

Abstract
Historically, association tests were limited to single variants, so that the allele was considered the basic unit for association testing. As marker density increases and indirect approaches are used to assess association through linkage disequilibrium, association is now frequently considered at the haplotypic level. We suggest that there are difficulties in replicating association findings at the single-nucleotide–polymorphism (SNP) or the haplotype level, and we propose a shift toward a gene-based approach in which all common variation within a candidate gene is considered jointly. Inconsistencies arising from population differences are more readily resolved by use of a gene-based approach rather than either a SNP-based or a haplotype-based approach. A gene-based approach captures all of the potential risk-conferring variations; thus, negative findings are subject only to the issue of power. In addition, chance findings due to multiple testing can be readily accounted for by use of a genewide-significance level. Meta-analysis procedures can be formalized for gene-based methods through the combination of P values. It is only a matter of time before all variation within genes is mapped, at which point the gene-based approach will become the natural end point for association analysis and will inform our search for functional variants relevant to disease etiology.
Introduction
The past decade has seen a dramatic increase in the use of association studies for the genetic analysis of complex disorders (Lander and Schork 1994; Risch 2000). The introduction of the transmission/disequilibrium test was one important landmark in the popularization of association studies (Spielman et al. 1993), followed by the demonstration by Risch and Merikangas (1996) of the potential feasibility of genomewide association studies and the comparatively greater power of association over linkage for detecting genes of minor or modest effect size. The enormous promise of association analysis is beginning to be realized through the improved detail and resolution of genetic maps, including the imminent completion of the International Haplotype Mapping (HapMap) Project (Couzin 2002; Stumpf and Goldstein 2003) and the rapid development of high-throughput genotyping technologies (Collins et al. 1997).
This explosion of association studies has, however, given rise to some controversy concerning study design, statistical analysis, and interpretation of findings. Many of these issues have been the subject of recent reviews (Risch and Merikangas 1996; Terwilliger and Weiss 1998; Schork et al. 2000; Cardon and Bell 2001; Clayton and McKeigue 2001; Reich and Lander 2001; Lewis 2002). In the present article, we discuss the fundamental question of what should constitute the basic genetic component to be considered for association with a complex disorder. Historically, association has referred primarily to allelic association, implicating the allele as the basic unit of analysis. With increasing marker density and the use of an indirect approach to association through linkage disequilibrium (LD), association is now often considered at the haplotypic level. These levels of analyses are, however, potentially problematic in the context of replication. Must a replication study obtain a pattern of association exactly the same as that of the original finding to count as supportive evidence? Conversely, can a negative finding be regarded as nonreplication if only the associated allele or haplotype from the initial study is examined? We argue that the current tendency to perform association analysis at the SNP or the haplotype level is problematic, and we suggest a move toward a gene-based approach in which all variants within a putative gene are considered jointly.
Complex Disorder/Complex Association
Risch and Merikangas (1996) identified SNPs as the putative genetic risk factors for association testing and proposed a genomewide-significance level set at the very low value of 10−8 to allow for the total number of intragenic SNPs in the human genome. Since most current studies are underpowered to achieve such a stringent level of significance, replications are usually necessary for the confirmation of an association finding. Sufficient data have been gathered to gain some insight into the fate of putative association findings, whether they are likely or unlikely to be confirmed subsequently. In a heroic study, Hirschhorn et al. (2002) conducted a meta-analysis of 166 initial association findings and their subsequent attempted replications, for a large number of complex disorders. They included putative association findings for which at least two subsequent replication attempts have been published, and they determined that only 6 of the 166 initial findings have been reliably replicated (with >75% of replication studies showing significant results). Of the other initial findings, 97 had at least one significant replication, and 63 have not been replicated. This excellent review is, however, restricted to only replications of precisely the same polymorphism as the initial finding and does not take account of supporting evidence from more-complex patterns of associations with other polymorphisms in the same gene. Similar surveys of the association literature have been conducted, yielding successful replication rates of 16%–30% (Ioannidis 2003; Ioannidis et al. 2003; Lohmueller et al. 2003).
The recent association findings on schizophrenia serve to illustrate the complexity of association findings that can arise from complex disorders. Following an initial study by Straub et al. (2002) that demonstrated an association with schizophrenia and dysbindin, located at 6p22.3, four attempts have been made at replication in six different populations (Morris et al. 2003; Schwab et al. 2003; Van Den Bogaert et al. 2003; Williams et al. 2004). All of these attempted replication studies examined more than just the most significant SNP from the initial study, but none chose exactly the same SNPs that comprise the original high-risk haplotype. The Schwab et al. (2003) and Van Den Bogaert et al. (2003) studies examined fewer SNPs than the original core high-risk haplotype. Schwab et al. (2003) concluded replication by finding an association signal in their subset of the markers, as did Van Den Bogaert et al. (2003) in their Swedish sample but not in their German or Polish samples. Given that only a subset of markers was used, however, it is not clear whether the absence of association between schizophrenia and dysbindin in the German or Polish samples should be regarded as nonreplication. Also of note is that the Schwab et al. study (2003) found evidence for association with the common haplotype, rather than the rarer haplotype identified by Straub et al. (2002). Morris et al. (2003) failed to replicate the association in an Irish sample, though, again, they did not use exactly the same markers as were used in the original study but typed additional markers across the gene. Given the increase in marker density in the study by Morris et al. (2003), this negative finding carried greater weight than did less extensive replication attempts. Continuing this pattern of thorough investigation, Williams et al. (2004) mapped a newly discovered region of dysbindin—the promoter—and, by doing so, found significant association in both the Irish sample of Morris et al. (2003) and another sample from Wales. By a parsing of the gene to capture more variation, a strong negative finding has now been transformed into a substantial positive, with the Irish sample (Morris et al. 2003) providing most of the evidence. For the location of the SNPs in the original and subsequent studies of dysbindin, see figure 1.
