

Abstract
Bietti crystalline corneoretinal dystrophy (BCD) is an autosomal recessive retinal dystrophy characterized by multiple glistening intraretinal crystals scattered over the fundus, a characteristic degeneration of the retina, and sclerosis of the choroidal vessels, ultimately resulting in progressive night blindness and constriction of the visual field. The BCD region of chromosome 4q35.1 was refined to an interval flanked centromerically by D4S2924 by linkage and haplotype analysis; mutations were found in the novel CYP450 family member CYP4V2 in 23 of 25 unrelated patients with BCD tested. The CYP4V2 gene, transcribed from 11 exons spanning 19 kb, is expressed widely. Homology to other CYP450 proteins suggests that CYP4V2 may have a role in fatty acid and steroid metabolism, consistent with biochemical studies of patients with BCD.
Introduction
Bietti crystalline corneoretinal dystrophy (BCD [MIM 210370]) is an autosomal recessive retinal dystrophy first described by Bietti (1937). The condition is characterized by numerous tiny glistening yellow-white crystals at the posterior pole of the retina associated with atrophy of the retinal pigment epithelium (RPE), pigment clumps, and choroidal sclerosis. Most cases have similar crystals at the corneoscleral limbus (Kaiser-Kupfer et al. 1994). Clinically, BCD is progressive; most patients develop decreased vision, nyctalopia, and paracentral scotomata between the 2nd and 4th decade of life. Later, patients develop peripheral visual field loss and marked visual impairment, usually progressing to legal blindness by the 5th or 6th decade of life (Kaiser-Kupfer et al. 1994).
Although patients with BCD from Europe, North and South America, the Middle East, and Africa have been described, BCD has been reported to be more common in East Asia, especially in Chinese and Japanese populations (Hu 1983). An epidemiological survey of genetic disease in a general population in China estimated the gene frequency to be 0.005 (Hu 1982). However, BCD accounted for ∼3% (3 of 104 index patients) of all nonsyndromic retinitis pigmentosa (RP) and 10% of nonsyndromic autosomal recessive “RP” in a recent series from Europe (Mataftsi et al., in press).
Histopathology shows advanced panchorioretinal atrophy, with crystals and complex lipid inclusions seen in choroidal fibroblasts, corneal keratocytes, and conjunctival and skin fibroblasts, as well as in circulating lymphocytes (Welch 1977; Wilson et al. 1989; Kaiser-Kupfer et al. 1994). These findings suggest that BCD may result from a systemic abnormality of lipid metabolism. Cells cultured from patients with BCD have abnormally high levels of triglycerides and cholesterol storage (Lee et al. 2001). Conversely, metabolism of labeled fatty acid precursors into n-3 polyunsaturated fatty acids (n-3PUFA) is decreased (Lee et al. 2001). Both findings suggest that a deficiency or dysfunction of a lipid metabolic enzyme or fatty acid–binding protein could cause decreased uptake and transfer between lipid pools. In addition, deficient elongation and desaturation of fatty acid precursors could cause the deposit of lipid within the diseased cells in crystalline form (Lee et al. 2001). In this regard, lymphocytes from patients with BCD lack two fatty acid–binding activities (Lee et al. 1998), but the relationship between the disease and these two absent binding activities remains unclear. In sum, these findings suggest that BCD may result from a systemic abnormality of lipid metabolism.
Previously, the BCD locus was linked to D4S426 (maximum LOD score [Zmax] = 4.81; recombination fraction [θ] = 0), D4S2688 (Zmax=3.98; θ=0), and D4S2299 (Zmax=5.31; θ=0) on chromosome 4q35-4qtel (Jiao et al. 2000). Multipoint analysis confirmed linkage to the region telomeric of D4S1652, with a maximum multipoint LOD score of 5.3 located 4 cM telomeric of marker D4S2930. The present study utilizes haplotype and linkage analysis to refine the critical region for the BCD locus to the region of chromosome 4q35 distal to D4S2924 and identifies mutations in the CYP4V2 gene in 23 of 25 unrelated patients with BCD. CYP4V2 is a previously uncharacterized gene that encodes a widely expressed 525–amino acid CYP450 family member, the structure of which suggests its involvement in fatty acid metabolism, consistent with metabolic studies of patients with BCD.
Material and Methods
Families
This project was approved by the National Eye Institute institutional review board. The patients admitted to the study gave their informed consent, and the study adhered to the tenets of the Declaration of Helsinki. Clinical diagnostic procedures, diagnostic criteria, and exclusion criteria have been described elsewhere (Jiao et al. 2000). Diagnosis was confirmed by the presence of lipid inclusions in lymphocytes in families 33001–33012 and 33015, as well as in one isolated European individual, 3300001. On the basis of the ethnic origin of the families, 50 unrelated and unaffected individuals (12 Chinese, 16 Japanese, and 22 European) were enrolled in this study as unaffected controls. DNA was extracted either directly from blood or from transformed lymphoblastoid cell lines (Smith et al. 1989).
