

Abstract
Although several genomewide scans have identified quantitative-trait loci influencing several obesity-related traits in humans, genes influencing normal variation in obesity phenotypes have not yet been identified. We therefore performed a genome scan of body mass index (BMI) on Mexican Americans, a population prone to obesity and diabetes, using a variance-components linkage analysis to identify loci that influence BMI. We used phenotypic data from 430 individuals (26% diabetics, 59% females, mean age ± SD = 43 ± 17 years, mean BMI ± SD = 30.0 ± 6.7, mean leptin (ng/ml) ± SD = 22.1 ± 17.1) distributed across 27 low-income Mexican American pedigrees who participated in the San Antonio Family Diabetes Study (SAFDS) for whom a 10–15-cM map is available. In this genomewide search, after accounting for the covariate effects of age, sex, diabetes, and leptin, we identified a genetic region exhibiting the most highly significant evidence for linkage (LOD 4.5) with BMI on chromosome 4p (4p15.1) at 42 cM, near marker D4S2912. This linkage result has been confirmed in an independent linkage study of severe obesity in Utah pedigrees. Two strong positional candidates, the human peroxisome proliferator-activated receptor gamma coactivator 1 (PPARGC1) and cholecystokinin A receptor (CCKAR) with major roles in the development of obesity, are located in this region. In conclusion, we identified a major genetic locus influencing BMI on chromosome 4p in Mexican Americans.
Introduction
Obesity is a complex phenotype resulting from the interaction of both genetic and environmental factors. Obesity is a risk factor for diseases such as type 2 diabetes (T2D), hypertension, and coronary heart disease in all populations, particularly in minority populations in the United States, such as Mexican Americans. Epidemiological studies have indicated that Mexican Americans are prone to complex diseases such as obesity, T2D, dyslipidemia, gallbladder disease, and other metabolic complications (Diehl and Stern 1989; Mitchell et al. 1990; Stern et al. 1990). The Mexican American population is also characterized by increased adiposity and a more centralized distribution of body fat (Joos et al. 1984; Haffner et al. 1986), and it has an increased prevalence of T2D, compared with that among non-Hispanic whites (Stern and Haffner 1990; Haffner et al. 1991).
Several studies have documented that genetic factors are involved in the development of obesity, and ∼30%–40% of normal variation is attributable to genetic factors (Bouchard 1997; Barsh et al. 2000; Comuzzie et al. 2001). Epidemiological studies involving twins, adoptees, and families showed that heritability of BMI or body fat varies from ∼25% in adoption studies to a high of 70% in twin studies, whereas estimates are ∼40% in family studies (Bouchard 1997; Rice et al. 1997; Borecki et al. 1998; Comuzzie and Allison 1998; Hager et al. 1998). Thus, genetic factors play a significant role in establishing an individual’s predisposition to developing obesity, whereas environmental and psychological factors contribute significantly to the expression of the obesity phenotype as well (Comuzzie and Allison 1998; Barsh et al. 2000; Kaplan 2000; Comuzzie et al. 2001).
Given the complexity of the obesity phenotype, it has been reported that several important obesity genes may be involved in the regulation of human adipose tissue function and its distribution. Several genes responsible for rare forms of monogenic obesity have been identified in animal models, but only a few of these genes may cause rare forms of monogenic obesity in humans (Barsh et al. 2000). However, despite the fact that knowledge of the genetics of obesity has increased substantially over the past few years, genetic control of common forms of human obesity is poorly understood. To date, on the basis of candidate genes studies and genomewide linkage studies, a number of genes and/or chromosomal regions have been reported to be linked or associated with human obesity phenotypes, but causative genes have not yet been identified (Chagnon et al. 2003).
BMI is a commonly used measure of obesity, and it exhibits a complex relationship with T2D and other measures of obesity (e.g., leptin). In view of these complex relationships and interactions between measures of obesity (BMI), leptin, and diabetes, it is hard to identify the trait-specific genes influencing various obesity phenotypes because these phenotypes are under the influence, not only of trait-specific genes, but shared genes (pleiotropic genes) as well (Mahaney et al. 1995; Arya et al. 2001b). One way to identify the trait-specific genes is to adjust the given phenotype for the effects of other correlated phenotypes and use the adjusted phenotype in the genetic analysis. Thus, in this study we scanned the genome and performed multipoint linkage analyses to identify the susceptibility loci influencing variation in common forms of obesity represented by the adjusted BMI phenotype in Mexican Americans.
Subjects and Methods
San Antonio Family Diabetes Study
Data were obtained from low-income Mexican American families living in San Antonio, TX, whose members were participants in the San Antonio Family Diabetes Study (SAFDS). Probands for the SAFDS were subjects with T2D identified in a prior epidemiological survey (Stern et al. 1989, 1993). Family members included all first-, second-, and third-degree relatives of probands aged ⩾18 years. In this study, we used 430 individuals from the 27 largest pedigrees for whom phenotypic and genotypic data were available. An ∼10-cM map was used for the multipoint linkage analyses. The Institutional Review Board of the University of Texas Health Science Center at San Antonio approved all procedures, and all subjects gave written informed consent.
Phenotypes
BMI was calculated as weight (in kilograms) divided by height squared (in meters). Leptin levels were measured by a commercial radioimmunoassay (Ma et al. 1996). T2D was diagnosed according to the WHO plasma glucose criteria (World Health Organization 1999). Individuals who reported a history of diabetes and stated that they were receiving insulin or oral antidiabetic agents were also considered to have diabetes. Since SAFDS families were ascertained on type 2 diabetic probands, we corrected for ascertainment bias by conditioning on likelihood of observing the BMI of the proband with T2D in all analyses. In this study, we conducted three types of analyses that differed by the number of covariates included in a given model. The first analysis (BMI1) included age and sex terms as covariates. The second analysis (BMI2) included age and sex terms, and T2D as covariates, and the third analysis (BMI3) included age, sex terms, T2D, and leptin as covariates.