Figure 1.

Adapted from figure 1 of Williams et al. (2004). Blackened boxes 1–13 represent the coding regions of dysbindin; adjacent unblackened boxes represent alternative splicing sites. P1–P4 represent the four hypothesized promoter regions of dysbindin. The numbered loci constitute the initial 8-marker haplotype specified by Straub et al. (2002). The lettered loci are the SNPs Williams et al. (2004) discovered and analyzed. The Roman numerals specify the additional markers typed by Schwab et al. (2003). Van Den Bogaert et al. (2003) typed 2, 4, 5, 7, and 8, whereas Schwab et al. (2003) typed 2, 3, 4, 6, 7, and 8.
Another example of a complex pattern of association is that between alcoholism and variants at the aldehyde dehydrogenase 2 (ALDH2) and alcohol dehydrogenase (ADH) genes. The low-activity variant ALDH2*487Lys, present in East Asian populations at 30% allele frequency, is responsible for the flush reaction to alcohol and is protective against alcoholism (Harada et al. 1981). However, this allele is extremely rare in European populations (Shibuya and Yoshida 1988; Peterson et al. 1999). The ADH genes, which cluster on chromosomal region 4q21-q25, can be grouped into five classes. The bulk of the known functional variation is restricted to the class I genes, consisting of three ADH genes in an 80-kb region. These ADH genes demonstrate strong associations with alcoholism, but there is much variability between the patterns of association in different populations. The reason may be that the frequencies of the alleles in these genes have been shown to be disparate in different populations, such as an enrichment of the ADH1B*47His allele in East Asian populations (Goedde et al. 1992; Thomasson et al. 1994; Neumark et al. 1998). Other population-based studies have detected significant differences in allele frequencies—as well as the pattern of LD—across a number of loci in this gene cluster (for review, see Osier et al. [2002]).
These examples illustrate some of possible reasons for the emergence of complex patterns of association findings in complex disorders. First, there may be important differences in allele frequency or LD structure across different populations. Thus, in different populations, the same high-risk allele may have a very different pattern of association with marker alleles and haplotypes. This may be particularly relevant under a common-disease-common-variant (CDCV) hypothesis (Chakravarti 1999; Weiss and Clark 2002), in which the age of the mutation allows for differential recombination histories in different populations (Pritchard and Przeworski 2001). The situation may be further complicated by the presence of hidden stratification in some populations, producing spurious association or altering the pattern of a true association (Morton and Collins 1998; Pritchard et al. 2000; Thomas and Witte 2002; Stumpf and Goldstein 2003; Freedman et al. 2004). Other sources of complexity are allelic heterogeneity, in which different alleles at the same locus are responsible for increased disease risk in different populations, and locus heterogeneity, in which alleles at different loci are responsible for increased disease risk in different populations. Both of these scenarios are likely when there are multiple rare variants (MRVs) that are fairly recent in origin. Note that, currently, little is known about the true nature of allelic heterogeneity with respect to disease (Pritchard 2001; Reich and Lander 2001). Aside from these genetically driven phenomena, study design and publication bias may also lead to complex patterns (Colhoun et al. 2003). Low-powered attempts at replication that conclude no evidence for association can be potentially misleading. Conversely, some spurious positive findings would be expected from multiple analyses from multiple small studies, especially when subgroups are identified by data exploration and then are pursued and reported.
Levels of Analysis and Replication
Some of these problems, we suggest, would be considerably reduced by performing association analyses and replications at the level of the gene. Currently, the levels of analysis and replication that are most common in the literature are SNP, haplotype, and functional variant. To clarify, SNP-based methods are predicated on genotyping only the most significant SNP from the prior study. Replication at the SNP level runs a high risk of false-negative results, because of the failure to include or tag relevant functional variants within the replication sample. Replication at the haplotype level uses the same markers as those in the initial study and may be potentially more powerful than analysis of a single SNP (Zhang et al. 2002a, 2003) because more variation is studied and tagged. However, use of the same markers as those in the initial study implicitly assumes that the allele frequencies and haplotype structure in the region are the same in the two populations, which may not be a valid assumption. The complexities of cross-population investigations are not fully understood, and only with more extensive data concerning population differences will we be able to accommodate the potential inconsistencies arising from such studies (Helmuth 2001; Couzin 2002; van den Oord and Neale 2003). The testing of only known functional variants has the advantage of biological plausibility but is predicated on full knowledge of gene function and how this is influenced by genetic variation.
In gene-based replication, the gene identified by the initial study is consequently examined for association with effectively all genetic variants in the intragenic and regulatory regions. There are several reasons for regarding gene-based replication as a gold standard. First and foremost, genes are the functional unit of the human genome, and the positions, sequence, and function of genes are highly consistent across diverse human populations. This universality is considerably greater than that of either a SNP or a haplotype. Furthermore, gene-based replication implies that each population will be studied with due account of local allele frequencies and LD structure and should therefore overcome many of the problems with nonreplication due to population differences, under both the CDCV and the MRV hypotheses. Third, gene-based replication simplifies the multiple-testing problem by conveniently dividing it into two stages, dealing first with the multiple variants within a gene and then with the multiple genes in the genome.
