

Abstract
The most frequent causes of death and disability in the Western world are atherosclerotic coronary artery disease (CAD) and acute myocardial infarction (MI). This common disease is thought to have a polygenic basis with a complex interaction with environmental factors. Here, we report results of a genomewide search for susceptibility genes for MI in a well-characterized U.S. cohort consisting of 1,613 individuals in 428 multiplex families with familial premature CAD and MI: 712 with MI, 974 with CAD, and average age of onset of 44.4±9.7 years. Genotyping was performed at the National Heart, Lung, and Blood Institute Mammalian Genotyping Facility through use of 408 markers that span the entire human genome every 10 cM. Linkage analysis was performed with the modified Haseman-Elston regression model through use of the SIBPAL program. Three genomewide scans were conducted: single-point, multipoint, and multipoint performed on of white pedigrees only (92% of the cohort). One novel significant susceptibility locus was detected for MI on chromosomal region 1p34-36, with a multipoint allele-sharing P value of <10−12 (LOD=11.68). Validation by use of a permutation test yielded a pointwise empirical P value of .00011 at this locus, which corresponds to a genomewide significance of P<.05. For the less restrictive phenotype of CAD, no genetic locus was detected, suggesting that CAD and MI may not share all susceptibility genes. The present study thus identifies a novel genetic-susceptibility locus for MI and provides a framework for the ultimate cloning of a gene for the complex disease MI.
Introduction
Atherosclerotic coronary artery disease (CAD) and its most important complication, acute myocardial infarction (MI), are the principal causes of death and disability in the United States and other Western countries (American Heart Association 2002). Recent work has illuminated the pathophysiology that atherosclerotic involvement of the coronary artery, with attendant inflammation of the arterial wall, can ultimately lead, in some patients, to plaque erosion, fissure, or rupture and induction of thrombosis, the clinical syndrome of acute MI (Lusis 2000; Lewis et al. 2002). Multiple risk factors for this disease have been identified (Lewis et al. 2002; Willett 2002)—including hypertension, hypercholesterolemia, obesity, smoking, and diabetes—but the epidemiologic factor that has attracted considerable attention is family history (Nora et al. 1980). Indeed, the relative risk of death from coronary disease for MZ twins was 8.1, compared with 3.8 for DZ twins (Slack and Evans 1966; Marenberg et al. 1994). A genetic-epidemiologic study with a Colorado population of 207 white patients who were affected with MI aged <55 years has suggested that the polygenic heritability of liability for early onset MI is 0.56–0.63 (Nora et al. 1980). For premature MI, on the binary scale, the estimated sample heritability in our ascertained population is 0.31 (data not shown).
Two major approaches have thus far been utilized to identify the susceptibility genes associated with CAD and MI. Current genetic studies of complex CAD and MI are largely focused on population-based case-control studies of candidate genes. Many gene variants have been investigated for their effects on CAD and MI risks. Polymorphisms in genes encoding ApoE, Lp(a) or Apo(a), ApoAI, ApoCIII, ApoIV, t-PA, fibrinogen, PAI-1 (plasminogen activator inhibitor), vWF (von Willebrand factor), platelet glycoprotein IIIa, lipoprotein lipase, cholesterol ester hydrolase, CETP (cholesterol ester transfer protein), FV (factor V), FVII (factor VII), ACE (angiotensin-converting enzyme), AGT (angiotensinogen), endothelial nitric oxide synthase, connexin 37, matrix metalloproteinase-3, and other proteins have been associated in certain populations with high risk of atherosclerosis, CAD, and MI (Wang and Pyeritz 2000; Lewis et al. 2002; Wang and Chen 2003). A recent large-scale genetic association study examined 62 vascular biology genes and 85 novel SNPs, and statistically significant association was found between 3 SNPs in three members of the thrombospondin (TSP) family (TSP-1, TSP-2, and TSP-4) and MI (Topol et al. 2001). Ozaki et al. (2002) carried out a large-scale genomewide case-control association study in a Japanese population with 92,788 SNPs. They found that two functional SNPs in the LTA gene encoding lymphotoxin-α were associated with a high risk of MI (Ozaki et al. 2002). Another study by Yamada et al. (2002) identified association of SNPs in connexin 37, stromelysin-1, and PAI-1 with sporadic MI, on a sex-specific basis in a large Japanese cohort. However, the results from these studies will require independent replication and proof of cause and effect.
A genomewide linkage scan is a comprehensive and unbiased approach for identifying CAD and MI genes (Olson et al. 1999; Wang et al. 2001). It may lead to the identification of novel CAD and MI genes and can define unrecognized genetic pathways for the pathophysiology of coronary atherosclerosis and MI. In 2000, a genomewide linkage scan of 156 families with CAD (364 patients; average sibship size 2.3) from Finland suggested two genetic loci for CAD, one on chromosome 2q21.1-22 (LOD=3.2) and the other on chromosome Xq23-26 (LOD=3.5) (Pajukanta et al. 2000). In 2001, using 535 individuals from 99 families of northeastern Indian origin, Francke et al. (2001) suggested a CAD locus on chromosome 16p13-pter (LOD=3.06; P=.00017). In contrast, only one genomewide linkage scan was carried out for MI. In 2002, Broeckel et al. (2002) finished a total-genome scan of 513 families in the German data set, and they identified a genetic locus for MI on chromosome 14, with a maximum LOD score of 3.9 (P=.00015). To our knowledge, none of these CAD/MI loci have yet been replicated, and the specific genes also remain to be identified.