Genotyping of Markers
A total of 439 individuals from the 27 largest SAFDS pedigrees were initially selected for genotyping. DNA was extracted from white blood cells by use of proteinase K digestion/phenol extraction and alcohol precipitation in a semiautomated fashion on an ABI 341 RNA/DNA extractor. Genotypic data were collected mainly by PCR assays with radiolabeled oligonucleotide primers, whereas data for some of the markers were collected using fluorescent dye-labeled primers purchased from Research Genetics. These markers were PCR amplified and loaded onto an Applied Biosystems Model 373 sequencer, and the data were analyzed with Applied Biosystems Genotyper software, as described elsewhere (Duggirala et al. 1999). The details of our genotypic data, including the procedures for checking Mendelian discrepancies, were as described elsewhere (Duggirala et al. 2000). In this study, we used a set of 326 highly polymorphic microsatellite markers, which generated a 10-cM map.
Statistical Genetic Analysis
Multipoint Mapping Procedure
This method uses information on all available markers separated by known map distances and all possible biological relationships simultaneously in deciphering the genetic architecture of a given quantitative phenotype. Since the method requires the distances between the markers to be known, the order of the loci spanning a given chromosome and the likelihood sex-averaged map distances between the loci were obtained employing Kosambi’s mapping function using the program CRI-MAP (Green et al. 1990). The advantage of the multipoint approach is that it not only identifies the location of a susceptibility locus but also places CIs on the location parameter, which makes it more powerful than two-point analysis.
Variance Components Linkage Analysis
We used a pedigree-based multipoint variance-components approach to test for linkage between marker loci and the obesity phenotype, using a maximum-likelihood method (Amos 1994; Almasy and Blangero 1998). In this method, the expected genetic covariances between relatives are specified as a function of the IBD relationships at a marker locus, which is assumed to be closely linked to a locus influencing the quantitative trait in question. The covariance matrix for a pedigree can be written as shown in the following equation:
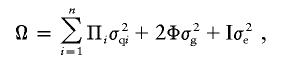 |
where Ω is the covariance matrix for a given pedigree; Π is a matrix with elements (πijl) providing the expected proportion of alleles at the specific chromosomal location of the quantitative trait locus that individuals j and l share IBD, which is estimated using genetic marker data; σ2q is the additive genetic variance due to the major locus; Φ is the kinship matrix; σ2g is the variance due to residual additive genetic effects; I is an identity matrix; and σ2e is the variance due to random environmental effects. Multipoint IBD matrices were estimated using Simwalk2 (Sobel et al. 2001) and were imported into SOLAR (Almasy and Blangero 1998). All multipoint variance-components linkage analyses were performed according to the procedures outlined in SOLAR (Almasy and Blangero 1998).
The null hypothesis that, for a given phenotype, the additive genetic variance due to the QTL, σ2q, equals zero (no linkage) can be tested using the likelihood-ratio test. The likelihood of a restricted model, in which σ2q is constrained to 0, is compared with the likelihood of the general model, in which genetic variance due to the QTL (σ2q) is estimated. The ln likelihood values of the general model and restricted model are then compared using the likelihood-ratio test. Twice the difference between the ln likelihood values of these models yields a test statistic that is asymptotically distributed as a 1/2: 1/2 mixture of a χ21 and a point mass at 0 (Self and Liang 1987). The likelihood value can be converted into a logarithm (base 10) of odds to obtain a LOD score that is equivalent to the classical LOD score of linkage analysis. A LOD score of ⩾3.0 was considered to be strong evidence in support of linkage, which is equivalent to a P value of .0001, suggesting that evidence for linkage is 1,000 times greater than the support for no linkage. Other chromosomal regions across the genome with nominal P values of .01 or less (i.e., LOD scores ⩾1.175) and LOD scores of ⩾1.9 are considered to show potential linkages and evidence suggestive of linkage, respectively.
Oligogenic Linkage Analysis
We also used a sequential strategy to identify multiple loci affecting the variation in BMI. In this approach, the genome was scanned for linkage and the chromosomal location that yielded the largest marginal LOD score was retained for further conditional analyses. Given the putative location of the first locus, we scanned the genome again and examined the resulting conditional LOD scores. In the two-locus model, the position of the first locus is fixed, and the location of the second locus is allowed to vary through the whole genome. The QTL variance (h2q) for the first locus is reestimated along with the second locus. Similarly, in the three-locus model, the positions of the first two loci are fixed, but the effect sizes are reestimated along with the third locus. LOD scores from the oligogenic models can be either joint, if they are compared to the polygenic null model, or conditional, if they are compared to the linkage model containing n−1 loci (Blangero and Almasy 1997).
Robust LOD (LODR) Score Estimation
To verify our major finding on chromosome 4, we performed simulation analysis. In the simulation analysis, a fully informative marker was simulated that was not linked to the QTL influencing variation in BMI. For this simulated marker, IBD information was calculated, and then linkage analysis was conducted. By use of simulations, the empirical distribution of the LOD scores under the assumption of multivariate normality was determined, on the basis of information obtained from 100,000 replicates by the SOLAR program (Almasy and Blangero 1998). The empirical distribution of the simulated LOD scores was used to assign percentiles to each replicate, and an expected test statistic was calculated on the basis of the percentile. The correction constant was obtained by regressing the expected LOD scores on the observed simulated LOD score, which is used to adjust the observed original LOD scores to obtain robust LOD (LODR) scores (Blangero et al. 2001).
Results
Descriptive statistics of the SAFDS subjects used in these analyses are presented in table 1. The patterns of phenotypic, genetic, and environmental correlations (ρ) between trait pairs BMI, leptin, and T2D in Mexican Americans are presented in table 2. The genetic correlations between BMI and leptin and between BMI and T2D were significant (P<.0001), whereas the genetic correlation between leptin and T2D was not significant (P>.05). Overall, polygenic heritability of BMI was estimated to be 48.6% ± 9.9%, which is highly significant (P<.0001), after the effects of age and sex are accounted for (table 3). Given the large sizes of these pedigrees, some related individuals may live in different households. We therefore estimated a household variance component (i.e., household effect was estimated as a surrogate measure of shared environmental influences), which was not significant (P=.24).
Table 1.
Characteristics of SAFDS Subjects
| Variable | Value |
| Males | 41.2% |
| Females | 58.8% |
| T2D | 26.3% |
| Age, mean ± SD (years) | 43.1 ± 17.1 |
| BMI, mean ± SD (kg/m2) | 30.0 ± 6.7a |
| Leptin, mean ± SD (ng/ml): | |
| Males and Females | 22.1 ± 17.1 |
| Males | 11.1 ± 7.9 |
| Females | 29.7 ± 17.6 |
Skewness = 0.9; kurtosis = 1.3.
