

Abstract
The calpain-10 gene (CAPN10) on chromosome 2q37.3 was the first candidate gene for type 2 diabetes (T2D) identified through a genomewide screen and positional cloning. One polymorphism (UCSNP-43: G→A) and a specific haplotype combination defined by three polymorphisms (UCSNP-43, -19, and -63) were linked to an increased risk of T2D in several populations. To quantitatively assess the collective evidence for the effects of CAPN10 on risk of T2D, we conducted a meta-analysis of both population-based and family-based association studies. We retrieved data from the MEDLINE, PubMed, and Online Mendelian Inheritance in Man databases, as well as from other relevant reports and abstracts published up to July 2003. From a total of 26 studies with primary data (21 population-based studies: 5,013 cases and 5,876 controls; 5 family-based studies: 487 parent-offspring trios), we developed a summary database that contains variables of study design, study population/ethnicity, specific polymorphisms and haplotype combinations in CAPN10, and diabetes-related metabolic phenotypes. For population-based studies, we used both fixed-effects and random-effects models to calculate the pooled odds ratio (OR) and 95% confidence interval (CI) for the associations of CAPN10 genotypes with the risk of T2D. We also calculated weighted mean differences for the associations between CAPN10 and diabetes-related quantitative traits. Under either an additive or a dominant effect model, we found no statistically significant relation between CAPN10 genotypes in the UCSNP-43 locus and T2D risk. However, under a recessive model, individuals homozygous for the common G allele had a statistically significant 19% higher risk of T2D than carriers of the A allele (OR 1.19; 95% CI 1.07–1.33). The association between the 112/121 haplotype combination and T2D risk appeared to be overestimated by several initial small studies with positive findings (OR 1.38; 95% CI 1.04–1.84). After we removed these initial studies, this association became nonsignificant (OR 1.11; 95% CI 0.91–1.35). Moreover, we found no evidence for the associations between the UCSNP-43 G/G genotype and the 112/121 haplotype combination and metabolic phenotypes. Our meta-analysis of family-based studies showed only an overtransmission of the rare allele C in UCSNP-44 from heterozygous parents to their affected offspring with T2D. Our analysis indicates that inadequate statistical power, racial/ethnic differences in frequencies of alleles, haplotypes and haplotype combinations, potential gene-gene or gene-environment interactions, publication bias, and multiple hypothesis testing may contribute to the significant heterogeneity in previous studies of CAPN10 and T2D. Our findings also suggest that both large-scale, well-designed association studies and functional studies are warranted to either reliably confirm or conclusively refute the initial hypothesis regarding the role of CAPN10 in T2D risk.
Introduction
As a complex metabolic disease, type 2 diabetes mellitus (T2D [MIM 125853]) is characterized by hyperglycemia and dyslipidemia and often leads to serious micro- and macrovascular complications, including cardiovascular disease. Although insulin resistance and progressive pancreatic β-cell dysfunction have been established as the two fundamental features in the pathogenesis of T2D (Kahn 1994), the specific molecular defects affecting insulin sensitivity and/or β-cell function remain largely undefined. The high prevalence of this disease in certain racial/ethnic groups (Lillioja et al. 1993) and the high concordance in MZ twin pairs (63%–90%) (Newman et al. 1987) suggest that susceptibility to T2D is highly heritable. Linkage analyses have identified several specific gene loci in a handful of families with diabetes that segregates as an autosomal dominant trait, termed “maturity-onset diabetes of the young” (MODY-1, -2, -3, -4, -5, and -6) (Florez et al. 2003). However, much remains unknown about the genetic determinants of the common (>95%) late-onset form of T2D.
For the common form of T2D in the general population, the calpain-10 (CAPN10 [MIM 605286]) gene is the first T2D candidate gene identified through genomewide linkage and positional cloning (Horikawa et al. 2000). CAPN10, located on chromosome 2q37.3, comprises 15 exons spanning 31 kb of genomic sequence that encodes a 672-amino-acid intracellular protease. Calpain-10 is primarily expressed in liver, skeletal muscle, and pancreatic islets and is one of the nonlysosomal cysteine proteases essential for calcium-regulated intracellular signaling (Goll et al. 2003).
Horikawa et al. (2000) first reported that homozygosity of the G allele in UCSNP-43 and a “high risk” haplotype combination (multilocus genotype) defined by the UCSNP-43 (MIM 605286.0001), -19 (two→three repeats of a 32-bp sequence in intron 6) (MIM 605286.0002), and -63 (C→T in intron 13) (MIM 605286.0003) polymorphisms were associated with a two- to threefold increased risk of T2D in a sample of Mexican Americans and in two samples of European populations. However, results from subsequent population-based association studies have been inconsistent; several studies found only a modest effect of CAPN10 (Schwarz et al. 2001; Cassell et al. 2002; Garant et al. 2002; Orho-Melander et al. 2002), whereas others found no association between CAPN10 variants and T2D risk (Baier et al. 2000; Evans et al. 2001; Hegele et al. 2001; Tsai et al. 2001; Xiang et al. 2001a; Daimon et al. 2002; Fingerlin et al. 2002; Malecki et al. 2002; Rasmussen et al. 2002; Sun et al. 2002; Horikawa et al. 2003; Iwasaki et al. 2003). Many studies showed a trend toward significance but lacked sufficient power individually to detect a modest genetic effect (Evans et al. 2001; Tsai et al. 2001; Xiang et al. 2001a; Fingerlin et al. 2002; Malecki et al. 2002; Rasmussen et al. 2002; Sun et al. 2002). Likewise, data from family-based studies were sparse and provided little or no conclusive evidence to either confirm or refute the initial findings. To provide a comprehensive and quantitative assessment of the impact of this gene on risk of T2D, we conducted a meta-analysis of both population and family-based studies with available primary data. To further identify issues that may aid in the design and analysis of future genetic association studies, we also explored potential sources of heterogeneity in these studies.
Methods
Study Selection
Relevant studies were identified by searching the MEDLINE, PubMed, and Online Mendelian Inheritance in Man (OMIM) databases for all published genetic association studies up to July 2003, using the search terms “calpain-10,” “calpain-10 gene,” “CAPN,” “CAPN gene,” “CAPN-10,” “CAPN-10 genotype,” “insulin resistance,” “glucose metabolism,” “type 2 diabetes,” and “diabetes.” Additional studies were retrieved through a hand search of references from original reports. We also reviewed abstracts presented at the American Diabetes Association from 2000 to 2003. All studies on this topic were considered eligible if they had data on the relationship between CAPN10 genotypes and diabetes-related quantitative traits and T2D risk. Only one non–English language article was identified and included (Xiang et al. 2001a).
Two of us (Y.S. and S.L.) independently reviewed each published paper and extracted relevant information examining the associations of CAPN10 gene polymorphisms or haplotype combinations with risk of T2D or metabolic phenotypes. Interobserver differences, if any existed, were reconciled through group discussion. In general, we included studies that provided estimates of relative risks (RRs) or odds ratios (ORs) of T2D or data that permitted estimation of these parameters. When a study reported results on different subpopulations, we considered each subpopulation as a separate study in the meta-analysis.
Of the 28 published articles that were identified and abstracted, 6 studies were excluded. These included two redundant reports of the same population (Xiang et al. 2001b; Stumvoll et al. 2002) and four reports that did not provide relevant data on the risk estimates (Hoffstedt et al. 2002a, 2002b; Engelman et al. 2003; Schwarz et al. 2003). The final data set of our meta-analyses included 21 independent case-control studies of unrelated subjects (see table A1 in the appendix), and five independent family-based studies (Evans et al. 2001; Cassell et al. 2002; Elbein et al. 2002; Orho-Melander et al. 2002; Sun et al. 2002). Data on CAPN10 genotypes and diabetes-related metabolic traits were also available in nine studies (Baier et al. 2000; Stumvoll et al. 2001; Xiang et al. 2001a; Daimon et al. 2002; Garant et al. 2002; Lynn et al. 2002; Orho-Melander et al. 2002; Rasmussen et al. 2002; Shore et al. 2002).