Premature CAD or MI is known to be the most aggressive form of the disease, affecting men aged <45 years and women aged <50 years. In the current study, we recruited 428 multiplex families with premature CAD and MI (table 1), predominantly American whites, and carried out a genomewide scan to identify novel genetic loci for CAD and MI.
Table 1.
Clinical and Demographic Features of the Study Population
| Feature | Finding |
| Age at onset (mean ± SD) | 44.4 ± 9.7 years |
| Age at exam (mean ± SD) | 54.4 ± 11.3 years |
| No. male/no. female | 1,152/878 |
| Ethnicity: | |
| African American | 2.3% |
| Asian or Pacific Islander | .8% |
| White | 91.7% |
| Hispanic | 1.4% |
| Native American or Alaskan | 1.7% |
| Mixed ethnicity | .8% |
| Unknown ethnicity | 1.3% |
| Pedigree structure: | |
| No. of pedigrees | 428 |
| No. of individuals genotyped | 1,163 |
| Pedigree size (mean ± SD) | 4.7 ± 1.3 |
| No. affected with CAD | 974 |
| No. affected with MI | 712 |
| No. of relative pairs: | |
| Sibling/sibling | 1,303 |
| Sister/sister | 258 |
| Brother/brother | 476 |
| Brother/sister | 569 |
| Half sibling/half sibling | 25 |
Subjects and Methods
Ascertainment of Patients and Families
The study participants have been enrolled in the United States. Four hundred twenty-eight families with premature CAD or MI were recruited in the present study through the Department of Cardiovascular Medicine at the Cleveland Clinic Foundation and 10 other institutions (Topol et al. 2001). The present study has been approved by each participating center’s institutional review board, and informed consent was obtained from all participants.
Each family in the cohort has at least two affected siblings with premature CAD, and the majority of families also have one unaffected sibling. “Premature CAD” was defined as any previous or current evidence of significant atherosclerotic CAD (defined as MI, percutaneous coronary angioplasty [PTCA], coronary artery bypass graft [CABG], or coronary angiography with >70% stenosis) occurring in males at age ⩽45 years or in females at age ⩽50 years. The probands, who had to have a living sibling meeting the same criteria, were selected from patients undergoing coronary angiography or intervention for clinically suspected CAD. Individuals with hypercholesterolemia, insulin-dependent diabetes, childhood hypertension, substance abuse, and congenital heart disease were excluded from this study.
For patients with premature CAD, the distribution of those classified by the diagnostic criteria was 54.9% MI, 15.4% CABG, 14.9% angiography with >70% stenosis, 12.2% PTCA, and 2.6% other causes. It is important to note that a large number of recruited patients with CAD in this cohort had an MI as their initial clinical manifestation. The epidemiological and demographic features of the participants in the study and the features of the pedigrees with multiplex premature CAD are shown in table 1. In total, 974 affected persons were recruited, with an average age at onset of 44.4 years. The epidemiological and demographic features of the affected individuals were similar to those of the unaffected participants. Our cohort was predominantly male (n=1,152) and white (91.7% for all the participants and 93.6% for those affected). Owing to the characteristic of onset at a relatively late age in life, the majority (423 pedigrees) of the recruited multiplex pedigrees with premature CAD consist of only two generations, with an average family size of 4.7 members (with a range of 2–10 members; average sibship size 2.7). The data were derived predominantly from first-degree relative pairs (parent/offspring and sibling/sibling); extended relative pairs (grandparent/grandchild, avuncular, and half sibling) were rare in our cohort.
Clinical phenotypic evaluation of premature CAD for probands and other participants in the study was performed by a panel of cardiologists. In most cases, the diagnosis of premature CAD had already been documented prior to recruitment in the study. The presence or absence of premature CAD manifestations was assessed according to a previous diagnosis of MI (on the basis of the existence of at least two of the following: chest pain of ⩾30-min duration, electrocardiogram [ECG] patterns consistent with acute MI, and significant elevation of cardiac enzymes); history of revascularization procedures, such as angioplasty or coronary artery bypass grafts; or current treatment for angina pectoris. When patients who had not previously received a diagnosis of premature CAD presented with clinical symptoms suggestive of ischemic heart disease and/or ECG patterns suggestive of myocardial ischemia or of old MI, they were referred to a cardiologist who either confirmed or rejected the diagnosis. For linkage analysis of MI, we defined the patients without MI as “unaffected” if they were men aged >45 years or women aged >50 years. Individuals without MI who were younger than these sex-specific age cutoffs were classified as “uncertain” and were excluded from the analysis. Three pairs of MZ twins were found in the GeneQuest population and were also excluded from statistical analysis.
Isolation of Human Genomic DNA and Genotyping
Genomic DNA was prepared from whole blood with the Puregene Kits (Gentra). Genotyping was performed by the National Heart, Lung, and Blood Institute (NHLBI) Mammalian Genotyping Service (directed by Dr. James L. Weber), Center for Medical Genetics, by use of the Screening Set 11, with 408 markers that span the entire human genome at every 10 cM (Weber and Broman 2001). DNA samples were genotyped for 1,163 well-characterized participants from 428 families.