Methodological Issues of a Gene-Based Approach
A gene-based approach would ideally consider all variation within a gene and its regulatory region for association with the phenotype. The presence of LD between some nearby variants means that full information often can be obtained by genotyping only a subset of the variants and by considering their joint haplotypes (Daly et al. 2001; Johnson et al. 2001; Patil et al. 2001; Gabriel et al. 2002; Ardlie et al. 2002). By use of this haplotype-tagging approach, it is possible to study all but rare genetic variants by genotyping a limited number of relatively common SNPs. A number of groups have developed methods for selecting haplotype-tagging SNPs from a larger set of SNPs (Byng et al. 2003; Chapman et al. 2003; Weale et al. 2003; Lowe et al. 2004; Zhang et al. 2004). A genewide approach also requires the analysis of multilocus genotype data; methods for doing this, taking into account phase uncertainty, are being rapidly developed (Schaid et al. 2002; Zaykin et al. 2002; Zhang et al. 2002b; North et al. 2003; Seltman et al. 2003; Tanck et al. 2003; Thomas et al. 2003; Zhao et al. 2003; Sham et al. 2004).
Direct versus Indirect Association
At present, direct association mapping of functional variants is limited by incomplete knowledge about functional variation. Most current association mapping is indirect, with reliance on LD between a disease susceptibility allele and either a single marker allele or a multilocus haplotype. Much recent methodological work has been conducted to optimize this indirect approach, including the investigation of haplotype-block structure and techniques for selecting haplotype-tagging SNPs. The systematization of the indirect approach is the aim of the HapMap Project, through a genomewide study of haplotype-block structure in several populations. As we approach saturation of genomic variation, it will become feasible for association studies to examine all the variants within and around putative genes, including the functional variants. This would represent a shift toward a direct approach to association analysis, with some important consequences. First, haplotype tagging would become less relevant and would be supplanted by tests of the main effects of the variants. In other words, the analysis would shift from a haplotype-scoring to a locus-scoring framework (Chapman et al. 2003). Second, haplotype methods would assume a new role in association studies, that of testing for cis-interactions in addition to main effects.
The haplotype-tagging approach is based on the existence of strong LD and limited haploptype diversity in small genomic regions. Thus, increasing the number of typed SNPs beyond the haplotype-tagging SNPs does not yield additional unique haplotypes and therefore does not add to the dimensionality (degrees of freedom) of the association test. In a similar fashion, a test for allele-specific main effects will also cease to increase in dimensionality, at or before the point when haplotype diversity is fully captured by the typed markers. In a regression-modeling framework, the maximum dimension is restricted by colinearity between alleles, requiring the removal of some alleles. Similarly, in the case of the multilocus score test of Chapman et al. (2003), colinearity will be manifested in a variance-covariance matrix that is short of full rank.
When few markers are examined, the dimensionality of the haplotype-scoring test is greater than that of the locus-scoring test (Chapman et al. 2003). However, as the number of typed markers increases toward the point of saturation, the dimensionality of the locus-scoring test might, in some cases, converge to that of the haplotype-scoring test. In the event that each haplotype is uniquely tagged by a single allele, the two tests will converge. When this is not the case, the difference between the two tests represents cis-interactions between alleles. Until marker saturation is achieved, a disease association that is stronger with a haplotype than with the constituent alleles is consistent with both cis-interaction and the tagging of a rare variant by the haplotype. The extent and importance of cis-interactions in the human genome are as yet unknown.
Currently, complete sequencing of candidate genes is feasible only on a limited scale and is seldom performed comprehensively on entire genes or on an adequate number of individuals. Indirect association via haplotype tagging is an efficient approach at present, and should remain so for the near future, especially when screening a large number of candidate genes. However, haplotype tagging in the absence of complete knowledge of genetic variation runs the risk of not detecting some rare variants, thereby missing an association signal and underestimating the dimensionality of the test for the entire gene.
Genewide Significance
The use of a gene-based approach to association would require some adjustment of our testing procedures and redefinition of significance levels. By analogy to the convention in linkage analysis of adopting a genomewide-significance level—even for studies that focus on restricted parts of the genome (Morton 1955; Lander and Kruglyak 1995)—it can be argued that one should adopt a significance level that reflects all the variation present in and around the gene, whether or not the study achieved complete coverage of the variation. This level of association testing could be termed “genewide significance,” whereas “genomewide significance” would also take into account the examination of all the genes in the genome. To determine the genewide-significance level, it is necessary to know the dimensionality of both the employed association test (Dtest) and the complete genewide-association test (Dgene). The ratio
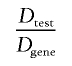 |
(abbreviated as “r”) is an indication of the proportion of all possible tests in the entire gene that have been performed for the study. By use of the Bonferroni procedure, the genewide significance is obtained by multiplication of the nominal P value of the test by the ratio r. More accurately, the corrected genewide significance is given by 1-(1-P)r, as is important for meta-analysis (see the “Meta-Analysis for Gene-Based Studies” section).
A fundamental issue in the assessment of Dgene is that variability will increase almost indefinitely by including progressively rarer variants and by sequencing larger samples. In practice, however, very rare alleles do not contribute to dimensionality, since they cannot contribute to the evidence for association, at least in a conventional test (e.g., a χ2 test). There is a minimum number of copies of an allele present in a sample, below which it is impossible to demonstrate a significant association between the allele and disease. For example, when the numbers of cases and controls are equal, the most extreme association occurs when all copies of the variants are found among cases or among controls, and the nominal P value associated with such a scenario with c copies of the variant is ∼.5c−1. To reach a nominal significance of .05, the minimum value of c must be 6. Figure 2 shows the minimal sample allele frequency that can be potentially significant at nominal, genewide, and genomewide significance (see the “Genomewide Significance” section). We define our genewide- and, consequently, genomewide-significance level in accordance with the findings of Crawford et al. (2004), who sequenced 100 candidate genes in two populations, one of African descent and the other of European descent, and reported ∼3,000 haplotypes across the 100 genes, formed by common SNPs with a minor-allele frequency of at least 5%. These data suggest an average genewide dimensionality of 30, yielding a Bonferroni correction of 30 when a single SNP is tested. It is clearly more satisfactory to determine genewide dimensionality for the specific genes being investigated, rather than an average value, but this requires a detailed sequencing effort to find all the variants and to delineate their LD relationships.