We developed a database that included the first author’s name, year of publication, study population, sample size, mean age for cases and controls, mean age at diagnosis, mean BMI, fasting plasma glucose levels, comparisons of allele frequencies between cases and controls, genotype distribution in cases and controls, and the estimates of RRs or ORs of T2D associated with the CAPN10 genotypes. Two studies that provided adjusted ORs made adjustment for age and sex (Baier et al. 2000; Garant et al. 2002). For family-based association studies, we collected all counts for transmission status of the allele examined (transmitted or not transmitted from a heterozygous parent to the affected offspring with T2D) through use of transmission/disequilibrium testing (TDT). We also extracted the number of subjects and the means and SDs of quantitative metabolic phenotypes, including the following: BMI; waist-to-hip ratio (WHR); plasma levels of high-density lipoprotein (HDL), low-density lipoprotein, and triglyceride; fasting plasma glucose and insulin levels; plasma levels of glucose, insulin, and C-peptide at 2 h after a 75-g oral glucose tolerance test; homeostasis model assessment of insulin resistance (HOMA-IR) index; and early insulin response (EIR).
Statistical Analyses
Population-based association analysis
We calculated the ORs and 95% CIs for the associations between CAPN10 genotypes and risk of T2D, through use of both the fixed-effects and the random-effects models. To explore the inheritance model for the effect of UCSNP-43 polymorphism, we evaluated the following genotype contrasts: G/G versus G/A and A/A combined (a recessive effect); G/G and G/A combined versus A/A (a dominant effect); G/G versus G/A; G/A versus A/A; and G/G versus A/A (additive or dose-response effect).
In the fixed-effects model, the summary OR was obtained by averaging the natural logarithms of the ORs from individual studies, weighted by the inverses of their variances. When the adjusted ORs were not available from the article, we used the crude ORs reported or calculated from the raw data. To incorporate both within-study and between-study variability, we used DerSimonian and Laird’s (1986) random-effects model. To further test the robustness of results obtained from both fixed-effects and random-effects models, we also applied the empirical Bayes method in a sensitivity analysis. Empirical Bayes estimates are calculated by shrinking the study-specific estimates towards the overall random-effects estimate by a factor that depends on the relative magnitude of the estimated within- and between-study variances (Normand 1999). Formal tests of heterogeneity were assessed by a χ2 statistic.
We also computed the population-attributable risk (PAR) of UCSNP-43, which indicates the fraction of T2D cases in the population that can be attributed to the independent effect of the G/G genotype. PAR was calculated, on the basis of case-control studies, as 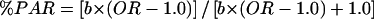 (Greenland 1998), where b is the prevalence of the G/G genotype in the source population, and the OR is the estimated pooled risk of T2D in individuals with the G/G genotype relative to that of those carrying the A allele.
(Greenland 1998), where b is the prevalence of the G/G genotype in the source population, and the OR is the estimated pooled risk of T2D in individuals with the G/G genotype relative to that of those carrying the A allele.
Family-based association analysis
For family-based studies, we calculated a transmission ratio from heterozygous parents in the trio population from each TDT, which is equivalent to an OR calculated from the McNemar test. The χ2 statistic was used to calculate P values for the TDT, and 95% CIs were calculated on the basis of a binomial distribution.
Assessment of publication bias
We assessed publication bias through use of Begg’s modified funnel plots, in which the OR was plotted on a logarithmic scale against its SE from each study (Egger et al. 2001). Publication bias was also assessed by two formal tests: Begg’s adjusted rank correlation test and Egger’s regression asymmetry test. The former was used to examine whether there is significant correlation between the effect estimates and their variances, and the latter uses an inverse-variance weighted regression of the effect sizes on their precision (the inverse of SE) to test whether the intercept deviates significantly from 0 (Egger et al. 2001). Meta-analyses, including tests of publication bias, were performed using Stata statistical software (version 7.0; Stata Web site).
Sample size estimation
To evaluate the sufficiency of statistical power in previous studies, we calculated the necessary sample size to achieve a given power for the CAPN10-T2D relation in both the population-based case-control design and the family-based association study design (TDT) (Gauderman 2002).
Results
Characteristics of Association Studies
Of the 21 studies involving a total of 5,013 cases and 5,876 controls, one-third involved white Europeans, one-third involved Americans, and one-third involved Asians (details of characteristics of included studies are given in the appendix). American populations included Mexican Americans, Pima Indians, African Americans, and Samoans. The mean age at diagnosis for affected individuals ranged from 46 to 56 years. Overall, subjects with diabetes had higher BMIs and fasting insulin levels than did control subjects. Five studies also reported independent family-based studies that examined the transmission of alleles or haplotypes from heterozygous parents to affected offspring in populations that included Chinese (Sun et al. 2002), Utah whites (Elbein et al. 2002), Finns (Orho-Melander et al. 2002), British/Irish whites (Evans et al. 2001), and South Indians (Cassell et al. 2002).
Allele Frequencies of UCSNP-44, -43, -19, -63 Polymorphisms in CAPN10
Table 1 shows significant differences in the allele frequencies of UCSNP-44, -43, -19, and -63 polymorphisms across different populations (dbSNP IDs rs2975760, rs3792267, rs3842570, and rs5030952, respectively [dbSNP Home Page]). In particular, the G allele frequency of UCSNP-43 in controls varied from 0.62 (Pima Indians) to 0.96 (Japanese). However, the allele frequencies of these four polymorphisms were not significantly different between cases and controls. In only two Finnish populations did T2D cases have a significantly higher frequency for the G allele in UCSNP-43 than controls (case vs. control: 0.77 vs. 0.67, P<.05; and 0.77 vs. 0.67, P=.005) (Horikawa et al. 2000; Orho-Melander et al. 2002). The rare allele T in UCSNP-63 was more frequent in cases than controls in one Finnish population (Horikawa et al. 2000). On the basis of studies that presented only allele data, we estimated that the pooled ORs were 0.96 (95% CI 0.80–1.15) for UCSNP-44 (T carriers vs. C carriers), 0.97 (95% CI 0.88–1.07) for -19 (carriers of two repeats of 32-bp sequence vs. carriers of three repeats), and 0.99 (95% CI 0.86–1.15) for -63 (C carriers vs. T carriers).
Table 1.
Allele Frequencies of UCSNP-44, -43, -19, and -63 Polymorphisms in the CAPN10 Gene Reported in 11 Diverse Populations
|
Frequency ofb |
|||||||||
| UCSNP-44 (T/C) T Allele |
UCSNP-43 (G/A) G Allele |
UCSNP-19 (del/ins)cdel Allele |
UCSNP-63 (C/T) C Allele |
||||||
| Study Population | No. of Cases/No. of Controlsa | Cases | Controls | Cases | Controls | Cases | Controls | Cases | Controls |
| British/Irish | 444/424 | .84 | .86 | .73 | .72 | NA | NA | NA | NA |
| Finnd | 395/298 | .80 | .77 | .75 | .71 | .51 | .50 | .89 | .92 |
| German | 232/88 | NA | NA | .72 | .66 | .66 | .73 | .92 | .94 |
| Polish | 229/148 | NA | NA | .73 | .69 | .66 | .65 | .93 | .91 |
| Scandinavian | 409/200 | NA | NA | .73 | .72 | .62 | .60 | NA | NA |
| African Americans | 166/993 | NA | NA | .89 | .87 | NA | NA | NA | NA |
| Mexican Americans | 233/101 | NA | NA | .80 | .75 | .42 | .43 | .77 | .77 |
| Samoan | 172/96 | NA | NA | .91 | .91 | .67 | .63 | .85 | .84 |
| Pima Indian | 298/677 | NA | NA | .63 | .62 | NA | NA | NA | NA |
| Chinesee | 297/296 | .87 | .91 | .90 | .89 | .70 | .67 | .80 | .78 |
| Japanesee | 382/866 | .88 | .90 | .96 | .96 | .62 | .63 | .74 | .72 |
| Total | 3,257/4,187 | .83 | .85 | .78 | .77 | .59 | .58 | .88 | .87 |
Total numbers of cases and controls from all studies that provided allele data.