Statistical Analysis
Prior to linkage scanning, obvious pedigree errors, data errors, genotyping errors, and locus-order errors that commonly occur with a large-scale linkage analysis were corrected. Allele frequencies for all the markers in the cohort were estimated by maximum likelihood methods using the SAGE program FREQ (SAGE 2003). Pedigree relationships were checked using RELTEST, which employs a Markov process model of allele sharing along the chromosome and classifies pairs of pedigree members according to their true relationship by use of genome-scan data (Olson 1999). Pedigree errors were found in 58 pedigrees, and 27 pedigrees with uncorrectable errors were excluded from the linkage analysis. After relationships were corrected, the SAGE program MARKERINFO was used to detect any Mendelian inheritance inconsistencies. Three additional pedigrees with a high incidence of inheritance errors were eliminated from the study. A genomewide linkage analysis was performed using allele sharing identical by descent, as estimated by the SAGE program GENIBD. Two covariates (sex and race) deemed to be important for cardiovascular diseases were modeled in this linkage analysis.
Linkage analysis of the genotyping data was performed with two of the modified Haseman-Elston regression models (Haseman and Elston 1972), as implemented in the SAGE program SIBPAL, using data from 428 multiplex families with premature CAD. We took the dependent variable to either the centered cross-product (Elston et al. 2000) or a weighted combination of squared trait difference and squared mean-corrected trait sum (Shete et al. 2003), rather than the original squared trait difference in the Haseman-Elston regression. We treated the binary MI phenotype as quantitative, without loss of generality, giving the phenotypes “affected” and “unaffected” different quantitative scores, 2 and 1, respectively. Each chromosome was analyzed separately. A statistical test of linkage (t test) compares the estimate of the regression parameter (β) with 0, its value under the null hypothesis of no linkage. Three different genomewide scans (single-point linkage analysis, multipoint linkage analysis, and multipoint analysis of white pedigrees only) were conducted.
Asymptotic P values were converted to asymptotic pointwise LOD scores for significant linkage regions identified by the modified Haseman-Elston regression. On the assumption that the Haseman-Elston regression t statistic follows a normal distribution, the P value from the Haseman-Elston regression analysis was converted to the corresponding χ2 statistic, with 1 df, and divided by a constant (4.6) to transform to an asymptotic LOD score.
Because the t test is valid only asymptotically, permutation tests were performed, as implemented in the SAGE program SIBPAL. We performed up to 100,000 permutations to obtain empirical pointwise P values. For all nominal P values (<.05), we did sufficient permutations to have 90% confidence that the estimated value would be within 20% of the true value.
Results
We considered any genetic region with a −log10 P value (pP) (Schick et al. 2003; Slager et al. 2003) >3.13 (LOD>2.2) as “potentially interesting,” which corresponds to suggestive linkage by the criteria of Lander and Kruglyak (1995). Potentially interesting linkage results of the three separate genomewide linkage scans for MI are shown in table 2. The three separate analyses are single-point Haseman-Elston linkage analysis with the centered cross-product as the dependent variable, multipoint analysis with the centered cross-product as the dependent variable, and multipoint analysis with a weighted combination of squared trait difference and squared mean-corrected trait sum as the dependent variables (W2 option, SAGE), for which weights are chosen proportional to the inverses of the residual variances of regressing the squared differences and sums. Throughout the whole genome, 25 chromosomal regions generated potentially interesting results in one or more linkage scans (fig. 1). No evidence for linkage was found for any marker on chromosomes 6, 8, 15, 17–19, and 22. The three linkage analyses were generally in good agreement and identified multiple chromosomal regions showing highly significant and/or significant asymptotic P values (pP=4.66; LOD>3.6). For the multipoint analysis with the centered cross-product, the linkage regions include chromosomes 1p34-36 (pP>12; LOD=11.68), 2p11 (pP=4.85; LOD=3.82), 4q32 (pP=6.17; LOD=5.08), 5p14 (pP=5.35; LOD=4.32), 7q22 (pP=4.77; LOD=3.74), 12q24 (pP=6.72; LOD=5.62), 13q32 (pP=4.89; LOD=3.86), and 14q24 (pP=4.68; LOD=3.64). Six regions were replicated using W2 and by single-point linkage analyses (but the highest peaks might be on the nearby markers) (table 2). It is important to note that the multipoint identity-by-descent analysis revealed that the genomewide highest peak at 1p34-36 spans a 32-cM region, with P values for four markers exceeding the significant linkage level (pP=6.52; LOD=5.4) (fig. 2).
Table 2.