Figure 2.

Minimum sample allele frequency for achieving different levels of significance. We assume equal numbers of cases and controls in an association sample and plot the behavior of the minimum-allele frequency capable of demonstrating significant association at the nominal (.05), genewide (.00167), and genomewide level (5.56×10-8) against total sample size. The genewide significance assumes 30 detectable haplotypes across the gene, in accordance with the Crawford et al. (2004) estimate, and the genomewide level assumes 30,000 genes. The minimum-allele frequency is derived from the instance in which all copies of the allele (c) are found in either the cases (disease predisposing) or controls (disease protective). The significance is defined as 0.5c-1, since, under the null hypothesis, the first copy of the allele must be in either the cases or controls, and each subsequent allele is regarded as independent and has .5 probability of being in the same group as the first allele. Note the convergence of the nominal and genewide frequencies as sample size increases.
Whereas complete marker saturation is not yet achieved, data from the HapMap Project are already providing a lower limit to the dimensionality of many genes. Differences in the extent of genetic variation exist between populations (Crawford et al. 2004) and should be taken into account in determining the dimensionality of genewide tests.
Genomewide Significance
The conversion of genewide to genomewide significance is relatively straightforward, since the approximate number of genes in the human genome is known, and these genes are likely to represent largely independent units. The different approaches to the problem of multiple testing in association studies have been reviewed by several authors (Altshuler et al. 1998; Colhoun et al. 2003; van den Oord and Sullivan 2003; Thomas and Clayton 2004; Wacholder et al. 2004). Under a classical frequentist approach, a genomewide significance (P*) can be obtained simply from a genewide significance (P) by the formula P*=1-(1-P)m (≈mp for small values of P, where m is the number of potential contributory genes in the genome). The most conservative approach, under the assumption of total ignorance, would set the value of m at the total number of genes in the genome, currently estimated at ∼30,000. Taking this most conservative approach would convert a nominal genewide-significance level of 1.5×10-7 to a genomewide-significance level of .05. However, if the set of plausible contributory genes can be restricted—for example, to only that expressed in a particular tissue or organ—then it may be reasonable to reduce the value of m accordingly.
From a Bayesian point of view, the critical question at the outset is that of prior probability of association. A simple view of this prior probability is that it is the proportion of all potential genes that we expect to have a detectable impact on the phenotype. From van den Oord and Sullivan (2003), the threshold value for a genewide significance to be considered significant at a genomewide level is given by
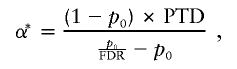 |
where PTD is power to detect association, p0 is the proportion of genes expected to have no effect, and FDR is the false discovery rate. For example, if we assume 10 genes to be contributing to a phenotype and that all 30,000 human genes are potential candidates, then p0=29,990/30,000≈.9997. If we further assume a minimum 80% power to detect and an FDR of .05—which means that, of all tests deemed significant, 5% will be false discoveries—then α*≈.000014. In other words, a genewide significance of ∼1.4×10-5 would be considered to be significant genomewide. For a replication attempt, it might be appropriate to consider a smaller value for p0, so that the threshold for genomewide significance would be lower. Ideally, p0 should reflect the posterior probability of association, given the initial finding. If, for example, an appropriate value of p0—considering the strength of the initial finding—is 0.8, then α*≈.01. Therefore, at this strength of prior evidence, the nominal genewide significance requires little correction.
Another approach to the problem of multiple testing at the genomewide level is to adopt an empirical Bayes approach in which the proportion of tests deviating from the null hypothesis is estimated internally from the data (Greenland and Robins 1991). This approach is particularly attractive when a large number of genes have been tested, as in a microarray experiment. Examples of this approach include those proposed by Benjamini and Hochberg (1995), Storey and Tibshirani (2003), and Gadbury et al. (in press).
Meta-Analysis for Gene-Based Studies
Meta-analysis of genetic association studies, as for epidemiological association studies, usually looks to the combination of odds ratios to give an overall odds ratio and to the variation between the same odds ratios to give a test for heterogeneity. This presents some difficulties for a gene-based approach, since, in general, the association cannot be summarized by a single odds ratio. A meta-analysis can, however, be conducted on the basis of the combination of P values, by use of a method proposed by Fisher (1954). The test involves calculating the χ2 statistic
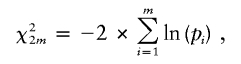 |
where m is the number of P values, pi is the P value of the ith study, and χ2 is distributed as χ2 with 2m df, under the null hypothesis. Justification for such a method has been derived for linkage (Allison and Heo 1998).
One important issue in conducting such a meta-analysis is whether the P values should be corrected for multiple testing prior to combination. Correction to the genewide level of significance prior to combination is strongly recommended, since failure to do so can lead to liberal combined P values, even if these are subsequently adjusted in accordance with the maximum dimension of the contributing tests. The conversion of a genewide significance level obtained from such a meta-analysis to a genomewide-significance level is, however, complicated by the poorly understood relationship between meta-analysis and multiple-testing correction.
Strategies for Screening a Large Number of Genes
For a complex disorder of uncertain pathophysiology, the number of genes that might be involved in etiology can be enormous, and a thorough study of all genetic variation in every potential candidate gene would be a very costly exercise. Budgetary constraints may therefore necessitate efficient methods for screening a large number of genes to select a subset of candidate genes for more intensive study.
One obvious way of screening a large number of genes is to adopt an indirect approach by selecting a small number of markers in each gene for genotyping, aiming to capture most but not all the variation in the gene. This is likely to be a cost-effective strategy, because the relationship between the number of markers genotyped and the proportion of tagged variation is likely to be nonlinear, such that the marginal increase in saturation tends to be less for each additional typed marker. Thus, genotyping four well-chosen SNPs within a gene may enable the tagging of 50% of all the variants within that gene, but genotyping an additional four SNPs may provide only an extra 25% saturation. Therefore, when a large number of genes has to be screened, it does not make sense to insist on complete saturation of selected genes, but to increase the number of genes studied by adopting an incomplete yet reasonably high level of saturation.