Table 2.
Phenotypic (ρP ), Genetic (ρG), and Environmental (ρE ) Correlations between Phenotype Pairs[Note]
|
Mean Correlation ± SEa |
|||
| PhenotypePair | ρP | ρG | ρE |
| BMI and leptin | .66 ± .03* | .71 ± .09* | .66 ± .06* |
| BMI and T2D | .27 ± .07* | .52 ± .23* | .06 ± .16 |
| Leptin and T2D | .00 ± .08 | −.03 ± .34 | .02 ± .15 |
Note.— Each trait is adjusted for age and sex terms.
Significant at P<.0001.
Table 3.
BMI Heritability (h2) in SAFDS Individuals
BMI1 adjusted for age and sex terms; BMI2 adjusted for age and sex terms and for diabetes; BMI3 adjusted for age and sex terms, diabetes, and leptin.
Significant at P<.0001.
Following the estimation of heritability, we performed three multipoint univariate linkage analyses across 22 autosomes for BMI. In the first analysis (BMI1), after accounting for the effects of age and sex terms, we found evidence for linkage (LOD 2.1, P=.0019) of a genetic region at 39 cM on chromosome 4 near marker D4S2912. In the second analysis (BMI2), after accounting for the covariate effects of age and sex terms and T2D, we again found evidence for linkage (LOD 2.8, P=.0003) at the same location. Finally, in the third analysis (BMI3), we removed the effects of leptin in addition to those of age, sex, and T2D. We again found a significant linkage of BMI (LOD 4.5, P=.000003) to a nearby genetic location at 42 cM on chromosome 4p, near marker D4S2912. The robust LOD score that corresponds to our maximum observed LOD (4.5) is LODR = 4.1, P=.000014, indicating that type 1 error rates are consistent with asymptotic expectation (i.e., the inflation of type 1 error rate is minimal). Thus, the implicated marker region (at 2 cM centromeric to marker D4S2912) appears to harbor a major susceptibility locus for variation in BMI in Mexican Americans. The LOD scores obtained for BMI from the three analyses were plotted against map positions on chromosome 4, as shown in figure 1. The other chromosomal regions with multipoint LOD scores (>1.175) from only the BMI3 analysis are presented in table 4, and the multipoint LOD profiles across the genome are reported in figure 2.
Figure 1.

Linkage of BMI to a genetic location on chromosome 4p in Mexican Americans
Table 4.
Chromosomal Regions (LOD ⩾1.175, P⩽.01) Linked to Obesity Phenotype (BMI3)
|
Distance from pter, in |
||||
| Marker Region | Chromosome | Kosambi cM | Haldane cM | LODScore |
| D2S293–D2S383 | 2 | 115 | 135 | 2.9 |
| D4S2912 | 4 | 36 | 42 | 4.5 |
| D4S1548–D4S1090 | 4 | 175 | 195 | 1.3 |
| D7S506–D7S653 | 7 | 90 | 102 | 1.9 |
| D9S299–D9S930 | 9 | 113 | 124 | 2.1 |
| D11S1984–D11S988 | 11 | 2 | 3 | 2.5 |
| D11S4464 | 11 | 150 | 175 | 2.3 |
| D18S858–D18S51 | 18 | 80 | 90 | 1.4 |
Figure 2.

Chromosomal regions linked to BMI in a genome scan (LOD ⩾1.175)
Since multiple linkage peaks with some evidence for linkage were observed in our genomewide search, we extended variance-components linkage analysis to oligogenic models to incorporate two or more loci simultaneously (Blangero and Almasy 1997; Blangero et al. 1999). As shown in table 5, genetic determinants of BMI3 involve possibly three loci that are found on three different chromosomes. In a joint two-locus analysis, the two-locus model with a LOD score of 6.1 (corresponding to a conditional LODR of 1.5 for the second QTL, P=.009) accounted for 35% and 23% of variance due to the QTLs on chromosomes 4 (D4S2912) and 11 (D11S1984–D11S988), respectively. The three-locus model with a total LOD score of 7.1 (corresponding to a conditional LODR of 0.9 for the third QTL, P=.03) accounted for 29%, 20%, and 16% of variance due to the QTLs on chromosomes 4 (D4S2912), 11 (D11S1984–D11S988), and 7 (D7S506–D7S653), respectively. These variance-component estimates for the three-QTL model are likely to be more accurate than those for the simpler models (Blangero and Almasy 1997; Blangero et al. 1999).
Table 5.
Results of the Oligogenic Analysis of Variation in Obesity Phenotype (BMI3)
| % of VariationExplained by Locus |
||||||
| Model and QTL Region | Chromosome | Location(cM) | SequentialLOD | First | Second | Third |
| One QTL: | ||||||
| D4S2912 | 4 | 42 | 4.5 | 47 | … | … |
| Two QTL: | ||||||
| D4S2912 | 4 | |||||
| D11S1984–D11S988 | 11 | 5 | 6.1 | 35 | 23 | … |
| Three QTL: | ||||||
| D4S2912 | 4 | |||||
| D11S1984–D11S988 | 11 | |||||
| D7S506–D7S653 | 7 | 105 | 7.1 | 29 | 20 | 16 |
Discussion
The association between obesity and T2D has been recognized for decades, and several epidemiological studies have indicated that obesity is a major risk factor for the development of T2D (Kahn and Flier 2000; Nadler and Attie 2001). Although it is clear that obesity influences glucose metabolism through several physiological mechanisms, the cause and effect relationship between obesity and diabetes is still not well understood (Kopelman 2000). Thus, it is often difficult to identify which obese individuals are more likely to become diabetics, since ∼80% of individuals with T2D are obese and only 10% of obese individuals may become diabetics (Nadler et al. 2000). On the other hand, the relationship between adiposity (i.e., BMI) and leptin levels has been found to be significant in both men and women of various populations (Maffei et al. 1995; Considine et al. 1996). However, the relationship between leptin and T2D is not clear. Some studies have indicated a complex interrelationship between leptin and insulin resistance (Tritos and Mantzoros 1997; Donahue et al. 1999), whereas some have failed to find any association. For example, Haffner et al. (1996) reported no association between leptin levels and diabetic status even though leptin levels are significantly associated with BMI in Mexican Americans. Although there is considerable residual variation in leptin levels at a given BMI value, diabetic status does not account for this variability. In this study, we therefore used the BMI adjusted for the effects of leptin and T2D to identify the genes that influence the common forms of obesity, on the premise that power to localize a trait-specific major gene may be increased by removing some of the background noise due to the phenotypic associations between the correlated traits (Mahaney et al. 1995; Arya et al. 2001b).