Summary of Regions Linked to MI (Asymptotic −log10 [P] or pP Values)
|
Linkage in |
||||||
| All Pedigrees |
||||||
| Chromosomeand Marker | Location | Map Position(cM) | Single-PointProduct | MultipointProduct | MultipointW2a | White Pedigrees Only,Multipoint Product |
| 1: | ||||||
| ATA47D07 | 1p36+3 | 46.6 | >12.00 | >12.00 | 11.70 | >12.00 |
| GATA124C08 | 1p21+3 | 129.4 | 3.29 | 1.85 | 1.59 | 1.72 |
| 2: | ||||||
| GATA88G05 | 2p11+2 | 103.0 | 1.82 | 4.85 | 4.66 | 4.33 |
| 3: | ||||||
| GATA131D09 | 3p24.3 | 19.3 | 4.24 | 1.64 | .89 | .92 |
| 4: | ||||||
| 4PTEL04 | 4p16 | .0 | 5.30 | 4.19 | 3.10 | 2.55 |
| GATA8A05 | 4q31+1 | 158.0 | 5.01 | 4.41 | 3.68 | 4.41 |
| GGAA19H07 | 4q32 | 176.2 | 4.55 | 6.17 | 5.05 | 5.48 |
| 5: | ||||||
| GATA84E11 | 5p15+1 | 14.3 | 6.74 | 2.28 | 1.46 | 2.39 |
| GATA134B03 | 5p14 | 36.0 | 4.57 | 5.35 | 4.74 | 4.77 |
| GATA21D04 | 5p12 | 59.0 | 5.33 | 2.85 | 2.15 | 2.03 |
| GATA52A12 | 5q14 | 85.0 | 4.64 | 2.42 | 3.18 | 2.39 |
| GATA62A04 | 5q22 | 130.0 | 3.60 | 3.09 | 2.85 | 3.40 |
| 7: | ||||||
| GATA23F05 | 7q22 | 114.0 | 9.37 | 4.77 | 4.48 | 4.62 |
| 9: | ||||||
| SNP9558 | 9p21 | 33.1 | 4.28 | 3.68 | 3.59 | 3.96 |
| 10: | ||||||
| ATCC001 | 10p14 | 17.9 | 3.96 | 1.77 | 1.74 | 1.70 |
| GATA121A08 | 10q21 | 88.0 | 3.82 | .85 | .82 | .85 |
| 11: | ||||||
| GATA63F09 | 11q12 | 58.0 | 3.40 | 1.21 | 1.12 | 1.24 |
| GATA30G01 | 11q13 | 85.0 | 1.12 | 4.17 | 4.07 | 4.34 |
| GATA64D03 | 11q23 | 123.0 | 3.96 | 3.35 | 3.19 | 3.31 |
| 12: | ||||||
| ATA29A06 | 12q24+3 | 161.0 | 7.26 | 6.72 | 5.77 | 5.52 |
| 13: | ||||||
| GATA51B02 | 13q32 | 94.0 | 3.82 | 4.89 | 4.55 | 4.64 |
| 14: | ||||||
| ATA19H08 | 14q22 | 67.0 | 3.80 | 4.68 | 5.29 | 5.30 |
| SNP672053 | 14q24.1 | 86.9 | 5.19 | 4.06 | 4.04 | 4.04 |
| 16: | ||||||
| GGAA3G05 | 16q11+2 | 58.0 | 4.15 | 1.29 | 1.26 | 1.31 |
| 20: | ||||||
| GATA51D03 | 20p12 | 12.0 | 3.54 | 3.20 | 3.00 | 3.18 |
| 21: | ||||||
| GATA70B08 | 21q22 | 57.8 | 3.33 | 2.68 | 2.27 | 2.64 |
W2, weighted square trait sum and difference (W2 option [SAGE 2003]), for which weights are chosen proportional to the inverses of the residual variances of the regression equations for the squared differences and sums.
Figure 1.

Haseman-Elston sib-pair regression analysis for scanning loci segregating with MI. The vertical axis of each plot is -log(P) or pP, where the P is the significance level from each of two analyses. The solid line indicates the multipoint linkage profile for all pedigrees; the dashed line indicates the multipoint linkage profile for white pedigrees only. The horizontal solid line in each subfigure indicates P=2.2×10-5 (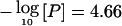 ; LOD=3.6). The X-axis denotes marker map positions. Note that the dashed line (P values for white pedigrees only) is often not visible because it frequently overlaps with the solid line (P values for all study families).
; LOD=3.6). The X-axis denotes marker map positions. Note that the dashed line (P values for white pedigrees only) is often not visible because it frequently overlaps with the solid line (P values for all study families).
Figure 2.

Identification of a novel genetic linkage for MI on chromosome 1p34-36. The vertical axis represents 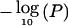 or pP, and the horizontal axis denotes the distance (cM) for markers along the chromosome. The results were obtained by multipoint linkage analysis of 2-cM intervals, through use of the SAGE package. The asymptotic P values were quoted accurately to only 12 decimal places, resulting in a flat pP peak.
or pP, and the horizontal axis denotes the distance (cM) for markers along the chromosome. The results were obtained by multipoint linkage analysis of 2-cM intervals, through use of the SAGE package. The asymptotic P values were quoted accurately to only 12 decimal places, resulting in a flat pP peak.
Three genomewide scan studies have been performed elsewhere for CAD, and suggestive susceptibility loci were identified on chromosomes 2q21.1-22 and Xq23-26 in a Finnish population (Pajukanta et al. 2000), on chromosome 16p13-pter in a population with northeastern Indian origin (Francke et al. 2001), and on chromosome 2q36-37.3 in an Australian population (Harrap et al. 2002). However, for a less-restrictive phenotype of CAD, no suggestive or significant linkage was identified in the population studied here.