Another approach to efficiently screen a large number of candidate genes is through DNA pooling, which allows allele frequencies to be measured directly in groups rather than calculated from individual genotypes (Sham et al. 2002). The reduction of genotyping cost per marker makes it possible to screen a much larger number of markers. The application of microarray genotyping technology on pooled DNA is a particularly promising way forward.
What to Do with Very Rare Variants?
The scenario in which the disorder is influenced by MRVs in a gene, each of minor or modest effect, is potentially problematic for association studies. The problem is that each risk-increasing variant may be so rare that there is only a handful of copies in a sample, making it impossible to achieve even a nominal level of significance. In this scenario, an indirect tagging approach may still be effective, since there is likely to be an uneven distribution of risk-increasing variants across the marker haplotypes (Sham et al. 2000). When a direct approach that aims to capture all genetic variants in cases and controls is adopted, evidence for the involvement of the gene may be gleaned from a greater number of variants in cases than in controls or from different distributions of the variants in the gene, with clustering in certain positions among the cases. This is an area of methodology that requires further development, as is the combination of information from common and rare variants.
Functional Considerations
Statistical association is an important—but not the only—consideration for concluding whether variation in a gene plays a role in causing individual differences in disease susceptibility. Other relevant considerations include biological plausibility, animal models, and gene-expression studies (Page et al. 2003). Since genetic variation must be ultimately translated into differences in gene function, to affect disease risk, a gene-based approach provides a convenient framework for integrating statistical and functional sources of information.
Ultimately, when we have full knowledge of functional consequences of genetic variation at a molecular and cellular level, the gene-based approach could be restricted to functional genetic variation only. However, the acquisition of this level of knowledge will be a gradual process, and a systematic approach covering all genetic variation within and around genes will likely remain efficient for the genetic dissection of complex disorders.
Conclusions
Adoption of a gene-based approach to association analysis and replication is becoming feasible and has many advantages. In contrast to SNP-based and haplotype-based approaches, a gene-based approach is less susceptible to erroneous findings due to genetic differences between populations. By capturing all of the potential risk-conferring variations, a gene-based approach is capable of excluding association, subject only to the issue of power. Use of a genewide-significance level should reduce the problem of chance findings due to multiple testing. A gene-based approach lends itself to meta-analysis of combined data from multiple studies. As our knowledge of the variation in genes grows, a gene-based approach will become the natural end point for association analysis and will provide pointers for genetic analysis at the functional level.
A gene-based approach requires detailed knowledge of genetic variation in coding sequences as well as regulatory and other regions affecting gene function. This level of knowledge is not generally available, at present. In the future, when knowledge of genetic variation allows a gene-based approach to be routinely employed, gene-based studies may become the preferred option for the genetic dissection of complex traits. At present, a gene-based approach should ideally be used for the replication of previous positive-association findings or for the study of very strong candidate genes. The gene-based approach may not be necessary when there is very detailed knowledge about functional variation in the gene and would not be efficient for screening a very large number of genes.
Acknowledgments
The work was supported by a Distinguished Visiting Professorship from the University of Hong Kong, by U.K. Medical Research Council grant G9901258 (to P.C.S.), by Wellcome Trust grant 055379 (to P.C.S.), and by National Eye Institute grant EY-12562 (to P.C.S.). The authors also thank Edwin van den Oord and Peter Visscher for helpful discussion. In addition, the authors thank the anonymous reviewer for insightful comments.
Electronic-Database Information
The URL for data presented herein is as follows:
- HapMap, http://www.hapmap.org/
References
- Allison DB, Heo M (1998) Meta-analysis of linkage data under worst-case conditions: a demonstration using the human OB region. Genetics 148:859–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altshuler D, Kruglyak L, Lander E (1998) Genetic polymorphisms and disease. N Engl J Med 338:1626 10.1056/NEJM199805283382214 [DOI] [PubMed] [Google Scholar]
- Ardlie KG, Kruglyak L, Seielstad M (2002) Patterns of linkage disequilibrium in the human genome. Nat Rev Genet 3:299–309 10.1038/nrg777 [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc Ser B 57:289–300 [Google Scholar]