Genotyping and Linkage Analyses
Two-point linkage analyses were performed with the FASTLINK implementation of the MLINK program in the LINKAGE program package (Lathrop and Lalouel 1984; Cottingham et al. 1993). BCD was analyzed as a fully penetrant autosomal recessive trait. The gene frequency of BCD was set at 0.005 (Hu 1983). For linkage and haplotype analysis, the markers in the BCD region were arranged according to the physical map (fig. 1; and see below). Haplotypes were constructed with 14 microsatellite markers and the mutation locus found in families with BCD to minimize recombinational events.
Figure 1.

Pedigrees of selected families with BCD with haplotypes of markers in the BCD region. Family 33001 was used in the genomewide scan. Recombinants in individual 4 of family 33010 and individual 6 of family 33011 set the centromeric boundary at D4S2924, as does the lack of homozygosity in D4S3047, D4S3032, and D4S2924 in individual 3 of consanguineous family 33005.
Physical Mapping
A YAC contig was constructed as a first step, followed by BAC and P1-derived artificial chromosome (PAC) maps in the candidate region (fig. 2). Total yeast genomic/YAC DNA was isolated and analyzed (Ayyagari et al. 1996). PAC clones were screened from a human genomic DNA PAC library, RPCI-1 (Incyte Genomics), by PCR with eight microsatellite markers in the linkage study (D4S3051, D4S2921, D4S426, D4S2688, D4S2299, D4S2930, D4S1652, and D4S2283) and another 20 STS markers located in the BCD region (D4S254, D4S187, D4S862, D4S3214, D4S130, D4S2930, D4S2441, D4S845, D4S592, D4S1369, D4S3186, D4S3212, D4S3239, WI5353, WI140, T78431, SGC38233, SHGC67241, D4S1621, and D4S139). Twenty BAC clones (RP11-308K2, 264F11, 237D3, 565A3, 713C19, 174N10, 219G10, 16M7, 16L12, 765P10, 553E4, 354H17, 818C3, 45F23, 462G22, 215O6, 30B7, 629I23, 690G11, and 600B8) were purchased from Research Genetics and regrown. Fifty-eight STS markers located in the BCD region were used for clone verification and contig construction. Assembly of YACs, PACs, and BACs spanning the BCD region was accomplished by a combination of PCR analysis of all clones for the presence or absence of the STS markers and EcoRI and NotI restriction digestion of YAC clones, followed by Southern blots of the digested YAC clones probed with the PCR-amplified products of the STS markers (Ayyagari et al. 1996).
Figure 2.

A, Physical map of BCD region at 4q35.1. The BCD region is covered contiguously by 6 overlapping YACs and 32 BACs, representing the shortest path. The STS marker positions are from the NCBI database. B, CYP4V2 cDNAs. Five overlapping cDNA fragments obtained by direct PCR and 5′ and 3′ RACE from human retinal cDNA and RPE cDNA provide a 2,041-bp cDNA. Sequence homology searches show partial sequence overlap with three NCBI predicted cDNA sequences: XM209611, XM114391, and XM209612. C, CYP4V2 gene structure. CYP4V2 gene structure is assembled by alignment of the cDNA sequence to genomic DNA sequence. The 11 identified exons are flanked by recognizable splice sites. The coding region extends as one ORF from the first Met (ATG) codon in exon 1 to the stop (TAA) codon in exon 11. This novel gene consists of 11 exons and spans 19 kb. Thirteen mutations of the CYP4V2 gene found in 23 of 25 unrelated patients with BCD are indicated below the gene structure.
Gene Structure and Sequence Analysis
Primer sequences that amplify each exon, including the intron-exon boundaries, are available as table A1 of appendix A (online only). cDNA structure was confirmed by direct PCR and 3′ and 5′ RACE from Marathon-ready human retina cDNA (BD Biosciences) and human RPE cDNA, shown in figure 2 with three sets of primers for RT-PCR of transformed lymphoblastoid lines and primary and nested primers for 3′ and 5′ RACE (available as table A1 of appendix A [online only]). DNA was sequenced with the ABI BigDye Terminator cycle sequencing kit v3.1, according to the manufacturer’s recommendations (Applied Biosystems), through use of an ABI 3100 sequencer. The sequence information was imported into SeqMan 5.01 (DNASTAR), and sequences of affected and normal individuals and consensus sequence were aligned to identify variations.
Expression Analysis
Expression of the novel gene CYP4V2 was tested by PCR of cDNA from multiple human normal tissues with primers for cDNA913, covering exons 5–11. Tissues included human retina, RPE, lymphocytes, heart, brain, placenta, lung, liver, skeletal muscle, kidney, and pancreas from the Multiple Human Tissue cDNA Panels (BD Biosciences). Expression status of CYP4V2 in patients with BCD was tested by RT-PCR with total RNA isolated from patient lymphoblastoid lines and was compared with the expression in unaffected carriers and normal control individuals.