We analyzed family data collected from Mexican American subjects as part of the San Antonio Family Diabetes Study to identify the trait-specific genes for a single obesity phenotype (i.e., adjusted BMI) in Mexican Americans. As our study reveals, BMI adjusted for the effects of leptin and T2D, which are strong correlates of BMI, is highly and significantly heritable. In addition, our genome scan linkage analysis revealed that there are loci with appreciable influences on BMI. More importantly, this genome scan provided strong evidence for the presence of a novel locus influencing variation in BMI on chromosome 4p. We observed the highest LOD score of 4.5 for BMI3 near marker D4S2912 at 42 cM from pter on chromosome 4p15.1, which provides highly significant support for linkage in a genomewide scan. In fact, the linkage signal (LOD 4.5) was slightly improved after saturation of the region with additional markers between flanking markers D4S2639 and D4S1581, compared with our initial findings (LOD 4.3) in the same region (Arya et al. 2001a).
This linkage finding has been replicated in a recent study of severe obesity in women by Stone et al. (2002), who found a highly significant linkage to high BMI in female patients at D4S2632 (4p15.1), with a multipoint heterogeneity LOD of 6.1 and a nonparametric linkage score of 5.3, located ∼5 cM telomeric to our region (D4S2912, 4p15.1). Furthermore, this region on chromosome 4p corresponds (∼5 cM apart) to the region previously reported to be linked (D4S2397, 4p15.2, LOD 2.3) to abdominal subcutaneous fat in the Quebec family data (Perusse et al. 2001). The region on chromosome 4p near marker D4S2912 (4p15.1) where we found a significant linkage signal contains two positional candidate genes: peroxisome proliferator activated receptor gamma coactivator 1 (PPARGC1 [previously called “PGC-1”], 4p15.1 [GenBank accession number NT_006316; MIM 604517]) and cholecystokinin A receptor (CCKAR, 4p15.1-p15.2 [NT_006316, MIM 118444]) (de Weerth et al. 1993; Huppi et al. 1995).
The gene for PPARGC1 comprises 13 exons spanning a genomic region of ∼98 kb and has a cytogenetic location of 4p15.1 (LocusLink). PPARGC1 is a novel transcriptional coactivator that coordinates the activities of many transcriptional factors that play an important role in adaptive thermogenesis (Lowell 1998; Puigserver et al. 1998; Spiegelman et al. 2000), mitochondrial biogenesis (Wu et al. 1999a), mitochondrial fatty acid oxidation (Vega et al. 2000), hepatic gluconeogenesis (Herzig et al. 2001; Yoon et al. 2001), and glucose uptake (Michael et al. 2001). PPARGC1 coactivates various nuclear receptors, including PPARγ, and promotes expression of mitochondrial proteins such as UCP1, which plays a major role in thermogenesis, both cold-induced and diet-induced, in brown adipose tissue and skeletal muscle, which is a key component of energy homeostasis and a metabolic defense against obesity (Puigserver et al. 1998; Larrouy et al. 1999).
PPARGC1 increases total cellular respiration and mitochondrial biogenesis through the coactivation of nuclear respiratory factor-1 (NRF-1) (Puigserver et al. 1998; Wu et al. 1999b). It promotes mitochondrial biogenesis through its ability to turn on the expression of both NRF-1 and -2 and coactivates NRF-1 through protein-protein interactions (Wu et al. 1999a; Monsalve et al. 2000). Both NRF-1 and NRF-2 bind to and regulate the promoters of several genes encoding mitochondrial proteins. Furthermore, it has been shown that expression of PPARGC-1 is decreased as a function of both insulin resistance and obesity, which in turn leads to decreased expression of NRF-dependent genes and thus to metabolic disturbances such as insulin resistance and diabetes (Patti et al. 2003). Thus, PPARGC1 plays a key role in linking nuclear receptors to the transcriptional program of adaptive thermogenesis, and increasing PPARGC1 activity may be a potential mechanism for energy dissipation and a therapeutic target for weight loss (Puigserver et al. 1998). Recently, Esterbauer et al. (2002) reported that, in middle-aged Austrian women, two PPARGC1 polymorphisms have been associated significantly with BMI (0.006), with waist (0.01) and hip (0.03) circumferences, and marginally with visceral and subcutaneous fat, suggesting a role of PPARGC1 in obesity.
Another candidate gene near our peak signal on chromosome 4p is the cholecystokinin A receptor (CCKAR), a G protein-coupled 7-transmembrane spanning receptor belonging to the rhodopsin family, which consists of 5 exons covering 9,025 bp with a cytogenetic location at 4p15.1-15.2 (de Weerth et al. 1993; Huppi et al. 1995; Inoue et al. 1997; LocusLink). Both the PPARGC1 and CCKAR genes are thus located centrally on the same physical 22.5 Mb genomic sequence reference contig (NT_006316, accessed September 17, 2003) and are just 2.6 Mb apart, a distance containing at least 26 known or suspected genes. This contig is flanked by markers D4S2912 and D4S1587, which encompass our linkage peak for adjusted BMI (BMI3).
CCKAR plays a role in mediating gallbladder contraction and secretion of pancreatic enzymes. This gene has been implicated in food intake and satiety (Gutzwiller et al. 2000; Beglinger et al. 2001), and CCKAR gene promoter polymorphism is associated with body fat (Funakoshi et al. 2000). Although evidence suggests that preabsorptive factors, such as cholecystokinin, are important cofactors in the regulation of energy intake, little is known about the biochemical processes that control hunger and satiety. CCK has been shown to affect the short-term control of food intake in animals and humans, and is proposed to act as a hormonal satiety signal (Drewe et al. 1992; Lieverse et al. 1994; Geary 1996; Gutzwiller et al. 1999). For example, fat ingestion stimulates the secretion of a number of gastrointestinal hormones including CCK. In addition, CCK-like peptides are endogenous signals that are mediated by CCKA receptors, involved in the control of food intake in humans (Gutzwiller et al. 2000). Using a single-strand conformational polymorphism strategy, Inoue et al. (1997) identified five sequence variants, including two missense variants in patients with T2D and obesity, which may be influencing obesity and diabetes. Moran and coworkers (1998) showed that rats that do not express CCKA receptors develop obesity, hyperglycemia, and T2D.