- Byng MC, Whittaker JC, Cuthbert AP, Mathew CG, Lewis CM (2003) SNP subset selection for genetic association studies. Ann Hum Genet 67:543–556 10.1046/j.1529-8817.2003.00055.x [DOI] [PubMed] [Google Scholar]
- Cardon LR, Bell JI (2001) Association study designs for complex diseases. Nat Rev Genet 2:91–99 10.1038/35052543 [DOI] [PubMed] [Google Scholar]
- Chakravarti A (1999) Population genetics: making sense out of sequence. Nat Genet Suppl 21:56–60 9915503 [DOI] [PubMed] [Google Scholar]
- Chapman JM, Cooper JD, Todd JA, Clayton DG (2003) Detecting disease associations due to linkage disequilibrium using haplotype tags: a class of tests and the determinants of statistical power. Hum Hered 56:18–31 10.1159/000073729 [DOI] [PubMed] [Google Scholar]
- Clayton D, McKeigue PM (2001) Epidemiological methods for studying genes and environmental factors in complex diseases. Lancet 358:1356–1360 10.1016/S0140-6736(01)06418-2 [DOI] [PubMed] [Google Scholar]
- Colhoun HM, McKeigue PM, Davey Smith G (2003) Problems of reporting genetic associations with complex outcomes. Lancet 361:865–872 10.1016/S0140-6736(03)12715-8 [DOI] [PubMed] [Google Scholar]
- Collins FS, Guyer MS, Charkravarti A (1997) Variations on a theme: cataloguing human DNA sequence variation. Science 278:1580–1581 10.1126/science.278.5343.1580 [DOI] [PubMed] [Google Scholar]
- Couzin J (2002) Genomics: new mapping project splits the community. Science 296:1391–1393 10.1126/science.296.5572.1391 [DOI] [PubMed] [Google Scholar]
- Crawford DC, Carlson CS, Rieder MJ, Carrington DP, Yi Q, Smith JD, Eberle MA, Kruglyak L, Nickerson DA (2004) Haplotype diversity across 100 candidate genes for inflammation, lipid metabolism, and blood pressure regulation in two populations. Am J Hum Genet 74:610–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly MJ, Rioux JD, Schaffner SF, Hudson TJ, Lander ES (2001) High-resolution haplotype structure in the human genome. Nat Genet 29:229–232 10.1038/ng1001-229 [DOI] [PubMed] [Google Scholar]
- Fisher RA (1954) Statistical methods for research workers, 12th ed. Hafner, New York [Google Scholar]
- Freedman ML, Reich D, Penney KL, McDonald GJ, Mignault AA, Patterson N, Gabriel SB, Topol EJ, Smoller JW, Pato CN, Pato MT, Petryshen TL, Kolonel LN, Lander ES, Sklar P, Henderson B, Hirschhorn JN, Altshuler D (2004) Assessing the impact of population stratification on genetic association studies. Nat Genet 36:388–393 10.1038/ng1333 [DOI] [PubMed] [Google Scholar]
- Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly MJ, Altshuler D (2002) The structure of haplotype blocks in the human genome. Science 296:2225–2229 10.1126/science.1069424 [DOI] [PubMed] [Google Scholar]
- Gadbury GL, Page GP, Edwards J, Kayo T, Prolla TA, Weindruch R, Permana PA, Mountz J, Allison DB. Power and sample size estimation in high dimensional biology. Stat Meth Med Res (in press) [Google Scholar]
- Goedde HW, Agarwal DP, Fritze G, Meier-Tackmann D, Singh S, Beckman G, Bhatia K, Chen LZ (1992) Distribution of ADH2 and ALDH2 genotypes in different populations. Hum Genet 88:344–346 [DOI] [PubMed] [Google Scholar]
- Greenland S, Robins JM (1991) Empirical-Bayes adjustments for multiple comparisons are sometimes useful. Epidemiology 2:244–251 [DOI] [PubMed] [Google Scholar]
- Harada S, Agarwal DP, Goedde HW (1981) Aldehyde dehydrogenase deficiency as cause of facial flushing reaction to alcohol in Japanese. Lancet 2:982 6117742 [DOI] [PubMed] [Google Scholar]
- Helmuth L (2001) Genome research: map of the human genome 3.0. Science 293:583–585 [DOI] [PubMed] [Google Scholar]
- Hirschhorn JN, Lohmueller K, Byrne E, Hirschhorn K (2002) A comprehensive review of genetic association studies. Genet Med 4:45–61 10.1097/00125817-200203000-00002 [DOI] [PubMed] [Google Scholar]
- Ioannidis JP (2003) Genetic associations: false or true? Trends Mol Med 9:135–138 10.1016/S1471-4914(03)00030-3 [DOI] [PubMed] [Google Scholar]
- Ioannidis JP, Trikalinos TA, Ntzani EE, Contopoulos-Ioannidis DG (2003) Genetic associations in large versus small studies: an empirical assessment. Lancet 361:567–571 10.1016/S0140-6736(03)12516-0 [DOI] [PubMed] [Google Scholar]
- Johnson GC, Esposito L, Barratt BJ, Smith AN, Heward J, Di Genova G, Ueda H, Cordell HJ, Eaves IA, Dudbridge F, Twells RC, Payne F, Hughes W, Nutland S, Stevens H, Carr P, Tuomilehto-Wolf E, Tuomilehto J, Gough SC, Clayton DG, Todd JA (2001) Haplotype tagging for the identification of common disease genes. Nat Genet 29:233–237 10.1038/ng1001-233 [DOI] [PubMed] [Google Scholar]
- Lander E, Kruglyak L (1995) Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11:241–247 7581446 [DOI] [PubMed] [Google Scholar]
- Lander ES, Schork NJ (1994) Genetic dissection of complex traits. Science 265:2037–2048 [DOI] [PubMed] [Google Scholar]
- Lewis CM (2002) Genetic association studies: design, analysis and interpretation. Brief Bioinform 3:146–153 12139434 [DOI] [PubMed] [Google Scholar]
- Lohmueller KE, Pearce CL, Pike M, Lander ES, Hirschhorn JN (2003) Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat Genet 33:177–182 10.1038/ng1071 [DOI] [PubMed] [Google Scholar]
- Lowe CE, Cooper JD, Chapman JM, Barratt BJ, Twells RC, Green EA, Savage DA, Guja C, Ionescu-Tirgoviste C, Tuomilehto-Wolf E, Tuomilehto J, Todd JA, Clayton DG (2004) Cost-effective analysis of candidate genes using htSNPs: a staged approach. Genes Immun 5:301–305 15029236 [DOI] [PubMed] [Google Scholar]