Molecular Modeling
Modeling of the CYP4V2 wild-type protein was performed by homology modeling based on crystal coordinates for mammalian CYP2C5 (Protein Data Bank [PDB] ID 1dt6) and bacterial CYPBM3 (PDB ID 2hpd) as the structural templates (Abola et al. 1987). Although CYP2C5 oxidoreductase demonstrates an identity (similarity) of 23% (43%), which is lower than that of CYPBM3, 32% (51%), in regions containing the P450 domain, its homology covers a broader sequence range (residues 18–448, shown in fig. B1 of appendix B [online only]). Thus, CYPBM3 modeled the high-homology area surrounding the active site, and CYP2C5 modeled the amino terminal region, for which homology to CYPBM3 was low. Primary sequences were aligned by the method of Needleman and Wunsch (1970) incorporated in the program Look, version 3.5.2 (Lee and Subbiah 1991; Lee 1994) for 3-D structure prediction. The location of the transmembrane segment was predicted by the TMHMM2 program incorporated in SMART (Schultz et al. 1998). Finally, the monomeric CYP4V2 and exon 7 deleted mutant CYP4V2-exon7del were built by the automatic segment matching method in the program Look (Levitt 1992), followed by 500 cycles of energy minimization. The same program generated the conformation of the proteins with point mutations—W44R, G61S, I111T, S341P, and R508H—and refined them by self-consistent ensemble optimization (500 cycles) (Lee 1994). The predicted structure of CYP4V2 (fig. 3) was regularized by an energy minimization procedure in the presence of water on the final step through use of the program Insight II version 2000.1 (Accelrys), and the geometry of the predicted structures was tested with the program Procheck (Laskowski et al. 1993).
Figure 3.

Structure of CYP4V2 protein and selected mutations. A, CYP4V2 protein structure. α Helices are shown as red cylinders, β sheets are shown as yellow ribbons, and the porphyrin ring is shown in blue. B, The peptide preserved by G134X, which truncates the protein just within the main globular domain, is shown in red, and truncated sequences are shown in blue, with the exception of exon 7, which is shown in green. C, The 15-bp deletion, including the 3′ (acceptor) splice site for exon 7, which removes the G, H, and part of the central I helices (green) and their connecting loops and causes uncoiling and displacement of the central I helix (the normal position is shown in light blue, and the displaced position is shown in dark blue). D, Replacement of hydrophobic aromatic tryptophan near or in the transmembrane segment with positively charged arginine (W44R). E, R508H, which might influence heme coordination. F, H331P, predicted to disrupt the I helix. G, I111T, near the surface of the molecule. H, S341P, predicted to disrupt the I helix.
Splice Site and Mutation Analysis
Computer analysis of the strength of splice sites of genomic sequences spanning relevant nucleotides was performed by the Delila package (Rogan et al. 1998; Delila Program Web site). These programs use an information theory–based model of donor and acceptor splice sites to assign a bit value as a representation of the strength of interaction between the splice site and the spliceosome for both the wild-type and the mutant splice sites. Blosum scores (Henikoff and Henikoff 1992) were taken from the Bioccelerator at the European Molecular Biology Laboratory Web site. The blosum70 score was selected on the basis of the overall homology levels among members of CYP450 protein families, which tend to range from 60% to 80%.
Results
A physical map of the BCD region from D4S2924 to 4qter, with an approximate size of 4 Mb, shown in fig. 2A, is consistent with the current versions of the National Center for Biotechnology Information (NCBI Home Page) and Celera (myScience Web site) databases. As shown in table 1, two-point linkage analysis continues to suggest that the BCD gene is linked to markers D4S2688 (Zmax=4.14; θ=0), D4S426 (Zmax=4.82; θ=0), and D4S2299 (Zmax=5.43; θ=0). Obligate recombinants are seen on two-point analysis for all markers centromeric of D4S2924.
Table 1.