Several other chromosomal regions across the genome that exhibited some evidence for linkage (LOD >1.175) to BMI correspond with previously reported findings. The genomic region between markers D2S293 and D2S383 (2q12.2-2q14.3) on chromosome 2 (LOD 2.9) corresponds to a previously reported genomic region near markers D2S160 and D2S347 (2q13-2q14.3), which may contain a QTL influencing obesity-related phenotypes in a white population of European origin. Other genomic regions on chromosomes 7q and 11q correspond to findings from a number of studies. We have identified a broader region (7q22.1-7q35) on chromosome 7q that contains susceptibility gene(s) for obesity or its related phenotypes—such as extremity skinfolds, BMI, triglyceride, and high-density lipoprotein cholesterol (HDL-C) levels—in Mexican Americans (Duggirala et al. 1996, 2000; Arya et al. 2002). The same region is also implicated by several other studies as influencing BMI in other populations (Wu et al. 2002; Platte et al. 2003). Also, the two genomic regions near markers D11S1984 (11p15.5) and D11S4464 (11q24.1) on chromosome 11 correspond to previously reported genetic regions: a region near markers D11S1984–D11S988 (11p15.5) linked to clinical gallbladder disease (GBD) in Mexican Americans (Duggirala et al. 2003) and a second region, near markers D11S4464–D11S912 (11q24.1), linked to BMI in Pima Indians and other populations (Hanson et al. 1998; Atwood et al. 2002; Adeyemo et al. 2003).
Although ∼68 chromosomal regions have been identified as possibly harboring QTLs for human obesity-related phenotypes (Chagnon et al. 2003), specific genes influencing obesity are yet to be identified. Some of the genomic regions hitherto identified to influence obesity-related phenotypes involve chromosomes 2p (Comuzzie et al. 1997; Hager et al. 1998), 6q (Duggirala et al. 2001; Atwood et al. 2002), 7q (Duggirala et al. 1996), 10p (Hager et al. 1998; Hinney et al. 2000), 11q, and 20q (Norman et al. 1998) and have been replicated supporting the presence of genetic variants in these regions that influence the risk of obesity. Thus, genomewide linkage analyses of such obesity-related phenotypes suggest that the observed variation in obesity is attributable to more than one major gene.
Conclusions
Our genome scan results provide strong evidence for a major gene influencing BMI, adjusted for T2D and leptin, on chromosome 4p in Mexican Americans. This linkage was confirmed in an independent study of severe obesity in Utah pedigrees and corresponds with linkage findings from the Quebec Family Study. The implicated genomic region on chromosome 4p contains two strong positional candidate genes, PPARGC1 and CCKAR, which have been shown elsewhere to have a major functional role in the development of obesity. We have begun positional cloning efforts to identify functional variants in these positional candidate genes.
Acknowledgments
This research was supported in part by grants from National Institutes of Health (DK42273, DK47482, DK53889, and MH59490). R.A., the William K. Warren Diabetes Fellow of the ADA Mentor-Based Postdoctoral Fellowship, was supported by the William K. Warren Medical Research Institute. We wish to thank the participants of the SAFDS families for their support and cooperation. We also wish to thank S. Fowler, K. Williams, R. Granato, and T. D. Dyer for their help and support.
Electronic-Database Information
Accession numbers and URLs for data presented herein are as follows:
- LocusLink, http://www.ncbi.nlm.nih.gov/LocusLink/ (NCBI Build 33, assembled from Genbank genomic sequence data on April 28, 2003)
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for PPARGC1 and CCKAR)
References
- Adeyemo A, Luke A, Cooper R, Wu X, Tayo B, Zhu X, Rotimi C, Bouzekri N, Ward R (2003) A genome-wide scan for body mass index among Nigerian families. Obes Res 11:266–273 [DOI] [PubMed] [Google Scholar]
- Almasy L, Blangero J (1998) Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet 62:1198–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amos CI (1994) Robust variance-components approach for assessing genetic linkage in pedigrees. Am J Hum Genet 54:535–543 [PMC free article] [PubMed] [Google Scholar]
- Arya R, Blangero J, Almasy L, O’Connell P, Stern MP, Duggirala R (2001a) A major locus for body mass index (BMI) on chromosome 4p in Mexican Americans. Obes Res Suppl 9:70S [Google Scholar]
- Arya R, Duggirala R, Almasy L, Rainwater DL, Mahaney MC, Cole S, Dyer TD, Williams K, Leach RJ, Hixson JE, MacCluer JW, O’Connell P, Stern MP, Blangero J (2002) Linkage of high-density lipoprotein-cholesterol concentrations to a locus on chromosome 9p in Mexican Americans. Nat Genet 30:102–105 10.1038/ng810 [DOI] [PubMed] [Google Scholar]
- Arya R, Duggirala R, Williams JT, Almasy L, Blangero J (2001b) Power to localize the major gene for disease liability is increased after accounting for the effects of related quantitative phenotypes. Genet Epidemiol Suppl 21:S774–S778 [DOI] [PubMed] [Google Scholar]
- Atwood LD, Heard-Costa NL, Cupples LA, Jaquish CE, Wilson PW, D’Agostino RB (2002) Genomewide linkage analysis of body mass index across 28 years of the Framingham Heart Study. Am J Hum Genet 71:1044–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsh GS, Farooqi IS, O’Rahilly S (2000) Genetics of body-weight regulation. Nature 404:644–651 [DOI] [PubMed] [Google Scholar]