Population admixture is well known to be a thorny issue in linkage analysis of complex human diseases. In previous linkage scans, an attempt was made to remove its interference with linkage assessment by modeling race as a covariate in the Haseman-Elston regression. Alternatively, we might consider analyzing separately the data from the different races. Since our genotyping population is predominantly (>90%) of white origin, we can perform an analysis of white pedigrees only with marginal loss of available sibling pairs. The linkage profiles from the genomewide scans with all pedigrees and with white pedigrees only are presented in figure 1. The two linkage profiles, as demonstrated in figure 1, are very similar, strongly supporting the hypothesis that the linkage evidence on multiple regions is largely from the white pedigrees. The results for the eight chromosome regions with suggestive or significant asymptotic P values identified using the white pedigrees are: 1p34-36 (pP>12; LOD=11.15), 2p11 (pP=4.33; LOD=3.30), 4q32 (pP=5.48; LOD=4.41), 5p14 (pP=4.77; LOD=3.73), 7q22 (pP=4.62; LOD=3.59), 12q24 (pP=5.52; LOD=4.44), 13q32 (pP=4.64; LOD=3.61), and 14q24 (pP=5.30 or LOD=4.24) (also see table 2).
The P values described above were asymptotic P values. To estimate the significance level of the linkage results, we obtained empirical P values using a permutation test incorporated into the SAGE program SIBPAL. The permutation test results for all families or for white families only are shown in figure 3. A pointwise empirical P value of .00011 was obtained for the chromosome 1p34-36 MI locus. To assess the significance level of a genomewide analysis, we evaluated the linkage results using the criteria given by both Lander and Kruglyak (1995) and Wiltshire et al. (2002). For a genomewide scan with a 10-cM marker map and 15% missing genotypes, our permutation test linkage peak on chromosome 1p34-36 corresponds to a genomewide probability of P=.030–.038 (Wiltshire et al. 2002) for the experiment settings that match closely with ours. The empirical pointwise P values for the other seven MI loci are .0026 for chromosome 12 locus, .01 for chromosome 4 locus, and <.05 for chromosomes 2, 5, 7, 13, and 14 loci. Although these empirical pointwise P values are all <.05, they are too high to reach the genomewide significance of .05. These results indicate that the strongest linkage to MI in the population studied here is on chromosome 1p34-36. In general, the empirical permutation analysis yielded P values of lesser statistical significance than the asymptotic analysis, which may be attributed to the limited sample size, since the asymptotic sib-pair regression t statistics requires a large sample size. Nevertheless, the linkage profiles and the associated linkage peaks remain largely unchanged (compare fig. 1 and fig. 3).
Figure 3.

Evaluation of linkage results (asymptotic P value) by use of permutation tests. Up to 100,000 permutations were performed to obtain the empirical P value at each locus by use of the SAGE program SIBPAL. The vertical axis represents 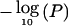 or pP, and the horizontal axis denotes the genetic distance (cM) for each chromosome. The solid curve indicates the empirical linkage profile for all pedigrees; the dashed line denotes the empirical linkage profile for white pedigrees only.
or pP, and the horizontal axis denotes the genetic distance (cM) for each chromosome. The solid curve indicates the empirical linkage profile for all pedigrees; the dashed line denotes the empirical linkage profile for white pedigrees only.
Discussion
To our knowledge, this study represents the first genomewide scan for genetic loci causing susceptibility to premature MI in a white American population. Our study provides strong evidence for a novel disease-susceptibility locus for MI on chromosome 1p34-36. Model-free multipoint linkage analysis revealed a plateau of genomewide significance in a 32-cM region between markers GATA27E01 (D1S1597) and ATA79C10. The permutation tests indicated an empirical pointwise P value of .00011 (genomewide P<.05 for a genomewide scan with a 10-cM marker map and 15% missing genotypes), validating the identification of the significant linkage to MI on chromosome 1p34-36.
The MI locus on 1p34-36 contains a strong candidate gene, connexin 37 (CX37). Connexin 37 is a gap-junction protein expressed in the arterial endothelium, including the human coronary artery (Yeh et al. 1998, 2000). Connexin 37 is required for vascular integrity, growth, regeneration after injury, and aging of endothelial cells, which is potentially important for the pathogenesis of vascular diseases, including CAD and MI (Yeh et al. 1998, 2000). The C allele of the SNP P319S (1019C/T) was associated elsewhere with thickening of the carotid intima in Swedish men (Boerma et al. 1999). The same allele was also associated with CAD in a Taiwanese population (Yeh et al. 2001). In a large case-control association study in a Japanese population, the T allele of CX37 SNP P319S was instead found to be associated with an increased risk for MI in men (Yamada et al. 2002). The colocalization of CX37 with the 1p34-36 MI susceptibility locus, along with positive associations between them, reported elsewhere, with CAD and MI in other populations, implicates this gene as a candidate gene for MI in the population we are studying (Yeh et al. 1998, 2000; Yamada et al. 2002).
Our successful identification of the chromosome 1p34-36 MI locus might be due to the unique selection strategy used in our study, which focused on clustering in families with well-diagnosed premature CAD and MI, and preempted common risk factors, such as hypercholesterolemia and insulin-dependent diabetes. Furthermore, compared with other reported genomewide scans in CAD and MI, our patient population has the youngest age at onset, <45 years in males and <50 years in females, which is expected to significantly increase the genetic component involved in these diseases.