Two-Point Linkage Results for Markers in the BCD Region of Chromosome 4q35
|
Position (4ptel) |
LOD Score at θ = |
||||||||||
| Marker | cM | Mb | 0 | .01 | .05 | .1 | .2 | .3 | .4 | Zmax | 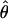 |
| D4S3047 | 190.25 | 185.99 | −∞ | −4.68 | −1.15 | −.06 | .42 | .29 | .08 | .42 | .2 |
| D4S3032 | 192.73 | 186.6 | −∞ | .45 | 1.30 | 1.36 | .96 | .52 | .13 | 1.37 | .06 |
| D4S2924 | 193.15 | 187.03 | −∞ | −1.5 | .66 | 1.05 | .91 | .45 | .10 | 1.09 | .09 |
| GDB186858 | 187.78 | 3.13 | 3.03 | 2.63 | 2.15 | 1.32 | .64 | .17 | 3.13 | 0 | |
| D4S3051 | 200.02 | 188.61 | −.09 | 1.35 | 1.68 | 1.47 | 1.01 | .45 | .15 | 1.7 | .06 |
| D4S3332 | 202.55 | 188.97 | 1.71 | 3.21 | 3.30 | 2.78 | 1.78 | .88 | .29 | 3.33 | .03 |
| D4S2921 | 202.69 | 189.48 | 1.19 | 2.54 | 2.82 | 2.55 | 1.68 | .83 | .26 | 2.82 | .05 |
| D4S2688 | 189.69 | 4.14 | 4.02 | 3.45 | 2.81 | 1.69 | .79 | .21 | 4.14 | 0 | |
| D4S426 | 202.69 | 189.69 | 4.82 | 4.58 | 4.04 | 3.30 | 2.03 | 1.00 | .27 | 4.82 | 0 |
| D4S2299 | 203.59 | 190.10 | 5.43 | 5.28 | 4.61 | 3.83 | 2.37 | 1.16 | .39 | 5.43 | 0 |
| D4S2390 | 204.18 | 190.34 | 2.57 | 4.04 | 4.01 | 3.45 | 2.15 | 1.02 | .20 | 4.2 | .03 |
| D4S1652 | 204.46 | 190.67 | 1.05 | 2.46 | 2.59 | 2.24 | 1.37 | .67 | .14 | 2.69 | .03 |
| D4S2930 | 204.47 | 190.68 | 1.85 | 3.36 | 3.45 | 2.93 | 1.90 | .88 | .30 | 3.53 | .02 |
| D4S2283 | 190.9 | 2.04 | 3.33 | 3.36 | 2.88 | 1.76 | .84 | .18 | 3.52 | .04 | |
Haplotype analysis refines the critical interval further (fig. 1). Recombinants in families 33010 and 33011 set the centromeric boundary at D4S2924, as does a lack of homozygosity in consanguineous family 33005. The telomeric boundary of the linked interval is indeterminate but places BCD telomeric of D4S2924.
One predicted gene in this region, Hs4-22948-33-121-2 in the NCBI database and HCG1787816 in the Celera database, is highly homologous to Mus musculus protein cytochrome 4V3 of the cytochrome P450 superfamily. PCR amplification and direct sequencing of the 11 putative exons and intron-exon boundaries shows mutations in 13 of 14 familial and 10 of 11 isolated BCD cases (table 2). Mutations found in familial cases were confirmed, in all available affected family members, to cosegregate with BCD. RT-PCR amplification and sequencing of lymphocyte mRNA from patients with the 15-bp homozygous deletion, including the exon 7 splice-acceptor site, confirmed skipping of exon 7 in these patients (fig. 4). In familial cases, heterozygous mutations were confirmed to reside on separate chromosomes by genotyping parents.
Table 2.
Mutations and Their Predicted Effects on CYP4V2 Structure and Function[Note]
| Gene Mutation | Exon | ProteinMutation (Blosum70 Score) | Location in Protein | Second Mutation (Ethnicity)a | Predicted Effect |
| 434T→A | 1 | W44R (−3) | Between transmembrane segment and A helix | I111T in individual 3300010 (E) | Increase the solubility of the transmembrane helix C cap |
| 485G→A | 1 | G61S (−1) | Between transmembrane segment and A helix | Exon9del in family 33014 (C) | Unknown |
| IVS2+1G→A (5′ [donor] splice site) | 2 | E72GNFNX | Truncation between transmembrane segment and A helix | ?IVS2+25 delT in individual 3300007 (J) | Eliminates entire globular structure of protein, no enzymatic activity |
| IVS2−1G→A (5′ [donor] splice site) | 2 | E72GNF4NX | Truncation between transmembrane segment and A helix | S341P in individual 3300013 (E) | Eliminates entire globular structure of protein, no enzymatic activity |
| 541G→T | 2 | E79D (1) | Between A and B helices | M123V in individual 3300005 (C) | Unknown |
| 636T→C | 3 | I111T (−1) | Between B and B’ helices (on protein surface) | I111T in family 33010 (E), G134X in family 33011 (E), W44R in individual 3300010 (E) | Changes interaction with water |
| 671A→G | 3 | M123V (1) | B’ helix | E79D in individual 3300005 (C) | Unknown |
| 704G→T | 3 | G134X | Truncation between B’ and C helices | I111T in family 33011 (E) | Complete loss of the cytochrome P450 function |
| IVS6−8delTCATACAGGTCATC (3′ [acceptor] splice site) | 7 | Exon7del | In-frame deletion excising all of H and parts of G and I helices | Exon7del in families 33002–33007 (J), 33008 (C), 33009 (C), and 33015 (C) and individuals 3300004 (C), 3300008 (C), and 3300009 (J); exon9del in individual 3300012 (C); H331P in family 33001 (C) | Decreased fatty acid binding, the cell attachment site is removed, shorter long helix |
| 1296A→C | 8 | H331P (−2) | I helix | Exon7del in family 33001 (C) | Change in conformation of central I helix; likely disruption of heme iron coordination |
| 1325T→C | 8 | S341P (−1) | I helix | E72GNFNX in individual 3300013 (E) | Change in conformation of the central I helix, as in the IVS6−8delTCATACAGGTCATC mutation |
| IVS8−2A→G (3′ [acceptor] splice site) | 9 | Exon9del | In-frame deletion excising 46 residues including the K helix | G61S in family 33014 (C), exon7del in individual 3300012 (C), exon9del in individual 3300011 (C) | Removes ERR triad and destabilizes the meander region |
| 1827G→A | 11 | R508H (0) | C terminus | R508H in individual 3300001 (E) | Change in fatty acid binding by reducing stability of loop 495-516 and altering coordination of heme iron |
Note.— Mutations are listed in exon order with the second mutation in individual patients given in column 5, so that heterozygous affected individuals are listed twice. No mutations were found in two isolated individuals of European descent (3301205 and 3300006).