- Beglinger C, Degen L, Matzinger D, D’Amato M, Drewe J (2001) Loxiglumide, a CCK-A receptor antagonist, stimulates calorie intake and hunger feelings in humans. Am J Physiol Regul Integr Comp Physiol 280:R1149–R1154 [DOI] [PubMed] [Google Scholar]
- Blangero J, Almasy L (1997) Multipoint oligogenic linkage analysis of quantitative traits. Genet Epidemiol 14:959–964 10.1002/(SICI)1098-2272(1997)14:6<959::AID-GEPI66>3.0.CO;2-K [DOI] [PubMed] [Google Scholar]
- Blangero J, Williams JT, Almasy L (2001) Variance component methods for detecting complex trait loci. Adv Genet 42:151–181 [DOI] [PubMed] [Google Scholar]
- Blangero J, Williams JT, Iturria SJ, Almasy L (1999) Oligogenic model selection using the Bayesian Information Criterion: linkage analysis of the P300 Cz event-related brain potential. Genet Epidemiol Suppl 17:S67–S72 [DOI] [PubMed] [Google Scholar]
- Borecki IB, Higgins M, Schreiner PJ, Arnett DK, Mayer-Davis E, Hunt SC, Province MA (1998) Evidence for multiple determinants of the body mass index: the National Heart, Lung, and Blood Institute Family Heart Study. Obes Res 6:107–114 [DOI] [PubMed] [Google Scholar]
- Bouchard C (1997) Genetics of human obesity: recent results from linkage studies. J Nutr 127:1887S–1890S [DOI] [PubMed] [Google Scholar]
- Chagnon YC, Rankinen T, Snyder EE, Weisnagel SJ, Perusse L, Bouchard C (2003) The human obesity gene map: the 2002 update. Obes Res 11:313–367 [DOI] [PubMed] [Google Scholar]
- Comuzzie AG, Allison DB (1998) The search for human obesity genes. Science 280:1374–1377 10.1126/science.280.5368.1374 [DOI] [PubMed] [Google Scholar]
- Comuzzie AG, Hixson JE, Almasy L, Mitchell BD, Mahaney MC, Dyer TD, Stern MP, MacCluer JW, Blangero J (1997) A major quantitative trait locus determining serum leptin levels and fat mass is located on human chromosome 2. Nat Genet 15:273–276 [DOI] [PubMed] [Google Scholar]
- Comuzzie AG, Williams JT, Martin LJ, Blangero J (2001) Searching for genes underlying normal variation in human adiposity. J Mol Med 79:57–70 10.1007/s001090100202 [DOI] [PubMed] [Google Scholar]
- Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, Ohannesian JP, Marco CC, McKee LJ, Bauer TL, Caro JF (1996) Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med 334:292–295 10.1056/NEJM199602013340503 [DOI] [PubMed] [Google Scholar]
- de Weerth A, Pisegna JR, Huppi K, Wank SA (1993) Molecular cloning, functional expression and chromosomal localization of the human cholecystokinin type A receptor. Biochem Biophys Res Commun 194:811–818 10.1006/bbrc.1993.1894 [DOI] [PubMed] [Google Scholar]
- Diehl AK, Stern MP (1989) Special health problems of Mexican-Americans: obesity, gallbladder disease, diabetes mellitus, and cardiovascular disease. Adv Intern Med 34:73–96 [PubMed] [Google Scholar]
- Donahue RP, Prineas RJ, Donahue RD, Zimmet P, Bean JA, De Court, Collier G, Goldberg RB, Skyler JS, Schneiderman N (1999) Is fasting leptin associated with insulin resistance among nondiabetic individuals? The Miami Community Health Study. Diabetes Care 22:1092–1096 [DOI] [PubMed] [Google Scholar]
- Drewe J, Gadient A, Rovati LC, Beglinger C (1992) Role of circulating cholecystokinin in control of fat-induced inhibition of food intake in humans. Gastroenterology 102:1654–1659 [DOI] [PubMed] [Google Scholar]
- Duggirala R, Dodd G, Fowler S, Schneider J, Arya R, Diehl AK, Almasy L, O’Connell P, Stern MP, Blangero J (2003) A major susceptibility locus for gallbladder disease is on chromosome 11p in Mexican Americans. Am J Hum Genet Suppl 73:195 [Google Scholar]
- Duggirala R, Blangero J, Almasy L, Arya R, Dyer TD, Williams KL, Leach RJ, O’Connell P, Stern MP (2001) A major locus for fasting insulin concentrations and insulin resistance on chromosome 6q with strong pleiotropic effects on obesity-related phenotypes in nondiabetic Mexican Americans. Am J Hum Genet 68:1149–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggirala R, Blangero J, Almasy L, Dyer TD, Williams KL, Leach RJ, O’Connell P, Stern MP (1999) Linkage of type 2 diabetes mellitus and of age at onset to a genetic location on chromosome 10q in Mexican Americans. Am J Hum Genet 64:1127–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggirala R, Blangero J, Almasy L, Dyer TD, Williams KL, Leach RJ, O’Connell P, Stern MP (2000) A major susceptibility locus influencing plasma triglyceride concentrations is located on chromosome 15q in Mexican Americans. Am J Hum Genet 66:1237–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggirala R, Stern MP, Mitchell BD, Reinhart LJ, Shipman PA, Uresandi OC, Chung WK, Leibel RL, Hales CN, O’Connell P, Blangero J (1996) Quantitative variation in obesity-related traits and insulin precursors linked to the OB gene region on human chromosome 7. Am J Hum Genet 59:694–703 [PMC free article] [PubMed] [Google Scholar]
- Esterbauer H, Oberkofler H, Linnemayr V, Iglseder B, Hedegger M, Wolfsgruber P, Paulweber B, Fastner G, Krempler F, Patsch W (2002) Peroxisome proliferator-activated receptor-gamma coactivator-1 gene locus: associations with obesity indices in middle-aged women. Diabetes 51:1281–1286 [DOI] [PubMed] [Google Scholar]
- Funakoshi A, Miyasaka K, Matsumoto H, Yamamori S, Takiguchi S, Kataoka K, Takata Y, Matsusue K, Kono A, Shimokata H (2000) Gene structure of human cholecystokinin (CCK) type-A receptor: body fat content is related to CCK type-A receptor gene promoter polymorphism. FEBS Lett 466:264–266 10.1016/S0014-5793(00)01080-2 [DOI] [PubMed] [Google Scholar]
- Geary N (1996) Failure of pulsatile infusion to increase glucagon’s satiating potency. Physiol Behav 59:613–616 10.1016/0031-9384(95)02121-3 [DOI] [PubMed] [Google Scholar]