Our successful identification of a susceptibility locus for MI may also reflect the strength of our analysis with a more stringent phenotype. If a broader definition of a disease like CAD—in which individuals with CAD or with MI were all classified as “affected”—were used, it may lead to a higher genetic heterogeneity and may generate many phenocopies. That might explain why no single significant linkage for CAD was detected. The importance of analysis with a more stringently defined phenotype has recently been demonstrated in a genomewide screen for migraine, in which a locus with significant linkage on 4q was detected when migraine with aura was defined as “affected,” but the linkage disappeared when a broader phenotyping—in which individuals with migraine with or without aura were all classified as “affected”—was used (Wessman et al. 2002). In another genomewide scan, of 460 white families, that identified ADAM33 to be a critical susceptibility gene for asthma, the initial linkage with a broader phenotype of asthma revealed only the 20p13 region to have a suggestive linkage, but the use of a more stringent phenotype (asthma plus bronchial hyperresponsiveness) increased the linkage to the genomewide significance level (Van Eerdewegh et al. 2002).
It had been anticipated that CAD and acute MI represent a continuum. However, our findings with MI, in contrast to the much less restrictive phenotype of CAD, are particularly noteworthy. Atherosclerotic coronary disease is nearly endemic in the Western world, but only a limited number of patients with this underlying condition progress to having an acute MI. A recent study of 65 teenagers and young adults (average age 25 years) who died in motor vehicle accidents demonstrated a nearly universal presence of coronary atheroma (Bertomeu et al. 2003). In the cohort of the present study, there were considerably fewer individuals who sustained MI, compared with those who had manifested coronary disease. This reflects the pivotal subgroup of patients with the disease who have a propensity to go beyond the development of coronary atheroma. In our high-throughput study of this population showing the association of TSP1, TSP2, and TSP4 SNPs with premature MI, we have noted that the level of statistical significance for coronary disease was considerably reduced (Topol et al. 2001). Similarly, the only successful high-throughput association studies in this field to date have reported only for MI and not for CAD (Ozaki et al. 2002; Yamada et al. 2002). All these results together may explain why, in our large-scale genomewide scans of >400 families with premature CAD and MI, no single significant linkage was detected for CAD, whereas a significant disease-susceptibility locus for MI was detected. Our findings suggest that the pathogenesis of CAD and MI may involve different sets of susceptibility genes and that MI may be considered a distinct, more restrictive phenotype.
Only one significant linkage region of 7 cM (123-130 cM on 14q, 1 LOD-unit support interval) for MI was reported in a German population with a mean age at onset 7 years older than that of the current cohort, without exclusion for insulin-dependent diabetes mellitus or hypercholesterolemia (Broeckel et al. 2002). This region does not overlap with the region of 50 cM on chromosome 14 (58–108 cM; 95% CI) with a significant asymptotic P value in this study. The ethnicity, age-at-onset, and concomitant risk-factor differences may account for the discrepancy between the results of our study and of the previous ones.
Although our genomewide scan revealed seven other chromosomal regions showing significant asymptotic multipoint P values (2p11, 4q32, 5p15, 7q22, 12q24, 13q32, and 14q24), later permutation tests generated empirical pointwise P values that were all <.05 but that were too high to reach a genomewide significance level of .05. Precautions are clearly needed for interpreting the significance levels on the basis of the asymptotic distributions often assumed in linkage analysis, especially with the new forms of Haseman-Elston regression testing. An advantage of using a permutation test is that it is based solely on the data that were observed and so serves as a stringent empirical strategy to cross-validate the asymptotic linkage results.
In conclusion, the present study identified one novel significant susceptibility locus for MI on 1p34-36; this should facilitate the identification of an underlying major (or minor) gene for complex disease MI. Identification of the MI gene(s) should uncover the molecular mechanism for the pathogenesis of MI and would ultimately provide a basis for improving prevention and medical management.
Acknowledgments
We thank J. Weber and the NHLBI Mammalian Genotyping Service, for genotyping; Edward Plow, from the Cleveland Clinic; JoAnne Meyer, Alex Parker, and Goeff Ginsburg, from Millennium, for their input; and other members of Wang Laboratory (S. Chen, A. Timur, C. Fan, S. You, L. Wu, M. Liu, R. Kadaba, and L. Wang), for preparing DNA samples for genotyping. This study was supported by the Cardiovascular Genetics Funds from the Cleveland Clinic Department of Cardiovascular Medicine, a Doris Duke Innovation in Clinical Research Award (to Q.W. and E.J.T.), and by U.S. Public Health Service resource grant RR03655 from the National Center for Research Resources and by research grant GM28356 from the National Institute of General Medicine Sciences (support to R.C.E.). Q.W. is also supported by National Institutes of Health grants R01 HL65630 and R01 HL66251. We thank the following participants in this study: Cleveland Clinic Foundation: David J. Moliterno, Gurunathan Murugesan, Patricia Welsh, and Monique Rosenthal; Emory University Hospital, Atlanta: Spencer B. King III, William Anderson, Joe Jean Borowski, and Kris Anderberg; Mayo Clinic, Rochester, MN: David R. Holmes, Jr., Charanjit Rihal, and Sharon McIntire-Langworthy; University of Alabama Medical Center, Birmingham: Ann Snider; Duke University Medical Center, Durham, NC: Laura Drew; the Lindner Center for Clinical Cardiovascular Research, Cincinnati: Dean Kereiakes, Eli Roth, and Louise Wohlford; LeBauer Cardiovascular Research Foundation, Greensboro, NC: Anthony De Franco and Teresa Schrader; St. Joseph Hospital, Savannah: Phillip Gainey and Sandra Arsenault; Lancaster Heart Foundation, Lancaster, PA: Paul Casale and Joann Tuzi; Latter Day Saints Hospital, Salt Lake City: Jeffrey Anderson, Juli Jerman, Rob Pearson, and Ann Allen; Diabetes and Glandular Associates, San Antonio: Sherwyn Schwartz and Sue Beasie; St. Louis University Hospital, St. Louis: Frank Aguirre, Sandra Aubuchon, and Kristin Weisbrod; the Heart Group, Saginaw: Jeffrey Carney and Muriel Harris; Michigan Heart and Vascular Institute, Ypsilanti: Jim Bengtson and Mary Adolphson; Oregon Cardiology Clinic, Portland: John Rudoff and Sue Williams.