Ethnicity abbreviations are as follows: E = European, J = Japanese, C = Chinese. Because of its uncertain consequences, the IVS2+25delT sequence change is entered only as second mutation and is preceded with a question mark (?).
Figure 4.

Expression pattern of CYP4V2. PCR was performed with cDNA from various tissues, using primers from exons 5 and 11. The lymphocyte (“ptnt”) sample is from patient 4 in family 33002 and displays a 727-bp fragment lacking exon 7. The negative control is H2O, and the positive control is pooled cDNA from human tissues (Universal cDNA pool [Clontech]).
An isolated European patient, 3300013, is heterozygous for an S341P mutation and an IVS2−1G→A change. Information-theory analysis of the splice site with the Delila program (Rogan et al. 1998; Delila Program Web site) shows a decrease in the bit value of this 5′ (donor) splice site from 8.6 to 5.6, consistent with a leaky mutation. In addition, a 5.5-bit cryptic splice site 10 bases downstream is likely to interfere with legitimate splicing at the mutant site. Transformed cell lines are not available to confirm this prediction. An isolated Japanese patient, 3300007, shows a heterozygous G→A transversion at position +1 of the intron 2 5′ (donor) splice site, with the only other change being IVS2+25delT. Fifty control samples, including 12 of Chinese, 16 of Japanese, and 22 of European origin, were screened for these mutations. Only one heterozygous AC transversion in exon 8 (1296A→C in the CYP4V2 mRNA, resulting in a putative H331P) was found in a single Chinese control sample.
Assembly of sequences from overlapping 3′ and 5′ RACE products and PCR fragments extending from exons 1–4, 1–7, and 5–11, all obtained from amplification of Marathon-Ready human retina cDNA, provides a 2,041-bp cDNA sequence extending from the 5′ UTR through the stop codon (TAA) and the 3′ UTR (fig. 2). It encodes a 525–amino acid protein for which BLASTP shows 80% identity and 89% positives to Mus musculus CYP4V3 (shown in fig. B1 of appendix B [online only]). The Motifs routine of the Wisconsin GCG program (Devereux et al. 1984) and SMART (Schultz et al. 1998) confirm that this protein belongs to the P450 heme-thiolate protein superfamily. The Cytochrome P450 Homepage lists CYP4V2 on chromosome 4q35.1, and, since the predicted amino acid sequence shows 98% identity with the listed sequence, this nomenclature is used. CYP4V2 is homologous to other members of CYP450 family 4, suggesting that it might act on fatty acids or steroids. We have previously shown abnormalities in n-3 polyunsaturated fatty acid metabolism in patients with BCD (Lee et al. 2001).
The CYP4V2 structure based on homology modeling is shown in figure 3, and homologous sequences are shown in fig. B1 of appendix B (online only). The predicted transmembrane segment resides near the amino terminus, followed by a globular structural domain typical of the CYP450 family. The globular domain of CYP4V2 comprises 18 helices and β structural segments (fig. 3A). The heme group, shown in blue, is located close to the surface of the protein, coordinated by the I helix towards the protein interior and the L helix superficially.
Possible effects of the BCD mutations based on the atomic model described above are shown in table 2 and figure 3. The most consequential are those in which large parts of the molecule are absent. The intron 2 +1G→A 5′ (donor) splice-site mutation is predicted by conceptual translation to result in skipping of exon 2, resulting in a frameshift in which the exon 1 sequence is followed by a novel four–amino acid sequence (GNFN) prematurely terminated before the conserved globular domain. A second pathogenic mutation, G134X, would be expected to truncate the peptide just within the globular domain (fig. 3B), removing the amino acids that coordinate the heme group and their supporting structures. A third pathogenic mutation is the 15-bp deletion including the 3′ (acceptor) splice site for exon 7, skipping exon 7. Exon 7 encodes amino acids 268–329, which form the G, H, and part of the central I helices and their connecting loops (fig. 3A). Their deletion not only eliminates these parts of the protein structure but also loosens and displaces the remaining I helix and would displace many amino acids co-coordinating the heme group, thus disrupting enzyme activity (fig. 3C). A fourth pathogenic mutation in the 3′ (acceptor) splice site, IVS8−2A→G, is predicted to cause skipping of exon 9, resulting in excision from the protein of the 46 amino acids encoded by exon 9, which include the J’ and K helices. This is predicted to remove the two residues of the ERR triad (E387 and R390 in the K helix), destabilizing the critical meander region. This should result in loss of heme binding, with concomitant loss of enzymatic activity.