- Green P, Falls K, Crooks S (1990) Documentation for CRI-MAP. Department of Genetics, School of Medicine, Washington University, St. Louis [Google Scholar]
- Gutzwiller JP, Drewe J, Ketterer S, Hildebrand P, Krautheim A, Beglinger C (2000) Interaction between CCK and a preload on reduction of food intake is mediated by CCK-A receptors in humans. Am J Physiol Regul Integr Comp Physiol 279:R189–R195 [DOI] [PubMed] [Google Scholar]
- Gutzwiller JP, Goke B, Drewe J, Hildebrand P, Ketterer S, Handschin D, Winterhalder R, Conen D, Beglinger C (1999) Glucagon-like peptide-1: a potent regulator of food intake in humans. Gut 44:81–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffner SM, Hazuda HP, Mitchell BD, Patterson JK, Stern MP (1991) Increased incidence of type II diabetes mellitus in Mexican Americans. Diabetes Care 14:102–108 [DOI] [PubMed] [Google Scholar]
- Haffner SM, Stern MP, Hazuda HP, Pugh J, Patterson JK, Malina R (1986) Upper body and centralized adiposity in Mexican Americans and non-Hispanic whites: relationship to body mass index and other behavioral and demographic variables. Int J Obes 10:493–502 [PubMed] [Google Scholar]
- Haffner SM, Stern MP, Miettinen H, Wei M, Gingerich RL (1996) Leptin concentrations in diabetic and nondiabetic Mexican Americans. Diabetes 45:822–824 [DOI] [PubMed] [Google Scholar]
- Hager J, Dina C, Francke S, Dubois S, Houari M, Vatin V, Vaillant E, Lorentz N, Basdevant A, Clement K, Guy-Grand B, Froguel P (1998) A genome-wide scan for human obesity genes reveals a major susceptibility locus on chromosome 10. Nat Genet 20:304–308 10.1038/3123 [DOI] [PubMed] [Google Scholar]
- Hanson RL, Ehm MG, Pettitt DJ, Prochazka M, Thompson DB, Timberlake D, Foroud T, Kobes S, Baier L, Burns DK, Almasy L, Blangero J, Garvey WT, Bennett PH, Knowler WC (1998) An autosomal genomic scan for loci linked to type II diabetes mellitus and body-mass index in Pima Indians. Am J Hum Genet 63:1130–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, Rudolph D, Schutz G, Yoon C, Puigserver P, Spiegelman B, Montminy M (2001) CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 413:179–183 10.1038/35093131 [DOI] [PubMed] [Google Scholar]
- Hinney A, Remschmidt H, Hebebrand J (2000) Candidate gene polymorphisms in eating disorders. Eur J Pharmacol 410:147–159 10.1016/S0014-2999(00)00812-8 [DOI] [PubMed] [Google Scholar]
- Huppi K, Siwarski D, Pisegna JR, Wank S (1995) Chromosomal localization of the gastric and brain receptors for cholecystokinin (CCKAR and CCKBR) in human and mouse. Genomics 25:727–729 10.1016/0888-7543(95)80018-H [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue H, Iannotti CA, Welling CM, Veile R, Donis-Keller H, Permutt MA (1997) Human cholecystokinin type A receptor gene: cytogenetic localization, physical mapping, and identification of two missense variants in patients with obesity and non-insulin-dependent diabetes mellitus (NIDDM). Genomics 42:331–335 10.1006/geno.1997.4749 [DOI] [PubMed] [Google Scholar]
- Joos SK, Mueller WH, Hanis CL, Schull WJ (1984) Diabetes alert study: weight history and upper body obesity in diabetic and non-diabetic Mexican American adults. Ann Hum Biol 11:167–171 [DOI] [PubMed] [Google Scholar]
- Kahn BB, Flier JS (2000) Obesity and insulin resistance. J Clin Invest 106:473–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan LM (2000) Genetics of obesity and body weight regulation. Curr Opin Endocrinol Diab 7:218–224 10.1097/00060793-200010000-00002 [DOI] [Google Scholar]
- Kopelman PG (2000) Obesity as a medical problem. Nature 404:635–643 [DOI] [PubMed] [Google Scholar]
- Larrouy D, Vidal H, Andreelli F, Laville M, Langin D (1999) Cloning and mRNA tissue distribution of human PPARgamma coactivator-1. Int J Obes Relat Metab Disord 23:1327–1332 10.1038/sj.ijo.0801106 [DOI] [PubMed] [Google Scholar]
- Lieverse RJ, Jansen JB, Masclee AM, Lamers CB (1994) Satiety effects of cholecystokinin in humans. Gastroenterology 106:1451–1454 [DOI] [PubMed] [Google Scholar]
- Lowell BB (1998) Adaptive thermogenesis: turning on the heat. Curr Biol 8:R517–R520 [DOI] [PubMed] [Google Scholar]
- Ma Z, Gingerich RL, Santiago JV, Klein S, Smith CH, and Landt M (1996) Radioimmunoassay of leptin in human plasma. Clin Chem 42:942–946 [PubMed] [Google Scholar]
- Maffei M, Halaas J, Ravussin E, Pratley RE, Lee GH, Zhang Y, Fei H, Kim S, Lallone R, Ranganathan S, Kern PA, Friedman JM (1995) Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med 11:1155–1161 [DOI] [PubMed] [Google Scholar]
- Mahaney MC, Blangero J, Comuzzie AG, VandeBerg JL, Stern MP, MacCluer JW (1995) Plasma HDL cholesterol, triglycerides, and adiposity: a quantitative genetic test of the conjoint trait hypothesis in the San Antonio Family Heart Study. Circulation 92:3240–3248 [DOI] [PubMed] [Google Scholar]
- Michael LF, Wu Z, Cheatham RB, Puigserver P, Adelmant G, Lehman JJ, Kelly DP, Spiegelman BM (2001) Restoration of insulin-sensitive glucose transporter (GLUT4) gene expression in muscle cells by the transcriptional coactivator PGC-1. Proc Natl Acad Sci USA 98:3820–3825 10.1073/pnas.061035098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell BD, Stern MP, Haffner SM, Hazuda HP, Patterson JK (1990) Risk factors for cardiovascular mortality in Mexican Americans and non-Hispanic whites. San Antonio Heart Study. Am J Epidemiol 131:423–433 [DOI] [PubMed] [Google Scholar]