NA = data not available.
UCSNP-19 alleles are two repeats of 32-bp sequence and three repeats (or 32-bp deletion/insertion).
Significant differences in allele frequencies between cases and controls are reported for UCSNP-43 in Finnish populations (P<.05 and P=.011) (Horikawa et al. 2000; Orho-Melander et al. 2002) and for UCSNP-63 in one Finnish population (P<.05) (Horikawa et al. 2000).
Multiple studies for the same study population were combined to form one case and one control group.
UCSNP-43 and T2D Risk
Analysis of population-based studies
Under a recessive model, the G allele appeared to confer a greater risk for T2D. Specifically, individuals homozygous for the G allele had a 19% increased risk of T2D compared with those with the G/A or A/A genotypes (OR 1.19; 95% CI 1.07–1.33) (fig. 1A). Under either an additive or a dominant effect model, however, we found no statistically significant relation between CAPN10 genotypes in the UCSNP-43 locus and T2D risk (table 2).
Figure 1.

Pooled estimates of OR of T2D and 95% CI, comparing individuals with the G/G genotype of UCSNP43 versus all carriers of the A allele (A) and the 112/121 haplotype combination versus all others (B). Black squares indicate the OR in each study, with the size of the square inversely proportional to its variance, and horizontal lines represent the 95% CIs. The pooled OR and its 95% CI are indicated by the unshaded diamond. Three studies reported results on different populations: “Horikawa 1, 2, and 3” indicate Mexican-American, German, and Finnish populations, respectively; “Schwarz 1 and 2” are German and Czech populations, respectively; and “Fingerlin 1 and 2” are two Finnish populations.
Table 2.
Pooled Estimates of ORs and 95% CIs for the Association between UCSNP-43 Polymorphism in the CAPN10 Gene and T2D Risk, from Population-Based Association Studies
|
OR (95% CI) under Model |
|||||
| GenotypeComparison | Na | No. ofCases/No. ofControlsb | Fixed-Effects | Random-Effects | P for Heterogeneity |
| Recessive model: | |||||
| G/G vs. G/A+A/A | 11 | 2,288/3,041 | 1.19 (1.07–1.33) | 1.18 (1.02–1.35) | .20 |
| Dominant model: | |||||
| G/G+G/A vs. A/A | 6 | 1,399/2,314 | 1.09 (.82–1.44) | 1.09 (.82–1.44) | .71 |
| Additive model: | |||||
| G/G vs. G/A | 7 | 1,480/2,395 | 1.13 (.95–1.33) | 1.13 (.95–1.33) | .95 |
| G/A vs. A/A | 6 | 1,399/2,314 | 1.03 (.77–1.39) | 1.03 (.77–1.39) | .71 |
| G/G vs. A/A | 6 | 1,399/2,314 | 1.14 (.85–1.53) | 1.14 (.85–1.53) | .72 |
Total number of studies with data available.
Total numbers of cases and controls from all studies that provided genotype data.
Analysis of family-based studies
In five family-based studies with a total of 487 trios in populations of Chinese (Sun et al. 2002), Utah whites (Elbein et al. 2002), Finns (Orho-Melander et al. 2002), British/Irish whites (Evans et al. 2001), and South Indians (Cassell et al. 2002), we found no evidence for overtransmission of the G alleles in UCSNP-43 from heterozygous parents to diabetic offspring (table 3).
Table 3.
Pooled Estimates of Transmission Ratios (from Heterozygous Parents to Their Affected Offspring), for UCSNP-44, -43, -19, and -63 Polymorphisms and Haplotypes Defined from Them, Based on Family-Based Association Studies (TDT)
|
No. of Alleles |
||||||
| CAPN10 Varianta | Nb | Transmitted | NotTransmitted | Transmission Ratio (95% CI)c | P Valued | P forHeterogeneity |
| UCSNP: | ||||||
| -44 | 3 | 77 | 117 | .66 (.49–.88) | .004 | .96 |
| -43 | 5 | 239 | 259 | .92 (.77–1.10) | .37 | .57 |
| -19 | 4 | 232 | 261 | .89 (.75–1.06) | .19 | .78 |
| -63 | 4 | 84 | 60 | 1.40 (1.00–1.95) | .05 | .80 |
| Haplotype (-43, -19, -63): | ||||||
| 111 | 3 | 133 | 129 | 1.03 (.81–1.32) | .80 | .22 |
| 112 | 3 | 31 | 42 | .74 (.50–1.19) | .18 | .18 |
| 121 | 3 | 167 | 191 | .87 (.71–1.08) | .21 | .82 |
| 221 | 3 | 113 | 103 | 1.10 (.84–1.43) | .50 | .76 |
| Haplotype (-44, -43, -19, -63): | ||||||
| 1111 | 3 | 91 | 93 | .98 (.73–1.31) | .89 | .30 |
| 2111 | 3 | 119 | 87 | 1.37 (1.04–1.80) | .03 | .59 |
| 1121 | 3 | 166 | 188 | .88 (.72–1.09) | .24 | .77 |
| 1221 | 3 | 113 | 102 | 1.11 (.85–1.45) | .46 | .80 |
| 1112 | 3 | 29 | 42 | .70 (.43–1.13) | .15 | .12 |
Allele designations in polymorphisms: UCSNP-44: allele 1 = T, allele 2 = C; UCSNP 43: allele 1 = G, allele 2 = A; UCSNP-19: allele 1 = two repeats of 32-bp sequence, allele 2 = three repeats (or 32 bp deletion/insertion); UCSNP-63: allele 1 = C, allele 2 = T. The transmission ratios in this table refer to the 1 allele.
TDT results are from five independent studies of different populations: British/Irish whites, Finn, Chinese, Utah whites with northern European ancestry, and South Indian populations. Haplotype data were provided by three studies (Evans et al. 2001; Cassell et al. 2002; Orho-Melander et al. 2002).
Transmission ratios and 95% CIs in the TDT were calculated using the McNemar test.
χ2 distributions were used to calculate P values for the pooled OR.
Haplotypes and Other SNPs in CAPN10 and T2D risk
Analysis of population-based studies
The haplotypes in CAPN10 were defined from three or four polymorphisms spanned across the gene: UCSNP-43 (G/A within intron 3; allele 1 = G and allele 2 = A), UCSNP-19 (two repeats of 32-bp sequence or three repeats of 32-bp sequence within intron 6; allele 1 = two repeats and allele 2 = three repeats), and UCSNP-63 (C/T within intron 13; allele 1 = C and allele 2 = T), without or with UCSNP-44 (T/C within intron 3; allele 1 = T and allele 2 = C) (fig. 2) (GenBank). UCSNP-43, -19, and -63 polymorphisms were used to define four most common haplotypes (i.e., 111, 112, 121, and 221). These polymorphisms plus UCSNP-44 defined five most common haplotypes: 1111, 2111, 1121, 1221, and 1112 (fig. 2). It is notable that the -44 and -43 loci are 11 bp apart but are not in complete linkage disequilibrium (LD). The common haplotypes span from intron 3 to intron 13 (a 11.5-kb region) and account for 99% of all haplotypes observed. However, there were considerable differences in the frequencies of haplotype combinations across different populations (table 4). The 112 haplotype was common in Asians but rare in whites, and the 121 haplotype was quite common in various populations. The frequencies of the 112/121 haplotype combination were extremely diverse, ranging from 0.01 in German populations (Horikawa et al. 2000) to 0.32 in Japanese populations (Horikawa et al. 2003; Iwasaki et al. 2003). All large ORs >2.00 with wide 95% CIs were reported by the first study in three populations and by the two smallest studies. Overall, the 112/121 haplotype combination had only a moderately increased risk of T2D for all studies combined (pooled OR 1.38; 95% CI 1.04–1.84), with significant between-study heterogeneity (χ213=24.1; P=.03) (fig. 1B).
Figure 2.