Electronic-Database Information
The URL for data presented herein is as follows:
- Center for Medical Genetics, http://research.marshfieldclinic.org/genetics/ (for Marshfield Genetic Database marker information)
References
- American Heart Association (2002) Heart disease and stroke statistics: 2003 update. American Heart Association, Dallas [Google Scholar]
- Bertomeu A, Garcia-Vidal O, Farre X, Galobart A, Vazquez M, Laguna JC, Ros E (2003) Preclinical coronary atherosclerosis in a population with low incidence of myocardial infarction: cross sectional autopsy study. BMJ 327:591–592 10.1136/bmj.327.7415.591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boerma M, Forsberg L, Van Zeijl L, Morgenstern R, De Faire U, Lemne C, Erlinge D, Thulin T, Hong Y, Cotgreave IA (1999) A genetic polymorphism in connexin 37 as a prognostic marker for atherosclerotic plaque development. J Intern Med 246:211–218 10.1046/j.1365-2796.1999.00564.x [DOI] [PubMed] [Google Scholar]
- Broeckel U, Hengstenberg C, Mayer B, Holmer S, Martin LJ, Comuzzie AG, Blangero J, Nurnberg P, Reis A, Riegger GA, Jacob HJ, Schunkert H (2002) A comprehensive linkage analysis for myocardial infarction and its related risk factors. Nat Genet 30:210–214 10.1038/ng827 [DOI] [PubMed] [Google Scholar]
- Elston RC, Buxbaum S, Jacobs KB, Olson JM (2000) Haseman and Elston revisited. Genet Epidemiol 19:1–17 [DOI] [PubMed] [Google Scholar]
- Francke S, Manraj M, Lacquemant C, Lecoeur C, Lepretre F, Passa P, Hebe A, Corset L, Yan SL, Lahmidi S, Jankee S, Gunness TK, Ramjuttun US, Balgobin V, Dina C, Froguel P (2001) A genome-wide scan for coronary heart disease suggests in Indo-Mauritians a susceptibility locus on chromosome 16p13 and replicates linkage with the metabolic syndrome on 3q27. Hum Mol Genet 10:2751–2765 10.1093/hmg/10.24.2751 [DOI] [PubMed] [Google Scholar]
- Harrap SB, Zammit KS, Wong ZY, Williams FM, Bahlo M, Tonkin AM, Anderson ST (2002) Genome-wide linkage analysis of the acute coronary syndrome suggests a locus on chromosome 2. Arterioscler Thromb Vasc Biol 22:874–878 10.1161/01.ATV.0000016258.40568.F1 [DOI] [PubMed] [Google Scholar]
- Haseman JK, Elston RC (1972) The investigation of linkage between a quantitative trait and a marker locus. Behav Genet 2:3–19 [DOI] [PubMed] [Google Scholar]
- Lander E, Kruglyak L (1995) Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11:241–247 [DOI] [PubMed] [Google Scholar]
- Lewis D, Wang Q, Topol EJ (2002) Ischaemic heart disease. Encyclopedia of Life Sciences 10:508–515 [Google Scholar]
- Lusis AJ (2000) Atherosclerosis. Nature 407:233–241 10.1038/35025203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marenberg ME, Risch N, Berkman LF, Floderus B, De Faire U (1994) Genetic susceptibility to death from coronary heart disease in a study of twins. N Engl J Med 330:1041–1046 10.1056/NEJM199404143301503 [DOI] [PubMed] [Google Scholar]
- Nora JJ, Lortscher RH, Spangler RD, Nora AH, Kimberling WJ (1980) Genetic-epidemiologic study of early-onset ischemic heart disease. Circulation 61:503–508 [DOI] [PubMed] [Google Scholar]
- Olson JM (1999) Relationship estimation by Markov-process models in a sib-pair linkage study. Am J Hum Genet 64:1464–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson JM, Witte JS, Elston RC (1999) Genetic mapping of complex traits. Stat Med 18:2961-2981 [DOI] [PubMed] [Google Scholar]
- Ozaki K, Ohnishi Y, Iida A, Sekine A, Yamada R, Tsunoda T, Sato H, Sato H, Hori M, Nakamura Y, Tanaka T (2002) Functional SNPs in the lymphotoxin-α gene that are associated with susceptibility to myocardial infarction. Nat Genet 32:650–654 10.1038/ng1047 [DOI] [PubMed] [Google Scholar]
- Pajukanta P, Cargill M, Viitanen L, Nuotio I, Kareinen A, Perola M, Terwilliger JD, Kempas E, Daly M, Lilja H, Rioux JD, Brettin T, Viikari JSA, Rönnemaa T, Laakso M, Lander ES, Peltonen L (2000) Two loci on chromosomes 2 and X for premature coronary heart disease identified in early- and late-settlement populations of Finland. Am J Hum Genet 67:1481–1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAGE (2003) Statistical analysis for genetic epidemiology. Statistical Solutions, Cork, Ireland [Google Scholar]