- Morris DW, McGhee KA, Schwaiger S, Scully P, Quinn J, Meagher D, Waddington JL, Gill M, Corvin AP (2003) No evidence for association of the dysbindin gene [DTNBP1] with schizophrenia in an Irish population-based study. Schizophr Res 60:167–172 10.1016/S0920-9964(02)00527-3 [DOI] [PubMed] [Google Scholar]
- Morton NE (1955) Sequential tests for the detection of linkage. Am J Hum Genet 7:277–318 [PMC free article] [PubMed] [Google Scholar]
- Morton NE, Collins A (1998) Tests and estimates of allelic association in complex inheritance. Proc Natl Acad Sci USA 95:11389–11393 9736746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumark YD, Friedlander Y, Thomasson HR, Li T-K (1998) Association of the ADH2*2 allele with reduced ethanol consumption in Jewish men in Israel: a pilot study. J Stud Alcohol 59:133–139 [DOI] [PubMed] [Google Scholar]
- North BV, Curtis D, Cassell PG, Hitman GA, Sham PC (2003) Assessing optimal neural network architecture for identifying disease-associated multi-marker genotypes using a permutation test, and application to calpain 10 polymorphisms associated with diabetes. Ann Hum Genet 67:348–356 10.1046/j.1469-1809.2003.00030.x [DOI] [PubMed] [Google Scholar]
- Osier MV, Pakstis AJ, Soodyall H, Comas D, Goldman D, Odunsi A, Okonofua F, Parnas J, Schulz LO, Bertranpetit J, Bonne-Tamir B, Lu R-B, Kidd JR, Kidd KK (2002) A global perspective on genetic variation at the ADH genes reveals unusual patterns of linkage disequilibrium and diversity. Am J Hum Genet 71:84–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page GP, George V, Go RC, Page PZ, Allison DB (2003) “Are we there yet?”: deciding when one has demonstrated specific genetic causation in complex diseases and quantitative traits. Am J Hum Genet 73:711–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil N, Berno AJ, Hinds DA, Barrett WA, Doshi JM, Hacker CR, Kautzer CR, Lee DH, Marjoribanks C, McDonough DP, Nguyen BT, Norris MC, Sheehan JB, Shen N, Stern D, Stokowski RP, Thomas DJ, Trulson MO, Vyas KR, Frazer KA, Fodor SP, Cox DR (2001) Blocks of limited haplotype diversity revealed by high-resolution scanning of human chromosome 21. Science 294:1719–1723 10.1126/science.1065573 [DOI] [PubMed] [Google Scholar]
- Peterson RJ, Goldman D, Long JC (1999) Nucleotide sequence diversity in non-coding regions of ALDH2 as revealed by restriction enzyme and SSCP analysis. Hum Genet 104:177–187 10.1007/s004390050932 [DOI] [PubMed] [Google Scholar]
- Pritchard JK (2001) Are rare variants responsible for susceptibility to complex diseases? Am J Hum Genet 69:124–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Przeworski M (2001) Linkage disequilibrium in humans: models and data. Am J Hum Genet 69:1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Rosenberg NA, Donnelly P (2000) Association mapping in structured populations. Am J Hum Genet 67:170–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich DE, Lander ES (2001) On the allelic spectrum of human disease. Trends Genet 17:502–510 10.1016/S0168-9525(01)02410-6 [DOI] [PubMed] [Google Scholar]
- Risch N, Merikangas K (1996) The future of genetic studies of complex human diseases. Science 273:1516–1517 [DOI] [PubMed] [Google Scholar]
- Risch NJ (2000) Searching for genetic determinants in the new millennium. Nature 405:847–856 10.1038/35015718 [DOI] [PubMed] [Google Scholar]
- Schaid DJ, Rowland CM, Tines DE, Jacobson RM, Poland GA (2002) Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am J Hum Genet 70:425–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schork NJ, Fallin D, Lanchbury JS (2000) Single nucleotide polymorphisms and the future of genetic epidemiology. Clin Genet 58:250–264 10.1034/j.1399-0004.2000.580402.x [DOI] [PubMed] [Google Scholar]
- Schwab SG, Knapp M, Mondabon S, Hallmayer J, Borrmann-Hassenbach M, Albus M, Lerer B, Rietschel M, Trixler M, Maier W, Wildenauer DB (2003) Support for association of schizophrenia with genetic variation in the 6p22.3 gene, dysbindin, in sib-pair families with linkage and in an additional sample of triad families. Am J Hum Genet 72:185–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seltman H, Roeder K, Devlin B (2003) Evolutionary-based association analysis using haplotype data. Genet Epidemiol 25:48–58 10.1002/gepi.10246 [DOI] [PubMed] [Google Scholar]
- Sham P, Bader JS, Craig I, O’Donovan M, Owen M (2002) DNA Pooling: a tool for large-scale association studies. Nat Rev Genet 3:862–871 10.1038/nrg930 [DOI] [PubMed] [Google Scholar]
- Sham PC, Rijsdijk FV, Knight J, Makoff A, North B, Curtis D (2004) Haplotype association analysis of discrete and continuous traits using mixture of regression models. Behav Genet 34:207–214 10.1023/B:BEGE.0000013734.39266.a3 [DOI] [PubMed] [Google Scholar]
- Sham PC, Zhao JH, Curtis D (2000) The effect of marker characteristics on the power to detect linkage disequilibrium due to single or multiple ancestral mutations. Ann Hum Genet 64:161–169 10.1046/j.1469-1809.2000.6420161.x [DOI] [PubMed] [Google Scholar]
- Shibuya A, Yoshida A (1988) Frequency of the atypical aldehyde dehydrogenase-2 gene (ALDH22) in Japanese and Caucasians. Am J Hum Genet 43:741–743 [PMC free article] [PubMed] [Google Scholar]