Schematic diagram of the human CAPN10 gene illustrating the location of polymorphisms used to define haplotypes and one coding polymorphism. The location of polymorphisms relative to the A of the start codon are 4841 (site 44), 4852 (site 43), 7917 (site 19), 9803 (site 110), and 16378 (site 63). The sequence length for CAPN10 gene is from GenBank (accession number AF158748). Frequencies of the haplotypes were provided in unrelated control groups.
Table 4.
Pooled Estimates of OR and 95% CIs for the Associations between Haplotype Combinations Defined by UCSNP-43, -19, and -63 Polymorphisms and Risk of T2D
| OR (95% CI)d under Model |
||||||
| HaplotypeCombinationa | Nb | No. ofCases/No. ofControls | Frequency Rangec | Fixed Effects | Random Effects | P Value for Heterogeneity |
| 111/111 | 9 | 184/143 | .03–.11 | 1.16 (.94–1.44) | 1.16 (.94–1.44) | .89 |
| 111/112 | 10 | 103/63 | .02–.09 | 1.03 (.79–1.34) | 1.03 (.79–1.34) | .94 |
| 111/121 | 10 | 389/257 | .11–.24 | .96 (.83–1.12) | .96 (.83–1.12) | .94 |
| 112/112 | 7 | 36/27 | .01–.08 | .74 (.46–1.19) | .74 (.46–1.19) | .91 |
| 112/121 | 14 | 1,848/1,237 | .01–.32 | 1.22 (1.01–1.47) | 1.38 (1.04–1.84) | .03 |
| 121/121 | 10 | 324/178 | .06–.33 | 1.12 (.94–1.34) | 1.14 (.90–1.46) | .06 |
| 221/221 | 8 | 121/95 | .00–.12 | .83 (.66–1.05) | .83 (.66–1.05) | .83 |
| 221/111 | 9 | 253/180 | .00–.24 | .93 (.79–1.08) | .93 (.79–1.09) | .42 |
| 221/112 | 10 | 82/55 | .01–.14 | .95 (.72–1.25) | .93 (.62–1.41) | .05 |
| 221/121 | 10 | 311/211 | .07–.22 | .92 (.78–1.08) | .92 (.78–1.08) | .52 |
Allele designation in haplotype combination: UCSNP 43: allele 1 = G, allele 2 = A; UCSNP-19, allele 1 = two repeats of 32-bp sequence, allele 2 = three repeats; UCSNP-63, allele 1 = C, allele 2 = T.
Total number of population-based studies included.
Frequency among control subjects from eight ethnic populations, including Japanese (Horikawa et al. 2003), Scandinavian (Rasmussen et al. 2002), Polish (Malecki et al. 2002), Samoan (Tsai et al. 2001), Mexican American (Horikawa et al. 2000), German (Horikawa et al. 2000), and two separate Finnish populations (Horikawa et al. 2000; Orho-Melander et al. 2002).
OR and 95% CI of each haplotype combination relative to its corresponding control group (all other haplotype combinations).
Analysis of family-based studies
In the pooled sample of 356 informative trios, the overall transmission ratio was statistically significant only for UCSNP-44 (pooled OR 0.66; 95% CI 0.49–0.88; P=.004) (table 3). Allele transmission from UCSNP-63 heterozygotes showed favorable transmission of the common allele “C,” although only the pooled estimated OR reached borderline significance (pooled OR 1.40; 95% CI 1.00–1.95; P=.05). In the pooled analysis of TDT for haplotypes defined by these three or four polymorphisms, a preferential transmission of the rare allele “C” in the UCSNP-44 locus linked with the haplotype 111 was evident (pooled OR 1.37; 95% CI 1.04–1.80; P=.03) (table 3).
CAPN10 and Diabetes-Related Quantitative Traits
Compared with carriers of the A allele, individuals homozygous for the G allele in the UCSNP-43 locus had slightly higher systolic blood pressure, but this finding was based on very limited data (from three studies) (table 5). In all other metabolic parameters—including BMI; WHR; plasma levels of lipids; fasting insulin and glucose levels; and plasma glucose, insulin, and C-peptide levels in response to a 2-h 75-g oral glucose tolerance test (OGTT)—there were no statistically significant differences between carriers of the G/G genotype and carriers of the A allele (table 5), and there were no significant differences in quantitative metabolic parameters between carriers of the 112/121 haplotype combination and all others.
Table 5.
Effects of the G/G Genotype of UCSNP-43 and the 112/121 Haplotype Combination on Metabolic Traits in Healthy Nondiabetic Controls
|
UCSNP-43 G/G versus G/C+C/C |
112/121 versus All Other Haplotype Combinations |
|||||
| MetabolicParametera | Nb | Weighted MeanDifference (95% CI) | P Value for Heterogeneity | Nb | Weighted MeanDifference (95% CI) | P Value for Heterogeneity |
| BMI | 7 | −.05 (−.15 to .05) | .16 | 2 | .30 (−.26 to .86) | .14 |
| WHR | 7 | .05 (−.04 to .15) | .51 | 2 | .04 (−.31 to .39) | .70 |
| SBP | 3 | .14 (.01 to .26) | .57 | NA | NA | NA |
| DBP | 3 | .11 (−.01 to .24) | .87 | NA | NA | NA |
| Total cholesterol | 2 | .32 (.01 to −.63) | .24 | NA | NA | NA |
| HDL-c | 2 | .07 (−.06 to .20) | .98 | NA | NA | NA |
| Triglyceride | 2 | .09 (−.04 to .22) | .39 | NA | NA | NA |
| Fasting insulin | 7 | .04 (−.06 to .14) | .41 | 2 | −.26 (−.62 to .10) | .17 |
| Fasting glucose | 8 | .06 (−.03 to .16) | .84 | 3 | .06 (−.21 to .33) | .08 |
| Fasting C-peptide | NA | NA | NA | 2 | .08 (−.28 to .43) | .27 |
| 2-h post-load glucose | 5 | .08 (−.07 to .22) | .46 | 3 | .15 (−.48 to .77) | .007 |
| 2-h post-load insulin | 3 | .05 (−.11 to .21) | .56 | 3 | .15 (−.13 to .42) | .005 |
| 2-h post-load C-peptide | NA | NA | NA | 2 | −.15 (−.51 to .21) | .05 |
| HOMA-IR | 4 | −.06 (−.22 to .11) | .81 | 3 | .03 (−.54 to .60) | .02 |
| EIR | 1 | .04 (−.61 to .69) | NA | 2 | −.18 (−.49 to .12) | .49 |
SBP = systolic blood pressure; DBP = diastolic blood pressure; OGTT = 75-g oral glucose tolerance test; NA = data not available. HOMA-IR (homeostasis model assessment of insulin resistance) was calculated as the product of fasting serum insulin and fasting plasma glucose. EIR = Early insulin response correlated with insulin secretion; determined as the ratio of the 30-min insulin increment to the 30-min increment in glucose levels following oral glucose loading.
Total number of studies included.
Publication Bias and Sensitivity Analyses
Figure 3 shows a funnel plot for the visual assessment of publication bias. In the absence of publication bias, one would expect studies of all sizes to be scattered equally above and below the line representing the pooled estimate of log OR. The results showed no evidence of the presence of substantial publication bias for the association between the homozygosity of the G allele of UCSNP-43 and T2D (P=.24 for Begg’s test and P=.28 for Egger’s test) (fig. 3A) (Stata Web site). Furthermore, our sensitivity analysis using the empirical Bayes estimation yielded the same results of the pooled OR of T2D comparing the homozygosity of the G allele to carriers of the A allele (OR 1.19; 95% CI 1.07–1.33) as those from both fixed-effects and random-effects models.
Figure 3.

Detecting publication bias using Begg’s funnel plot: the ORs for the individuals homozygous for the G allele versus carriers of the C allele in the UCSNP-43 locus (A) and for carriers of the 112/121 haplotype combination versus all others (B). The funnel plot shows the OR on a log scale by its SE, for each study included in the meta-analysis. The horizontal line indicates the pooled estimate of OR and 95% CI, with the sloping lines representing the expected 95% CI for a given SE, under the assumption of no heterogeneity between studies.