- Schick JH, Iyengar SK, Klein BE, Klein R, Reading K, Liptak R, Millard C, Lee KE, Tomany SC, Moore EL, Fijal BA, Elston RC (2003) A whole-genome screen of a quantitative trait of age-related maculopathy in sibships from the Beaver Dam Eye Study. Am J Hum Genet 72:1412–1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shete S, Jacobs KB, Elston RC (2003) Adding further power to the Haseman and Elston method for detecting linkage in larger sibships: weighting sums and differences. Hum Hered 55:79–85 10.1159/000072312 [DOI] [PubMed] [Google Scholar]
- Slack J, Evans KA (1966) The increased risk of death from ischaemic heart disease in first degree relatives of 121 men and 96 women with ischaemic heart disease. J Med Genet 3:239–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slager SL, Schaid DJ, Cunningham JM, McDonnell SK, Marks AF, Peterson BJ, Hebbring SJ, Anderson S, French AJ, Thibodeau SN (2003) Confirmation of linkage of prostate cancer aggressiveness with chromosome 19q. Am J Hum Genet 72:759–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topol EJ, McCarthy J, Gabriel S, Moliterno DJ, Rogers WJ, Newby LK, Freedman M, Metivier J, Cannata R, O'Donnell CJ, Kottke-Marchant K, Murugesan G, Plow EF, Stenina O, Daley GQ (2001) Single nucleotide polymorphisms in multiple novel thrombospondin genes may be associated with familial premature myocardial infarction. Circulation 104:2641–2644 [DOI] [PubMed] [Google Scholar]
- Van Eerdewegh P, Little RD, Dupuis J, Del Mastro RG, Falls K, Simon J, Torrey D, et al (2002) Association of the ADAM33 gene with asthma and bronchial hyperresponsiveness. Nature 418:426–430 10.1038/nature00878 [DOI] [PubMed] [Google Scholar]
- Wang Q, Bond M, Elston RC, Tian X (2001) Molecular genetics. In: Topol EJ (ed) Textbook of cardiovascular medicine, 2nd ed. Lippincott, Williams & Wilkins, Philadelphia [Google Scholar]
- Wang Q, Chen Q (2003) Cardiovascular disease and congenital heart defects. Nature Encyclopedia of Human Genome 1:396–411 [Google Scholar]
- Wang Q, Pyeritz RE (2000) Molecular genetics of cardiovascular disease. In: Topol EJ (ed) Textbook of cardiovascular medicine, 1st ed. Lippincott, Williams & Wilkins, New York, pp 1–12 [Google Scholar]
- Weber JL, Broman KW (2001) Genotyping for human whole-genome scans: past, present, and future. Adv Genet 42:77–96 [DOI] [PubMed] [Google Scholar]
- Wessman M, Kallela M, Kaunisto MA, Marttila P, Sobel E, Hartiala J, Oswell G, Leal SM, Papp JC, Hämäläinen E, Broas P, Joslyn G, Hovatta I, Hiekkalinna T, Kaprio J, Ott J, Cantor RM, Zwart J-A, Ilmavirta M, Havanka H, Färkkilä M, Peltonen L, Palotie A (2002) A susceptibility locus for migraine with aura, on chromosome 4q24. Am J Hum Genet 70:652–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett WC (2002) Balancing life-style and genomics research for disease prevention. Science 296:695–698 10.1126/science.1071055 [DOI] [PubMed] [Google Scholar]
- Wiltshire S, Cardon LR, McCarthy MI (2002) Evaluating the results of genomewide linkage scans of complex traits by locus counting. Am J Hum Genet 71:1175–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Y, Izawa H, Ichihara S, Takatsu F, Ishihara H, Hirayama H, Sone T, Tanaka M, Yokota M (2002) Prediction of the risk of myocardial infarction from polymorphisms in candidate genes. N Engl J Med 347:1916–1923 10.1056/NEJMoa021445 [DOI] [PubMed] [Google Scholar]
- Yeh HI, Chou Y, Liu HF, Chang SC, Tsai CH (2001) Connexin37 gene polymorphism and coronary artery disease in Taiwan. Int J Cardiol 81:251–255 10.1016/S0167-5273(01)00574-5 [DOI] [PubMed] [Google Scholar]
- Yeh HI, Lai YJ, Chang HM, Ko YS, Severs NJ, Tsai CH (2000) Multiple connexin expression in regenerating arterial endothelial gap junctions. Arterioscler Thromb Vasc Biol 20:1753–1762 [DOI] [PubMed] [Google Scholar]
- Yeh HI, Rothery S, Dupont E, Coppen SR, Severs NJ (1998) Individual gap junction plaques contain multiple connexins in arterial endothelium. Circ Res 83:1248–1263 [DOI] [PubMed] [Google Scholar]