- Spielman RS, McGinnis RE, Ewens WJ (1993) Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am J Hum Genet 52:506–516 [PMC free article] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R (2003) Statistical significance for genomewide studies. Proc Natl Acad Sci USA 100:9440–9445 10.1073/pnas.1530509100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub RE, Jiang Y, MacLean CJ, Ma Y, Webb BT, Myakishev MV, Harris-Kerr C, Wormley B, Sadek H, Kadambi B, O’Neill FA, Walsh D, Kendler KS (2002) Genetic variation in the 6p22.3 gene DTNBP1, the human ortholog of the mouse dysbindin gene, is associated with schizophrenia. Am J Hum Genet 71:337–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpf MP, Goldstein DB (2003) Demography, recombination hotspot intensity, and the block structure of linkage disequilibrium. Curr Biol 13:1–8 10.1016/S0960-9822(02)01404-5 [DOI] [PubMed] [Google Scholar]
- Tanck MW, Klerkx AH, Jukema JW, De Knijff P, Kastelein JJ, Zwinderman AH (2003) Estimation of multilocus haplotype effects using weighted penalised log-likelihood: analysis of five sequence variations at the cholesteryl ester transfer protein gene locus. Ann Hum Genet 67:175–184 10.1046/j.1469-1809.2003.00021.x [DOI] [PubMed] [Google Scholar]
- Terwilliger JD, Weiss KM (1998) Linkage disequilibrium mapping of complex disease: fantasy or reality? Curr Opin Biotechnol 9:578–594 10.1016/S0958-1669(98)80135-3 [DOI] [PubMed] [Google Scholar]
- Thomas DC, Clayton DG (2004) Betting odds and genetic associations. J Natl Cancer Inst 96:421–423 [DOI] [PubMed] [Google Scholar]
- Thomas DC, Stram DO, Conti D, Molitor J, Marjoram P (2003) Bayesian spatial modeling of haplotype associations. Hum Hered 56:32–40 10.1159/000073730 [DOI] [PubMed] [Google Scholar]
- Thomas DC, Witte JS (2002) Point: population stratification: a problem for case-control studies of candidate-gene associations? Cancer Epidemiol Biomarkers Prev 11:505–512 12050090 [DOI] [PubMed] [Google Scholar]
- Thomasson HR, Crabb DW, Edenberg HJ, Li T-K, Hwu H-G, Chen C-C, Yeh E-K, Yin S-J (1994) Low frequency of the ADH2*2 allele among Atayal natives of Taiwan with alcohol use disorders. Alcohol Clin Exp Res 18:640–643 [DOI] [PubMed] [Google Scholar]
- Van Den Bogaert A, Schumacher J, Schulze TG, Otte AC, Ohlraun S, Kovalenko S, Becker T, Freudenberg J, Jönsson EG, Mattila-Evenden M, Sedvall GC, Czerski PM, Kapelski P, Hauser J, Maier W, Rietschel M, Propping P, Nöthen MM, Cichon S (2003) The DTNBP1 (dysbindin) gene contributes to schizophrenia, depending on family history of the disease. Am J Hum Genet 73:1438–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Oord EJ, Neale BM (2003) Will haplotype maps be useful for finding genes? Mol Psychiatry 9:227–236 14610524 [DOI] [PubMed] [Google Scholar]
- van den Oord EJ, Sullivan PF (2003) False discoveries and models for gene discovery. Trends Genet 19:537–542 10.1016/j.tig.2003.08.003 [DOI] [PubMed] [Google Scholar]
- Wacholder S, Chanock S, Garcia-Closas M, Elghormli L, Rothman N (2004) Assessing the probability of false-positive reports in molecular epidemiology studies. J Natl Cancer Inst 96:434–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weale ME, Depondt C, Macdonald SJ, Smith A, Lai PS, Shorvon SD, Wood NW, Goldstein DB (2003) Selection and evaluation of tagging SNPs in the neuronal-sodium-channel gene SCN1A: implications for linkage-disequilibrium gene mapping. Am J Hum Genet 73:551–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss KM, Clark AG (2002) Linkage disequilibrium and the mapping of complex human traits. Trends Genet 18:19–24 10.1016/S0168-9525(01)02550-1 [DOI] [PubMed] [Google Scholar]
- Williams NM, Preece A, Morris DW, Spurlock G, Bray NJ, Stephens M, Norton N, Williams H, Clement M, Dwyer S, Curran C, Wilkinson J, Moskvina V, Waddington JL, Gill M, Corvin AP, Zammit S, Kirov G, Owen MJ, O’Donovan MC (2004) Identification in 2 independent samples of a novel schizophrenia risk haplotype of the dystrobrevin binding protein gene (DTNBP1). Arch Gen Psychiatry 61:336–344 10.1001/archpsyc.61.4.336 [DOI] [PubMed] [Google Scholar]
- Zaykin DV, Westfall PH, Young SS, Karnoub MA, Wagner MJ, Ehm MG (2002) Testing association of statistically inferred haplotypes with discrete and continuous traits in samples of unrelated individuals. Hum Hered 53:79–91 10.1159/000057986 [DOI] [PubMed] [Google Scholar]
- Zhang K, Calabrese P, Nordborg M, Sun F (2002a) Haplotype block structure and its applications to association studies: power and study designs. Am J Hum Genet 71:1386–1394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Qin ZS, Liu JS, Chen T, Waterman MS, Sun F (2004) Haplotype block partitioning and tag SNP selection using genotype data and their applications to association studies. Genome Res 14:908–916 15078859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Sha Q, Chen H-S, Dong J, Jiang R (2003) Transmission/disequilibrium test based on haplotype sharing for tightly linked markers. Am J Hum Genet 73:566–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Collins A, Abecasis GR, Cardon LR, Morton NE (2002b) Mapping quantitative effects of oligogenes by allelic association. Ann Hum Genet 66:211–221 12174212 [DOI] [PubMed] [Google Scholar]
- Zhao LP, Li SS, Khalid N (2003) A method for the assessment of disease associations with single-nucleotide polymorphism haplotypes and environmental variables in case-control studies. Am J Hum Genet 72:1231–1250 12704570 [DOI] [PMC free article] [PubMed] [Google Scholar]