For haplotype comparison, however, the ORs with large SEs tended to scatter above the horizontal line, indicating significant publication bias in favor of small studies with positive findings (P=.006 for Begg’s test and P=.001 for Egger’s test) (fig. 3B) (Stata Web site). After excluding the initial results, the pooled OR comparing the 112/121 haplotype combination to all others became nonsignificant (OR 1.11; 95% CI 0.91–1.35; P for heterogeneity = .14). If we excluded all results from the three smallest studies, the pooled OR was attenuated toward the null (OR 1.04; 95% CI 0.84–1.27; P for heterogeneity = .57) (fig. 1B).
Assessment of Statistical Power
To provide an accurate assessment of sufficiency of statistical power in previous studies, we calculated the sample size required to detect a modest genetic effect under the assumption of a recessive model. For population-based studies, an equal number of 2,188 cases and controls would be required to have 80% power to detect the OR of 1.19 (one-sided test: α=.05) (fig. 4A). Therefore, none of the previously published case-control studies achieved this sample size to confirm the modest (OR 1.19) but important (PAR 10%) genetic association between UCSNP-43 and T2D risk. For family-based studies (TDT), the number of parent-offspring trios required to detect an OR of 1.19 is estimated to be 1,617 for a power of 80% (one-sided test: α=.05) (fig. 4B). Thus, even our pooled TDT analysis with a total of 487 parent-offspring trios had <40% power to detect this transmission/disequilibrium ratio of 1.19.
Figure 4.

Sample size calculations for testing the effect of a susceptibility allele (or a haplotype) in a case-control study design (A) and a family-based association study design (B). All calculations assume a recessive model, and the prevalence is 5.4% for T2D. The required sample sizes are calculated to achieve a power of 80% for detecting various genetic effects at a one-sided type I error rate of 0.05. The frequency of susceptibility allele in controls is shown on the X-axis. The sample size in the case-control study is the number of subjects in each group, under the assumption of an equal number of cases and controls. The sample size in the family-based study (TDT) is the number of parents-offspring trios.
Discussion
On the basis of these 21 population-based studies including 5,013 T2D cases and 5,876 controls, our meta-analysis showed a significant 19% increased risk of T2D among individuals with the G/G genotype in the UCSNP-43 locus relative to all others. However, the association between the 112/121 haplotype combination and T2D risk appeared to be weak and to have significant between-study heterogeneity. This finding was not supported by family-based studies. In addition, we found no significant association between CAPN10 genotypes and metabolic phenotypes.
Failure to replicate the initial findings is a major concern in genetic association studies (Altshuler et al. 2000; Cardon and Bell 2001). False-positive and false-negative reports from underpowered studies may, in part, explain the inconsistent replication. In the present study, none of the previous studies were found to individually achieve the required statistical power to provide a robust estimate of the possible modest effect of CAPN10 on T2D risk; however, our pooled analysis of all available data suggests a modest but significant OR of 1.19 for carriers of the G/G genotype in the UCSNP-43 locus versus carriers of the A allele. Furthermore, recent evidence showed that the first study tends to overestimate the genetic effects, which often cannot be replicated in subsequent larger studies (Ioannidis et al. 2001). Nevertheless, the G/G genotype remained significantly associated with T2D even after removing the first study. In addition, our finding of a recessive inheritance model for the modest effect of the UCSNP-43 locus on T2D was compatible with results from several metabolic studies showing that the G/G genotype was associated with appreciable degrees of physiologic effects, compared with the A/A genotype alone or with the G/A genotype (Baier et al. 2000; Hoffstedt et al. 2002b). Taken together, these findings suggest that the common G allele in the UCSNP-43 locus may confer a modest susceptibility to T2D. Because of its high allele frequency, the modest genetic effect would be translated into a relatively large PAR. On the basis of the pooled data, we estimated that the G/G genotype accounts for 10% of total diabetes cases. Nevertheless, it is important to keep in mind that PAR is meaningful insofar as the relationship between UCSNP-43 and T2D risk is causal and that a more accurate estimate of PAR can be estimated only by studies using population-based controls.
In contrast, our analysis of five family-based studies provided little support for the association between the G allele and T2D but revealed a significant association between risk of T2D and the rare allele C in the UCSNP-44 locus alone or in combination with the 111 haplotype (Elbein et al. 2002). However, even this pooled analysis of family-based studies is not large enough to provide a reliable estimate of a modest genetic effect. Additional large-scale studies are needed to confirm this finding. To assess a genetic effect under a recessive model, a future modified TDT that tests allele transmission from parents to their affected offspring homozygous for the G allele may be more powerful than the traditional TDT used in previous studies.
Despite the observed association between the G/G genotype and the risk of T2D, the haplotype that includes this variant and other adjacent variants did not appear to increase susceptibility to T2D (Horikawa et al. 2000; Cox 2001, 2002). In contrast to the two- or threefold increased risk reported in the original study (Horikawa et al. 2000), we observed only a moderate effect of the 112/121 haplotype combination on risk of T2D (OR 1.38; 95% CI 1.04–1.84). Furthermore, when we excluded the initial positive results and/or the other two smallest studies, the evidence for heterogeneity disappeared, and there was no significant relation between the 112/121 haplotype combination and T2D risk. It seems likely that this observed significant association was overestimated by these underpowered studies. Thus, the haplotype-specific effects found in the initial study remain questionable.
Recent empirical data have suggested that LD patterns in the human genome are characterized by a “haplotype block” structure, in which a series of high-LD regions is interspersed with short, discrete segments of very low LD (which may represent recombination hotspots) (Daly et al. 2001; Gabriel et al. 2002). Given limited haplotype diversity within each block, a minimal set of SNPs, designated as “haplotype-tagging SNPs” (htSNPs), is thought to be sufficient to capture the LD variation in a population (Johnson et al. 2001). Therefore, increased understanding of LD patterns in the genomic sequences of interest has important implications for the design of practical haplotype-based association studies. In the original study, Horikawa et al. (2000) sequenced a 66-kb genomic region that encompasses CAPN10 in 10 diabetic Mexican Americans and then identified the high-risk haplotypes on the basis of the evidence from both association and linkage analysis. Subsequently, most studies have not determined the haplotype structure of CAPN10 in their own study samples but, rather, have genotyped the SNPs (UCSNP-43, -19, -63) originally defined in Mexican Americans (Horikawa et al. 2000). Such a strategy, though efficient, may have overlooked the population-specific LD pattern in characterizing recombination hotspots and choosing htSNPs in the genomic region near CAPN10. It is thus possible that the CAPN10 variants chosen for haplotype definition in previous studies may not be sufficient and that the haplotypes so defined may not capture the vast majority of genetic variability of CAPN10 in diverse populations. Furthermore, all of these variants used for haplotype definition are intronic and are very likely to be in LD with other unidentified functional variants (regulatory or coding polymorphisms) within or in the proximity of the CAPN10 locus (Horikawa et al. 2000; Cox 2002). Only UCSNP-44 has been observed to be in perfect LD with a coding polymorphism in CAPN10 (UCSNP-110: A→G in exon 10 [Thr504Ala], dbSNP ID rs7607759) (Evans et al. 2001; Schwarz et al. 2001). Several nonsynonymous polymorphisms of CAPN10 have been identified, but most of them appeared to have relatively rare frequencies (<5%) (Horikawa et al. 2000; Evans et al. 2001; dbSNP Home Page; OMIM Web site).
Geographic/ethnic differences in the frequencies of these known CAPN10 variants, haplotypes, and haplotype combinations are pronounced across multiple ethnic populations. A recent study of CAPN10 in 561 individuals from 11 populations also showed substantial degrees of diversity in the frequencies of CAPN10 haplotypes between African and non-African populations (Fullerton et al. 2002). The 121 haplotype was absent in two African populations, whereas the 112 haplotype appeared to be much more common in these two African populations (0.59–0.82) than in the non-African populations (Fullerton et al. 2002). Such genetic heterogeneity of CAPN10 in populations of various ethnic origins not only affects the power of individual studies but also indicates different patterns and magnitudes of LD between variants in this gene among different ethnic groups (i.e., population-specific LD). Therefore, population- or locus-specific LD patterns may contribute, at least in part, to inconsistent findings of previous studies. Future haplotype-based studies need to carefully survey the genomic region in and near CAPN10, such that the underlying structure of LD for each ethnic population of interest can be better characterized.