- Monsalve M, Wu Z, Adelmant G, Puigserver P, Fan M, and Spiegelman BM (2000) Direct coupling of transcription and mRNA processing through the thermogenic coactivator PGC-1. Mol Cell 6:307–316 [DOI] [PubMed] [Google Scholar]
- Moran TH, Katz LF, Plata-Salaman CR, Schwartz GJ (1998) Disordered food intake and obesity in rats lacking cholecystokinin A receptors. Am J Physiol 274:R618–R625 [DOI] [PubMed] [Google Scholar]
- Nadler ST, Attie AD (2001) Please pass the chips: genomic insights into obesity and diabetes. J Nutr 131:2078–2081 [DOI] [PubMed] [Google Scholar]
- Nadler ST, Stoehr JP, Schueler KL, Tanimoto G, Yandell BS, Attie AD (2000) The expression of adipogenic genes is decreased in obesity and diabetes mellitus. Proc Natl Acad Sci USA 97:11371–11376 10.1073/pnas.97.21.11371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman RA, Tataranni PA, Pratley R, Thompson DB, Hanson RL, Prochazka M, Baier L, Ehm MG, Sakul H, Foroud T, Garvey WT, Burns D, Knowler WC, Bennett PH, Bogardus C, Ravussin E (1998) Autosomal genomic scan for loci linked to obesity and energy metabolism in Pima Indians. Am J Hum Genet 62:659–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, Landaker EJ, Goldfine AB, Mun E, DeFronzo R, Finlayson J, Kahn CR, Mandarino LJ (2003) Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci USA 100:8466–8471 10.1073/pnas.1032913100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perusse L, Rice T, Chagnon YC, Despres JP, Lemieux S, Roy S, Lacaille M, Ho-Kim MA, Chagnon M, Province MA, Rao DC, Bouchard C (2001) A genome-wide scan for abdominal fat assessed by computed tomography in the Quebec Family Study. Diabetes 50:614–621 [DOI] [PubMed] [Google Scholar]
- Platte P, Papanicolaou GJ, Johnston J, Klein CM, Doheny KF, Pugh EW, Roy-Gagnon MH, Stunkard AJ, Francomano CA, Wilson AF (2003) A study of linkage and association of body mass index in the Old Order Amish. Am J Med Genet 121:71–80 10.1002/ajmg.c.20005 [DOI] [PubMed] [Google Scholar]
- Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM (1998) A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92:829–839 [DOI] [PubMed] [Google Scholar]
- Rice T, Despres JP, Perusse L, Gagnon J, Leon AS, Skinner JS, Wilmore JH, Rao DC, Bouchard C (1997) Segregation analysis of abdominal visceral fat: the HERITAGE Family Study. Obes Res 5:417–424 [DOI] [PubMed] [Google Scholar]
- Self SG, Liang K-Y (1987) Asymptotic properties of maximum likelihood estimators and likelihood ratio tests under nonstandard conditions. J Am Stat Assoc 82:605–610 [Google Scholar]
- Sobel E, Sengul H, Weeks DE (2001) Multipoint estimation of identity-by-descent probabilities at arbitrary positions among marker loci on general pedigrees. Hum Hered 52:121–131 10.1159/000053366 [DOI] [PubMed] [Google Scholar]
- Spiegelman BM, Puigserver P, Wu Z (2000) Regulation of adipogenesis and energy balance by PPARγ and PGC-1. Int J Obes Relat Metab Disord Suppl 24:S8–S10 [DOI] [PubMed] [Google Scholar]
- Stern MP, Haffner SM (1990) Type II diabetes and its complications in Mexican Americans. Diabetes Metab Rev 6:29–45 [DOI] [PubMed] [Google Scholar]
- Stern MP, Morales PA, Valdez RA, Monterrosa A, Haffner SM, Mitchell BD, Hazuda HP (1993) Predicting diabetes: moving beyond impaired glucose tolerance. Diabetes 42:706–714 [DOI] [PubMed] [Google Scholar]
- Stern MP, Patterson JK, Haffner SM, Hazuda HP, Mitchell BD (1989) Lack of awareness and treatment of hyperlipidemia in type II diabetes in a community survey. JAMA 262:360–364 [PubMed] [Google Scholar]
- Stern MP, Patterson JK, Mitchell BD, Haffner SM, Hazuda HP (1990) Overweight and mortality in Mexican Americans. Int J Obes 14:623–629 [PubMed] [Google Scholar]
- Stone S, Abkevich V, Hunt SC, Gutin A, Russell DL, Neff CD, Riley R, Frech GC, Hensel CH, Jammulapati S, Potter J, Sexton D, Tran T, Gibbs D, Iliev D, Gress R, Bloomquist B, Amatruda J, Rae PM, Adams TD, Skolnick MH, Shattuck D (2002) A major predisposition locus for severe obesity, at 4p15-p14. Am J Hum Genet 70:1459–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritos NA, Mantzoros CS (1997) Leptin: its role in obesity and beyond. Diabetologia 40:1371–1379 10.1007/s001250050838 [DOI] [PubMed] [Google Scholar]
- Vega RB, Huss JM, Kelly DP (2000) The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol 20:1868–1876 10.1128/MCB.20.5.1868-1876.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization, Department of Noncommunicable Disease Surveillance (1999) Definition, diagnosis, and classification of diabetes mellitus and its complications: report of a WHO consultation. Part 1: Diagnosis and classification of diabetes mellitus. World Health Organization, Geneva [Google Scholar]
- Wu X, Cooper RS, Borecki I, Hanis C, Bray M, Lewis CE, Zhu X, Kan D, Luke A, Curb D (2002) A combined analysis of genomewide linkage scans for body mass index from the National Heart, Lung, and Blood Institute Family Blood Pressure Program. Am J Hum Genet 70:1247–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM (1999a) Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98:115–124 [DOI] [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Spiegelman BM (1999b) Transcriptional activation of adipogenesis. Curr Opin Cell Biol 11:689–694 10.1016/S0955-0674(99)00037-X [DOI] [PubMed] [Google Scholar]
- Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, Adelmant G, Stafford J, Kahn CR, Granner DK, Newgard CB, Spiegelman BM (2001) Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 413:131–138 10.1038/35093050 [DOI] [PubMed] [Google Scholar]