Additional sequence changes, although not as dramatic as elimination of large parts of the protein sequence, seem highly likely to affect enzymatic activity or localization of CYP4V2. Two of these are H331P and S341P (fig. 3F and 3H), each occurring in the central I helix. Insertion of proline into this helix is predicted to influence the stability of the helical structure, on which coordination of the heme group depends. A third mutation, W44R (fig. 3D), replaces a hydrophobic aromatic tryptophan in or near the transmembrane segment with a positively charged arginine, which is likely to disrupt insertion of CYP4V2 into the membrane, interfering with protein localization. Finally, since R508 is predicted to coordinate the heme iron along with R465, R508H, although a relatively conservative substitution with a blosum70 score of 0, might influence heme coordination and substrate binding (fig. 3E). Although the remaining coding sequence changes result in alterations of the amino acid sequence and are absent from controls, their effects on CYP4V2 structure and function are more difficult to predict. Two, E79D and M123V, have positive blosum70 scores (+1), emphasizing their conservative nature, whereas two more, G61S and I111T, have negative blosum70 scores (−1).
Expression of CYP4V2 was observed by PCR of human tissue–specific cDNA from human heart, brain, placenta, lung, liver, skeletal muscle, kidney, pancreas, retina, RPE, and lymphocytes (fig. 4), consistent with earlier predictions of broad expression of this gene (Lee et al. 2001). Although the heaviest bands are seen in the retina, this analysis merely detects the presence of the CYP4V2 mRNA and is not quantitative.
Discussion
We have refined the position of the BCD locus to that part of chromosome 4 distal to D4S2924 and describe association of sequence changes in CYP4V2 with BCD. Previous linkage analysis had placed the BCD locus on chromosome 4q35 telomeric of D4S1652 (Jiao et al. 2000). Fine mapping with newly developed markers restricted the BCD critical region to the region distal to D4S2924. We have identified 13 putative mutations in the CYP4V2 gene. Three of these mutations result in termination of the peptide chain before the cytochrome P450 structure is formed, and two more cause deletion of large internal stretches of amino acids including parts of the I helix, which might be critical for protein structure and positioning of the heme group. Although we failed to detect mutations in two patients and detected only a single certain mutation in one patient, it is possible that some mutations were missed in the analysis, particularly those in promoter regions or in intronic regions active in splicing, or large intragenic deletions present in compound heterozygotes.
The CYP4V2 gene comprises 11 exons spanning 19 kb and is a novel member of the cytochrome P450 gene family. The coding sequence begins in exon 1 and continues through exon 11. Although different sequences corresponding to various parts of CYP4V2 are present in the NCBI and Celera databases, analysis of mRNA in the retina and RPE cells showed only the single sequence shown in figure 4. Thus, although alternative splicing or initiation may occur in a small fraction of transcripts in these tissues, the major gene product is the one used in this analysis. In addition, the protein predicted to result from the mRNA structure aligns well with other cytochrome P450 family members, and structural modeling is consistent with a member of the cytochrome P450 family. Thus, it seems likely that this sequence is the functional CYP4V2, although confirmation awaits expression and biochemical characterization.
Comparison of the CYP4V2 sequence with other members of the cytochrome P450 gene family suggests that, as is true for other members of the CYP4 family, it might act in fatty acid metabolism. This would be consistent with the results of Lee et al. (2001), which demonstrated that BCD is characterized by a lower than normal conversion of FA precursors into n-3PUFA. Both of these findings raise the possibility that abnormal lipid metabolism associated with BCD is the result of defects in fatty acid metabolism—possibly binding, elongation, or desaturation. The relationship between the absence of two fatty acid–binding activities in lymphocytes from patients with BCD (Lee et al. 1998) and the genetic cause and pathophysiology of the disease remains unexplained. The genes encoding these proteins do not reside in the BCD region on chromosome 4q35.1 (Jiao et al. 2000), suggesting that they may be involved in subsequent steps of the pathological process in this disease.
CYP4V2 is expressed in a wide variety of tissues, consistent with inclusions and their clinical sequelae occurring in the cornea, retina, and lymphocytes. Although there is no reported functional impairment of tissues other than the cornea and retina, it is unclear whether careful histological characterization of additional tissues has been performed. Just as the inclusions in lymphocytes do not appear to have any functional consequences, inclusions might accumulate in other tissues without causing clinically significant abnormalities.
Here we report refinement of the BCD region to chromosome 4q35 distal to D4S2924 and association of mutations in the CYP4V2 gene with BCD. The sequence changes in the CYP4V2 gene are predicted to have functional consequences on the basis of structural modeling of the gene product and its mutant forms and their absence from 100 control chromosomes. Although the structure of CYP4V2 suggests that it may be active in fatty acid metabolism, the specific enzymatic activity of this cytochrome P450 family member remains to be elucidated. Additional studies are under way to understand the function of CYP450 and to identify the pathogenic mechanisms underlying BCD.