Some essential issues related to genetic association studies also merit further consideration. First, population stratification may be a concern, especially when data from diverse populations are included. However, empirical evidence indicates that population stratification is likely to be minimal in well-designed population studies that match or control for ethnicity (Wacholder et al. 2000; Ardlie et al. 2002; Pankow et al. 2002). Furthermore, bias due to population stratification in different studies appears to be random. Thus, the pooled estimate from each independent study of ethnically homogeneous populations is less likely to be biased by the presence of population stratification. Second, differences in control selection of case-control studies may contribute to heterogeneity between studies and may need to be carefully considered in the design of future association studies. Epidemiologists have long noted the numerous advantages of the prospective study design over hospital-based case-control studies in minimizing selection bias or population stratification. Of the 21 case-control studies included in our analysis, only 1 used population-based controls with a prospective study design (Garant et al. 2002). In that study, the OR of incident diabetes among African American participants with the G/G genotype compared with those carrying the A allele was not statistically significant (OR 1.21; 95% CI 0.86–1.27), but its magnitude is similar to our pooled estimated OR. Third, publication bias may exist, although we have made every attempt to identify relevant reports. Although we found little evidence of publication bias regarding the association between UCSNP-43 and T2D, our sensitivity analyses showed that the excess risk of T2D with the haplotype combination 112/121 could be explained largely by publication bias from small studies with positive findings. Fourth, possible gene-gene or gene-environmental interactions could lead to the varying genetic effects of CAPN10 observed in different populations. For example, previous linkage analysis and a family-based test indicate an interaction between locus NIDDM1 (MIM 601283) on chromosome 2q37 and CYP19 (MIM 107910) on chromosome 15q21.1 in conferring increased susceptibility for T2D (Cox et al. 1999; Engelman et al. 2003). The UCSNP-19 was found to be associated with reduced β3-adrenoceptor function in overweight people (BMI >25 kg/m2) but not in lean ones (Hoffstedt et al. 2002a). Ultimately, further clarification of potential gene-gene or gene-environmental interactions can be achieved only by well-designed large-scale prospective studies. Fifth, although analysis of haplotypes is generally believed to offer more power to detect associations than does simply focusing on a single variant, the statistical power of haplotype analysis may still be reduced by multiple hypothesis testing, because of the potential large number of haplotypes or haplotype combinations. Finally, it should be noted that genetic evidence alone is not sufficient to confirm any causal genetic variants for disease susceptibility. If the observed relations between genotype and disease risk can be supported by biological plausibility, the causal arguments for the effect of CAPN10 on T2D can be strengthened. Since little is known about the genuine physiological functions of calpain-10 in insulin secretion and action, the underlying molecular mechanism whereby CAPN10 genetic variation influences risk of T2D remains uncertain and needs further investigation.
In conclusion, our analysis suggests that the G allele in the CAPN10 UCSNP-43 locus may have a modest effect on risk of T2D in a recessive model. The evidence for the association between the risk haplotype 112/121 and T2D risk, however, is complicated by the presence of significant between-study heterogeneity. Inadequate statistical power; racial/ethnic differences in frequencies of alleles, haplotypes, and haplotype combinations; gene-gene or gene-environment interactions; publication bias; and multiple hypothesis testing can reasonably contribute to the nonreplication of association in previous studies. Future large-scale, well-designed studies using a prospective design and population-based controls is needed to either reliably confirm or conclusively refute the hypothesized effect of CAPN10 on T2D. Additional functional data are also warranted to elucidate the potential biological role of CAPN10 in the pathogenesis of T2D.
Acknowledgments
We thank two anonymous reviewers for their critical comments on the manuscript. This work was supported by National Institutes of Health grant DK62290.
Appendix
Table A1.
Characteristics of Included Studies[Note]
|
Subjects with T2D |
Control Individuals |
||||||||
| Source (StudyPopulation) | No. ofIndividuals(Men/Women) | Age(years) | Age atDiagnosis(years) | BMI(kg/m2) | Plasma FastingGlucose(mmol/liter) | No. ofIndividuals(Men/Women) | Age(years) | BMI(kg/m2) | Plasma FastingGlucose(mmol/liter) |
| European (7 studies): | |||||||||
| Evans et al. 2001 (British/Irish) | 222 (106/116) | 35–70 | 56±8 | 28±5 | NA | 212 (71/141) | 50±14 | 27±5 | <5.5 |
| Schwarz et al. 2001 (German) | 291 | 62±11 | NA | 25±4 | NA | 88 | 51±12 | 29±5 | NA |
| Schwarz et al. 2001 (Czech) | 324 | 59±7 | NA | 30±5 | NA | 156 | 18±2 | 24±4 | NA |
| Fingerlin et al. 2002 (Finn) | 781 (438/343) | 64±8 | 51±8 | 30±5 | 10±3 | 408 (168/240) | 66 | 28±4 | 5.1±0.5 |
| Malecki et al. 2002 (Polish) | 229 (138/91) | 56±12 | 46±11 | 30±6 | NA | 148 (81/67) | 57±14 | 27±6 | NA |
| Orho-Melander et al. 2002 (Finn) | 395 (171/224) | 61±10 | 53±11 | 29±5 | 10±3 | 298 (140/158) | 60±9 | 27±4 | 5.5±0.6 |
| Rasmussen et al. 2002 (Scandinavian) | 409 | 61 (22–86) | NA | 29 | NA | 200 | 52 (30–88) | 25 | NA |
| American (7 studies): | |||||||||
| Baier et al. 2000 (Pima Indian) | 298 | NA | NA | NA | NA | 677 | NA | NA | NA |
| Horikawa et al. 2000 (Mexican-American) | 233 | NA | NA | NA | NA | 101 | NA | NA | NA |
| Horikawa et al. 2000 (Finn) | 192 (87/105) | 61±9 | 55±9 | 29±5 | 8.9±2.9 | 192 (88/104) | 61±9 | 27±4 | 5.0±0.5 |
| Horikawa et al. 2000 (German) | 232 (93/139) | 64±10 | 50±11 | 28±5 | 8.4±4.0 | 88 (31/57) | 51±12 | 25±4 | 5.9±2.1 |
| Hegele et al. 2001 (Canadian) | 121 (47/74) | NA | NA | NA | NA | 468 | NA | NA | NA |
| Tsai et al. 2001 (Samoan) | 172 | 55±12 | 48±11 | NA | NA | 96 | ⩾50 | <30 | <6.67 |
| Garant et al. 2002 (African-American) | 166 | 53±6 | NA | 31±6 | NA | 993 | 55±6 | 29±6 | 5.5±0.6 |
| Asian (7 studies): | |||||||||
| Ng et al. 2001 (Chinese) | 103 | NA | NA | NA | NA | 185 | NA | NA | NA |
| Xiang et al. 2001a (Chinese) | 124 (57/67) | 56±10 | NA | NA | NA | 144 (64/80) | 53±11 | NA | NA |
| Daimon et al. 2002 (Japanese) | 81 (45/36) | 65±11 | NA | 26±4 | NA | 81 (46/35) | 62±8 | 24±4 | NA |
| Cassell et al. 2002 (Indian) | 85 | 53 | NA | NA | NA | 323 | 45 | NA | NA |
| Sun et al. 2002 (Chinese) | 173 | NA | NA | NA | NA | 152 | NA | NA | NA |
| Horikawa et al. 2003 (Japanese) | 177 (114/63) | 62±11 | 50±11 | 24±3 | 9.3±3.8 | 172 (82/90) | 68±6 | 23±3 | NA |
| Iwasaki et al. 2003 (Japanese) | 205 | NA | NA | NA | NA | 694 | NA | NA | NA |
Note.— Data are the mean±SD or range; “NA” indicates that data were not available.
