Murray JC, Sheffield VC, White RL, Weber JL (1998) Comprehensive human genetic maps: individual and sex-specific variation in recombination. Am J Hum Genet 63:861–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty R, Stivers DN, Deka R, Yu LM, Shriver MD, Ferrell RE (1996) Segregation distortion of the CTG repeats at the myotonic dystrophy locus. Am J Hum Genet 59:109–118 [PMC free article] [PubMed] [Google Scholar]
- Crow JF (1991) Why is Mendelian segregation so exact? Bioessays 13:305–312 [DOI] [PubMed] [Google Scholar]
- Daw EW, Thompson EA, Wijsman EM (2000) Bias in multipoint linkage analysis arising from map misspecification. Genet Epidemiol 19:366–380 [DOI] [PubMed] [Google Scholar]
- Eaves IA, Bennett ST, Forster P, Ferber KM, Ehrmann D, Wilson AJ, Bhattacharyya S, Ziegler AG, Brinkmann B, Todd JA (1999) Transmission ratio distortion at the INS-IGF2 VNTR. Nat Genet 22:324–325 10.1038/11890 [DOI] [PubMed] [Google Scholar]
- Edmonds DK, Lindsay KS, Miller JF, Williamson E, Wood PJ (1982) Early embryonic mortality in women. Fertil Steril 38:447–453 [PubMed] [Google Scholar]
- Edwards JH (2003) Sib-pairs in multifactorial disorders: the sib-similarity problem. Clin Genet 63:1–9 10.1034/j.1399-0004.2003.630101.x [DOI] [PubMed] [Google Scholar]
- Evans K, Fryer A, Inglehearn C, Duvall-Young J, Whittaker JL, Gregory CY, Butler R, Ebenezer N, Hunt DM, Bhattacharya S (1994) Genetic linkage of cone-rod retinal dystrophy to chromosome 19q and evidence for segregation distortion. Nat Genet 6:210–213 [DOI] [PubMed] [Google Scholar]
- Girardet A, McPeek MS, Leeflang EP, Munier F, Arnheim N, Claustres M, Pellestor F (2000) Meiotic segregation analysis of RB1 alleles in retinoblastoma pedigrees by use of single-sperm typing. Am J Hum Genet 66:167–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood CM, Morgan K (2000) The impact of transmission-ratio distortion on allele sharing in affected sibling pairs. Am J Hum Genet 66:2001–2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong A, Gudbjartsson DF, Sainz J, Jonsdottir GM, Gudjonsson SA, Richardsson B, Sigurdardottir S, Barnard J, Hallbeck B, Masson G, Shlien A, Palsson ST, Frigge ML, Thorgeirsson TE, Gulcher JR, Stefansson K (2002) A high-resolution recombination map of the human genome. Nat Genet 31:241–247 [DOI] [PubMed] [Google Scholar]
- Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES (1996) Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet 58:1347–1363 [PMC free article] [PubMed] [Google Scholar]
- LeMaire-Adkins R, Hunt PA (2000) Nonrandom segregation of the mouse univalent X chromosome: evidence of spindle-mediated meiotic drive. Genetics 156:775–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyttle TW (1993) Cheaters sometimes prosper: distortion of Mendelian segregation by meiotic drive. Trends Genet 9:205–210 10.1016/0168-9525(93)90120-7 [DOI] [PubMed] [Google Scholar]
- Macklon NS, Geraedts JPM, Fauser BCJM (2002) Conception to ongoing pregnancy: the “black box” of early pregnancy loss. Hum Reprod Update 8:333–343 [DOI] [PubMed] [Google Scholar]
- Naumova AK, Greenwood CM, Morgan K (2001) Imprinting and deviation from Mendelian transmission ratios. Genome 44:311–320 10.1139/gen-44-3-311 [DOI] [PubMed] [Google Scholar]
- Naumova AK, Leppert M, Barker DF, Morgan K, Sapienza C (1998) Parental origin–dependent, male offspring–specific transmission-ratio distortion at loci on the human X chromosome. Am J Hum Genet 62:1493–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman DL, Abney M, Dytch H, Parry R, McPeek MS, Ober C (2003) Major loci influencing serum triglyceride levels on 2q14 and 9p21 localized by homozygosity-by-descent mapping in a large Hutterite pedigree. Hum Mol Genet 12:137–144 10.1093/hmg/ddg012 [DOI] [PubMed] [Google Scholar]
- Ober C (1998) HLA and pregnancy: the paradox of the fetal allograft. Am J Hum Genet 62:1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ober C, Hyslop T, Hauck WW (1999) Inbreeding effects on fertility in humans: evidence for reproductive compensation. Am J Hum Genet 64:225–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ober C, Tsalenko A, Parry R, Cox NJ (2000) A second-generation genomewide screen for asthma-susceptibility alleles in a founder population. Am J Hum Genet 67:1154–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell JR, Weeks DE (1998) PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet 63:259–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo-Manuel de Villena F, de la Casa-Esperón E, Sapienza C (2000) Natural selection and the function of genome imprinting: beyond the silenced minority. Trends Genet 16:573–579 10.1016/S0168-9525(00)02134-X [DOI] [PubMed] [Google Scholar]
- Pardo-Manuel de Villena F, Sapienza C (2001) Nonrandom segregation during meiosis: the unfairness of females. Mamm Genome 12:331–339 10.1007/s003350040003 [DOI] [PubMed] [Google Scholar]
- Polanski A, Chakraborty R, Kimmel M, Deka R (1998) Dynamic balance of segregation distortion and selection maintains normal allele sizes at the myotonic dystrophy locus. Math Biosci 147:93–112 10.1016/S0025-5564(97)00082-5 [DOI] [PubMed] [Google Scholar]
- Regan L, Rai R (2000) Epidemiology and the medical causes of miscarriage. Baillieres Best Pract Res Clin Obstet Gynaecol 14:839–854 10.1053/beog.2000.0123 [DOI] [PubMed] [Google Scholar]
- Risch N, deLeon D, Ozelius L, Kramer P, Almasy L, Singer B, Fahn S, Breakefield X, Bressman S (1995) Genetic analysis of idiopathic torsion dystonia in Ashkenazi Jews and their recent descent from a small founder population. Nat Genet 9:152–159 [DOI] [PubMed] [Google Scholar]
- Risch N, Spiker D, Lotspeich L, Nouri N, Hinds D, Hallmayer J, Kalaydjieva L, et al (1999) A genomic screen of autism: evidence for a multilocus etiology. Am J Hum Genet 65:493–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts CJ, Lowe CR (1975) Where have all the conceptions gone? Lancet 7907:498–499 10.1016/S0140-6736(75)92837-8 [DOI] [PubMed] [Google Scholar]
- Spielman RS, McGinnis RE, Ewens WJ (1993) Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am J Hum Genet 52:506–513 [PMC free article] [PubMed] [Google Scholar]
- Weber JL, Broman KW (2001) Genotyping for human whole-genome scans: past, present, and future. In: Rao DC, Province MA (eds) Advances in genetics, volume 42. Academic Press, San Diego, pp 77–96 [DOI] [PubMed] [Google Scholar]
- Westendorp RG, vanDunne FM, Kirkwood TB, Helmerhorst FM, Huizinga TW (2001) Optimizing human fertility and survival. Nat Med 7:873 10.1038/90868 [DOI] [PubMed] [Google Scholar]