Acknowledgments
We thank the subjects with BCD and their families, for their willing and continued participation in these studies. We also thank Dr. Ying Hu, National Cancer Institute Center for Bioinformatics, National Institutes of Health, for assistance with computer graphic design, and we acknowledge support from Swiss National Science Foundation grant #32-065250.01. R.A.L. is a Senior Scientific Investigator of Research to Prevent Blindness, New York. This work also was supported by a Grant in Aid for Scientific Research (B08457468) from the Ministry of Education, Science and Culture of Japan; by a Japanese Ministry of Health and Welfare grant for the study of chorioretinal degeneration; and, in part, by the Foundation Fighting Blindness and Research to Prevent Blindness.
Appendix A
Table A1.
PCR Primers for 11 Exons, 3 cDNA Fragments, and RACE of the CYP4V2 Gene
|
Sequence |
|||
| Primer | Forward | Reverse | AnnealingTemperature(°C) |
| Exon 1 | 5′CAACCTCGCAGCACCCTCAGAA | 5′ACTTTGGGATGGGGCACTAGCAGT | 66 |
| Exon 2 | 5′ACCTGGCTTCCTCTAACAGTAACA | 5′TTTTTGTGCTGAAATGGCTGAA | 53 |
| Exon 3 | 5′AGATTCGCCTCCTCCCACCTCAC | 5′ACCTGGACTCTTGGCCTCTTGACG | 57 |
| Exon 4 | 5′TGCCAAAAGCATTTGAGAACCTGT | 5′CGCGCTGAAGAGCCCGTCAC | 55 |
| Exon 5 | 5′AGGAAGAACAGGAACAGGGAGTAG | 5′CAACGCAGAAATTGTTAGCAATAA | 53 |
| Exon 6 | 5′GCTTCATGGGATGCGTAATAGC | GAAATGAACGGTGGGGATGGT | 54 |
| Exon 7 | 5′CCTATGTTGTCGAAATGTTGAAAT | 5′TCTGAAGAAGTTGAGCTGGTACTT | 51 |
| Exon 8 | 5′TTTGCAGTCACAGTGCAGTCATCA | 5′CCAGCATCCGGCCTAGTACAGTC | 57 |
| Exon 9–10 | 5′ATGCCATGCCTTGATCCACCTGT | 5′TGGGCAATGTCACATCACATCTCA | 58 |
| Exon 11 | 5′CTCTTCATCTTTAACAGGTGTTCC | 5′CAAAACTCAAAACTTTTTCTTTGT | 52 |
| cDNA752 | 5′GGGCCTCCCGTGCAATCACTAC | 5′ACATCTTTCTCCTGGAGCGCCATTTGTTTC | 58 |
| cDNA637 | 5′GGCCACGCGCTGCTGATGAAG | 5′GCCCGTTCCGCGATGACACTGT | 57 |
| cDNA913 | 5′TGATTCCGAGTATGTCCGTGCAGTTTATAG |
5′TCCTCTTCAACTTGATCCAGATGCCATTAC |
56 |
| Gene-Specific Primer |
Nested Gene-Specific Primer |
||
| 5′ RACE | 5′GCCCGTTCCGCGATGACACTGT | 5′TTTGTGTTCCCATCCTTCTTTAAACATAAG | 68 |
| 3′ RACE | 5′GGCCACGCGCTGCTGATGAAG | 5′GAAACAAATGGCGCTCCAGGAGAAAGATGT | 68 |
Appendix B
Figure B1.

Alignment of CYP4V2 with CYP450 family members. Mus musculus CYP4V3 shows the highest identify, whereas rabbit CYP2C5 and Bacillus mesenterium CYPBM3, whose structures have been determined crystallographically, are further removed. CYPBM3 was used to model the high-homology area surrounding the active site, and CYP2C5 was used to model the amino-terminal regions for which homology to CYPBM3 was low.