Electronic-Database Information
Accession numbers and URLs for data presented herein are as follows:
- dbSNP Home Page, http://www.ncbi.nlm.nih.gov/SNP/ (for UCSNP-44 [rs2975760], -43 [rs3792267], -19 [rs3842570], -63 [rs5030952], and -110 A→G: Thr504Ala [rs7607759])
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for genomic structure of CAPN10 [accession number AF158748])
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for T2D, CAPN10, UCSNP-43, UCSNP-19, UCSNP-63, NIDDM1, and CYP19)
- PubMed, http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=PubMed (for literature search)
- Stata Web site, http://www.stata.com/search.cgi?query=meta (for the Stata software for meta-analysis)
References
- Altshuler D, Daly M, Kruglyak L (2000) Guilt by association. Nat Genet 26:135–137 10.1038/79839 [DOI] [PubMed] [Google Scholar]
- Ardlie KG, Lunetta KL, Seielstad M (2002) Testing for population subdivision and association in four case-control studies. Am J Hum Genet 71:304–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baier LJ, Permana PA, Yang X, Pratley RE, Hanson RL, Shen GQ, Mott D, Knowler WC, Cox NJ, Horikawa Y, Oda N, Bell GI, Bogardus C (2000) A calpain-10 gene polymorphism is associated with reduced muscle mRNA levels and insulin resistance. J Clin Invest 106:R69–R73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardon LR, Bell JI (2001) Association study designs for complex diseases. Nat Rev Genet 2:91–99 10.1038/35052543 [DOI] [PubMed] [Google Scholar]
- Cassell PG, Jackson AE, North BV, Evans JC, Syndercombe-Court D, Phillips C, Ramachandran A, Snehalatha C, Gelding SV, Vijayaravaghan S, Curtis D, Hitman GA (2002) Haplotype combinations of calpain 10 gene polymorphisms associate with increased risk of impaired glucose tolerance and type 2 diabetes in South Indians. Diabetes 51:1622–1628 [DOI] [PubMed] [Google Scholar]
- Cox NJ (2001) Challenges in identifying genetic variation affecting susceptibility to type 2 diabetes: examples from studies of the calpain-10 gene. Hum Mol Genet 10:2301–2305 10.1093/hmg/10.20.2301 [DOI] [PubMed] [Google Scholar]
- ——— (2002) Calpain 10 and genetics of type 2 diabetes. Curr Diab Rep 2:186–190 [DOI] [PubMed] [Google Scholar]
- Cox NJ, Frigge M, Nicolae DL, Concannon P, Hanis CL, Bell GI, Kong A (1999) Loci on chromosomes 2 (NIDDM1) and 15 interact to increase susceptibility to diabetes in Mexican Americans. Nat Genet 21:213–215 10.1038/6002 [DOI] [PubMed] [Google Scholar]
- Daimon M, Oizumi T, Saitoh T, Kameda W, Yamaguchi H, Ohnuma H, Igarashi M, Manaka H, Kato T (2002) Calpain 10 gene polymorphisms are related, not to type 2 diabetes, but to increased serum cholesterol in Japanese. Diabetes Res Clin Pract 56:147–152 10.1016/S0168-8227(01)00372-2 [DOI] [PubMed] [Google Scholar]
- Daly MJ, Rioux JD, Schaffner SF, Hudson TJ, Lander ES (2001) High-resolution haplotype structure in the human genome. Nat Genet 29:229–232 10.1038/ng1001-229 [DOI] [PubMed] [Google Scholar]
- DerSimonian R, Laird NM (1986) Meta-analysis in clinical trials. Control Clin Trials 7:177–188 10.1016/0197-2456(86)90046-2 [DOI] [PubMed] [Google Scholar]
- Egger M, Smith GD, Altman DG (2001) Systematic reviews in health care: meta-analysis in context. The BMJ Publishing Group, London [Google Scholar]
- Elbein SC, Chu W, Ren Q, Hemphill C, Schay J, Cox NJ, Hanis CL, Hasstedt SJ (2002) Role of calpain-10 gene variants in familial type 2 diabetes in Caucasians. J Clin Endocrinol Metab 87:650–654 [DOI] [PubMed] [Google Scholar]
- Engelman C, Barmada M, Ferrell R, Norris J (2003) Investigation of an interaction between Calpain-10 and CYP 19 in the susceptibility to type 2 diabetes. Diabetes 52 Suppl:A510–A511 [Google Scholar]
- Evans JC, Frayling TM, Cassell PG, Saker PJ, Hitman GA, Walker M, Levy JC, et al (2001) Studies of association between the gene for calpain-10 and type 2 diabetes mellitus in the United Kingdom. Am J Hum Genet 69:544–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fingerlin TE, Erdos MR, Watanabe RM, Wiles KR, Stringham HM, Mohlke KL, Silander K, Valle TT, Buchanan TA, Tuomilehto J, Bergman RN, Boehnke M, Collins FS (2002) Variation in three single nucleotide polymorphisms in the calpain-10 gene not associated with type 2 diabetes in a large Finnish cohort. Diabetes 51:1644–1648 [DOI] [PubMed] [Google Scholar]
- Florez JC, Hirschhorn J, Altshuler D (2003) The inherited basis of diabetes mellitus: implications for the genetic analysis of complex traits. Annu Rev Genomics Hum Genet 4:257–291 10.1146/annurev.genom.4.070802.110436 [DOI] [PubMed] [Google Scholar]
- Fullerton SM, Bartoszewicz A, Ybazeta G, Horikawa Y, Bell GI, Kidd KK, Cox NJ, Hudson RR, Di Rienzo A (2002) Geographic and haplotype structure of candidate type 2 diabetes susceptibility variants at the calpain-10 locus. Am J Hum Genet 70:1096–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly MJ, Altshuler D (2002) The structure of haplotype blocks in the human genome. Science 296:2225–2229 10.1126/science.1069424 [DOI] [PubMed] [Google Scholar]
- Garant MJ, Kao WH, Brancati F, Coresh J, Rami TM, Hanis CL, Boerwinkle E, Shuldiner AR (2002) SNP43 of CAPN10 and the risk of type 2 Diabetes in African-Americans: the Atherosclerosis Risk in Communities Study. Diabetes 51:231–237 [DOI] [PubMed] [Google Scholar]
- Gauderman WJ (2002) Sample size requirements for association studies of gene-gene interaction. Am J Epidemiol 155:478–484 10.1093/aje/155.5.478 [DOI] [PubMed] [Google Scholar]
- Goll DE, Thompson VF, Li H, Wei W, Cong J (2003) The calpain system. Physiol Rev 83:731–801 [DOI] [PubMed] [Google Scholar]
- Greenland S (1998) Applications of stratified analysis methods. In: Rothman KJ, Greenland S (eds) Modern epidemiology. Lippincott Williams & Wilkins, Philadelphia, pp 281–300 [Google Scholar]
- Hegele RA, Harris SB, Zinman B, Hanley AJ, Cao H (2001) Absence of association of type 2 diabetes with CAPN10 and PC-1 polymorphisms in Oji-Cree. Diabetes Care 24:1498–1499 [DOI] [PubMed] [Google Scholar]
- Hoffstedt J, Naslund E, Arner P (2002a) Calpain-10 gene polymorphism is associated with reduced β3-adrenoceptor function in human fat cells. J Clin Endocrinol Metab 87:3362–3367 [DOI] [PubMed] [Google Scholar]
- Hoffstedt J, Ryden M, Lofgren P, Orho-Melander M, Groop L, Arner P (2002b) Polymorphism in the Calpain 10 gene influences glucose metabolism in human fat cells. Diabetologia 45:276–282 10.1007/s00125-001-0732-2 [DOI] [PubMed] [Google Scholar]