Electronic-Database Information
The URLs for data presented herein are as follows:
- Bioccelerator at European Molecular Biology Laboratory, http://eta.embl-heidelberg.de:8000/
- Cytochrome P450 Homepage, http://drnelson.utmem.edu/cytochromep450.html
- Delila Program, Laboratory of Computational and Experimental Biology, National Cancer Institute, National Institutes of Health, http://www.lecb.ncifcrf.gov/~toms/delila.html
- myScience, http://myscience.appliedbiosystems.com/
- National Center for Biotechnology Information, http://www.ncbi.nlm.nih.gov/
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for BCD) [PubMed]
- Protein Data Bank, Research Collaboratory for Structural Bioinformatics, http://www.rcsb.org/pdb/ (for mammalian CYP2C5 [ID 1dt6] and bacterial CYPBM3 [ID 2hpd])
References
- Abola E, Bernstein FC, Bryant SH, Koetzle TF, Weng J (1987) Protein data bank. In: Allen FH, Bergerhoff G, Sievers R (eds) Crystallographic databases: information content, software systems, scientific applications. Data Commission of the International Union of Crystallography, Cambridge, pp 107–132 [Google Scholar]
- Ayyagari R, Nestorowicz A, Li Y, Chandrasekharappa S, Chinault C, van Tuinen P, Smith RJH, Hejtmancik JF, Permutt MA (1996) Construction of a YAC contig encompassing the Usher syndrome type 1C and familial hyperinsulinism loci on chromosome 11p14-15.1. Genome Res 6:504–514 [DOI] [PubMed] [Google Scholar]
- Bietti G (1937) Ueber familiaeres vorkommen von “retinitis punctata albescens” (verbunden mit “dystrophia marginalis cristallinea corneae”), glitzern des glaskoerpers und anderen degenerativen augenveraenderungen. Klin Mbl Augenheilk 99:737–757 [Google Scholar]
- Cottingham RW, Idury RM, Schaffer AA (1993) Faster sequential genetic linkage computations. Am J Hum Genet 53:252–263 [PMC free article] [PubMed] [Google Scholar]
- Devereux J, Haeberli P, Smithies O (1984) A comprehensive set of sequence analysis programs for the VAX. Nucleic Acids Res 12:387–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, Henikoff JG (1992) Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci USA 89:10915–10919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu DN (1982) Genetic aspects of retinitis pigmentosa in China. Am J Med Genet 12:51–56 [DOI] [PubMed] [Google Scholar]
- ——— (1983) Ophthalmic genetics in China. Ophthal Paed Genet 2:39–45 [Google Scholar]
- Jiao X, Munier FL, Iwata F, Hayakawa M, Kanai A, Lee J, Schorderet DF, Chen MS, Kaiser-Kupfer M, Hejtmancik JF (2000) Genetic linkage of Bietti crystalline corneoretinal dystrophy to chromosome 4q35. Am J Hum Genet 67:1309–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser-Kupfer MI, Chan CC, Markello TC, Crawford MA, Caruso RC, Csaky KG, Guo J, Gahl WA (1994) Clinical biochemical and pathologic correlations in Bietti’s crystalline dystrophy. Am J Ophthalmol 118:569–582 [DOI] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst 26:283–291 10.1107/S0021889892009944 [DOI] [Google Scholar]
- Lathrop GM, Lalouel JM (1984) Easy calculations of lod scores and genetic risks on small computers. Am J Hum Genet 36:460–465 [PMC free article] [PubMed] [Google Scholar]
- Lee C (1994) Predicting protein mutant energetics by self-consistent ensemble optimization. J Mol Biol 236:918–939 10.1006/jmbi.1994.1198 [DOI] [PubMed] [Google Scholar]
- Lee C, Subbiah S (1991) Prediction of protein side-chain conformation by packing optimization. J Mol Biol 217:373–388 [DOI] [PubMed] [Google Scholar]
- Lee J, Jiao X, Hejtmancik JF, Kaiser-Kupfer M, Chader GJ (1998) Identification, isolation, and characterization of a 32-kDa fatty acid-binding protein missing from lymphocytes in humans with Bietti crystalline dystrophy (BCD). Mol Genet Metab 65:143–154 10.1006/mgme.1998.2723 [DOI] [PubMed] [Google Scholar]
- Lee J, Jiao X, Hejtmancik JF, Kaiser-Kupfer M, Gahl WA, Markello TC, Guo J, Chader GJ (2001) The metabolism of fatty acids in human Bietti crystalline dystrophy. Invest Ophthalmol Vis Sci 42:1707–1714 [PubMed] [Google Scholar]
- Levitt M (1992) Accurate modeling of protein conformation by automatic segment matching. J Mol Biol 226:507–533 [DOI] [PubMed] [Google Scholar]
- Mataftsi A, Zografos L, Milla E, Munier FL. Bietti’s crystalline corneoretinal dystrophy: a cross-sectional study. Retina (in press) [DOI] [PubMed] [Google Scholar]
- Needleman SB, Wunsch CD (1970) A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol 48:443–453 [DOI] [PubMed] [Google Scholar]
- Rogan PK, Faux BM, Schneider TD (1998) Information analysis of human splice site mutations. Hum Mutat 12:153–171 [DOI] [PubMed] [Google Scholar]
- Schultz J, Milpetz F, Bork P, Ponting CP (1998) SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci USA 95:5857–5864 10.1073/pnas.95.11.5857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RJH, Holcomb JD, Daiger SP, Caskey CT, Pelias MZ, Alford BR, Fontenot DD, Hejtmancik JF (1989) Exclusion of Usher syndrome gene from much of chromosome 4. Cytogenet Cell Genet 50:102–106 [DOI] [PubMed] [Google Scholar]
- Welch RB (1977) Bietti’s tapetoretinal degeneration with marginal corneal dystrophy crystalline retinopathy. Trans Am Ophthalmol Soc 75:164–179 [PMC free article] [PubMed] [Google Scholar]
- Wilson DJ, Weleber RG, Klein ML, Welch RB, Green WR (1989) Bietti’s crystalline dystrophy. A clinicopathologic correlative study. Arch Ophthalmol 107:213–221 [DOI] [PubMed] [Google Scholar]