- Horikawa Y, Oda N, Cox NJ, Li X, Orho-Melander M, Hara M, Hinokio Y, Lindner TH, Mashima H, Schwarz PE, del Bosque-Plata L, Oda Y, Yoshiuchi I, Colilla S, Polonsky KS, Wei S, Concannon P, Iwasaki N, Schulze J, Baier LJ, Bogardus C, Groop L, Boerwinkle E, Hanis CL, Bell GI (2000) Genetic variation in the gene encoding calpain-10 is associated with type 2 diabetes mellitus. Nat Genet 26:163–175 10.1038/79876 [DOI] [PubMed] [Google Scholar]
- Horikawa Y, Oda N, Yu L, Imamura S, Fujiwara K, Makino M, Seino Y, Itoh M, Takeda J (2003) Genetic variations in calpain-10 gene are not a major factor in the occurrence of type 2 diabetes in Japanese. J Clin Endocrinol Metab 88:244–247 10.1210/jc.2002-020847 [DOI] [PubMed] [Google Scholar]
- Ioannidis JP, Ntzani EE, Trikalinos TA, Contopoulos-Ioannidis DG (2001) Replication validity of genetic association studies. Nat Genet 29:306–309 10.1038/ng749 [DOI] [PubMed] [Google Scholar]
- Iwasaki N, Horikawa Y, Kitamura Y, Nakamura T, Tanizawa Y, Oka Y, Hara K, Kadowaki T, Honda M, Ogata M, Kamatani N, Cox NJ, Iwamoto Y (2003) Variations in the calpain 10 gene affect plasma glucose levels in non-diabetic subjects in Japanese. Diabetes 52 Suppl:A511–A512 [Google Scholar]
- Johnson GC, Esposito L, Barratt BJ, Smith AN, Heward J, Di Genova G, Ueda H, Cordell HJ, Eaves IA, Dudbridge F, Twells RC, Payne F, Hughes W, Nutland S, Stevens H, Carr P, Tuomilehto-Wolf E, Tuomilehto J, Gough SC, Clayton DG, Todd JA (2001) Haplotype tagging for the identification of common disease genes. Nat Genet 29:233–237 10.1038/ng1001-233 [DOI] [PubMed] [Google Scholar]
- Kahn CR (1994) Banting Lecture: insulin action, diabetogenes, and the cause of type II diabetes. Diabetes 43:1066–1084 [DOI] [PubMed] [Google Scholar]
- Lillioja S, Mott DM, Spraul M, Ferraro R, Foley JE, Ravussin E, Knowler WC, Bennett PH, Bogardus C (1993) Insulin resistance and insulin secretory dysfunction as precursors of non-insulin-dependent diabetes mellitus. Prospective studies of Pima Indians. N Engl J Med 329:1988–1992 10.1056/NEJM199312303292703 [DOI] [PubMed] [Google Scholar]
- Lynn S, Evans JC, White C, Frayling TM, Hattersley AT, Turnbull DM, Horikawa Y, Cox NJ, Bell GI, Walker M (2002) Variation in the calpain-10 gene affects blood glucose levels in the British population. Diabetes 51:247–250 [DOI] [PubMed] [Google Scholar]
- Malecki MT, Moczulski DK, Klupa T, Wanic K, Cyganek K, Frey J, Sieradzki J (2002) Homozygous combination of calpain 10 gene haplotypes is associated with type 2 diabetes mellitus in a Polish population. Eur J Endocrinol 146:695–699 [DOI] [PubMed] [Google Scholar]
- Newman B, Selby JV, King MC, Slemenda C, Fabsitz R, Friedman GD (1987) Concordance for type 2 (non-insulin-dependent) diabetes mellitus in male twins. Diabetologia 30:763–768 [DOI] [PubMed] [Google Scholar]
- Ng MCY, So W, Critchley JAJH, Cockram CS, Chan JCN (2001) Association of calpain 10 genetic polymorphisms with type 2 diabetes and insulin response in nondiabetic Chinese. Diabetes 50 Suppl 2:A234 [Google Scholar]
- Normand ST (1999) Meta-analysis: formulating, evaluating, combining, and reporting. Stat Med 18:321–359 [DOI] [PubMed] [Google Scholar]
- Orho-Melander M, Klannemark M, Svensson MK, Ridderstrale M, Lindgren CM, Groop L (2002) Variants in the calpain-10 gene predispose to insulin resistance and elevated free fatty acid levels. Diabetes 51:2658–2664 [DOI] [PubMed] [Google Scholar]
- Pankow JS, Province MA, Hunt SC, Arnett DK (2002) Regarding “Testing for population subdivision and association in four case-control studies.” Am J Hum Genet 71:1478–1480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SK, Urhammer SA, Berglund L, Jensen JN, Hansen L, Echwald SM, Borch-Johnsen K, Horikawa Y, Mashima H, Lithell H, Cox NJ, Hansen T, Bell GI, Pedersen O (2002) Variants within the calpain-10 gene on chromosome 2q37 (NIDDM1) and relationships to type 2 diabetes, insulin resistance, and impaired acute insulin secretion among Scandinavian Caucasians. Diabetes 51:3561–3567 [DOI] [PubMed] [Google Scholar]
- Schwarz P, Fischer S, Goergens H, Koehler C, Graessler J, Fuecker K, Julius U, Schulze J, Schackert H, Hanefeld M (2003) Haplotype 112 in CAPN10 is associated with increased proinsulin values during 75 g OGTT in newly diagnosed type 2 diabetes. Diabetes 52 Suppl:A255 [Google Scholar]
- Schwarz PEH, Horikawa Y, Vcelak J, Selisko T, Rietzsch H, Bendlova B, Schulze J, Cox NJ (2001) Genetic variation of CAPN10 affects susceptibility to type 2 diabetes in German and Czech population. Diabetes 50 Suppl 2:A232 [Google Scholar]
- Shore AC, Evans JC, Frayling TM, Clark PM, Lee BC, Horikawa Y, Hattersley AT, Tooke JE (2002) Association of calpain-10 gene with microvascular function. Diabetologia 45:899–904 10.1007/s00125-002-0847-0 [DOI] [PubMed] [Google Scholar]
- Stumvoll M, Fritsche A, Madaus A, Stefan N, Weisser M, Machicao F, Haring H (2001) Functional significance of the UCSNP-43 polymorphism in the CAPN10 gene for proinsulin processing and insulin secretion in nondiabetic Germans. Diabetes 50:2161–2163 [DOI] [PubMed] [Google Scholar]
- Stumvoll M, Wahl HG, Machicao F, Haring H (2002) Insulin sensitivity of glucose disposal and lipolysis: no influence of common genetic variants in IRS-1 and CAPN10. Diabetologia 45:651–656 10.1007/s00125-002-0793-x [DOI] [PubMed] [Google Scholar]
- Sun HX, Zhang KX, Du WN, Shi JX, Jiang ZW, Sun H, Zuo J, Huang W, Chen Z, Shen Y, Yao ZJ, Qiang BQ, Fang FD (2002) Single nucleotide polymorphisms in CAPN10 gene of Chinese people and its correlation with type 2 diabetes mellitus in Han people of northern China. Biomed Environ Sci 15:75–82 [PubMed] [Google Scholar]
- Tsai HJ, Sun G, Weeks DE, Kaushal R, Wolujewicz M, McGarvey ST, Tufa J, Viali S, Deka R (2001) Type 2 diabetes and three calpain-10 gene polymorphisms in Samoans: no evidence of association. Am J Hum Genet 69:1236–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacholder S, Rothman N, Caporaso N (2000) Population stratification in epidemiologic studies of common genetic variants and cancer: quantification of bias. J Natl Cancer Inst 92:1151–1158 10.1093/jnci/92.14.1151 [DOI] [PubMed] [Google Scholar]
- Xiang K, Fang Q, Zheng T, Jia W, Wang Y, Zhang R, Li J, Shen K (2001a) [The impact of calpain-10 gene combined-SNP variation on type 2 diabetes mellitus and its related metabolic traits]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 18:426–430 [PubMed] [Google Scholar]
- Xiang K, Zheng T, Fang Q, Jia W, Wang Y, Zhang R, Sheng K, Lee J, Lu J, Lu H (2001b) Calpain-10 SNP43 polymorphism is associated with insulin sensitivity and insulin levels during glucose load in Chinese non-diabetic subjects. Diabetes 50 Suppl:A232 [Google Scholar]