

Abstract
Lung cancer is a major cause of death in the United States and other countries. The risk of lung cancer is greatly increased by cigarette smoking and by certain occupational exposures, but familial factors also clearly play a major role. To identify susceptibility genes for familial lung cancer, we conducted a genomewide linkage analysis of 52 extended pedigrees ascertained through probands with lung cancer who had several first-degree relatives with the same disease. Multipoint linkage analysis, under a simple autosomal dominant model, of all 52 families with three or more individuals affected by lung, throat, or laryngeal cancer, yielded a maximum heterogeneity LOD score (HLOD) of 2.79 at 155 cM on chromosome 6q (marker D6S2436). A subset of 38 pedigrees with four or more affected individuals yielded a multipoint HLOD of 3.47 at 155 cM. Analysis of a further subset of 23 multigenerational pedigrees with five or more affected individuals yielded a multipoint HLOD score of 4.26 at the same position. The 14 families with only three affected relatives yielded negative LOD scores in this region. A predivided samples test for heterogeneity comparing the LOD scores from the 23 multigenerational families with those from the remaining families was significant (P=.007). The 1-HLOD multipoint support interval from the multigenerational families extends from C6S1848 at 146 cM to 164 cM near D6S1035, overlapping a genomic region that is deleted in sporadic lung cancers as well as numerous other cancer types. Parametric linkage and variance-components analysis that incorporated effects of age and personal smoking also supported linkage in this region, but with somewhat diminished support. These results localize a major susceptibility locus influencing lung cancer risk to 6q23–25.
Introduction
There are more individuals who die from lung cancer than from breast, colon, and prostate cancer combined, with an estimated 173,700 new cases and 160,440 deaths expected in the United States in 2004 (Jemal et al. 2004). The overall 5-year survival rate of lung cancer is 15% (Jemal et al. 2004). Advances in the detection and treatment of this disease have only resulted in a marginal improvement in mortality rates. In the last 50 years, lung cancer incidence increased by 249%, and mortality increased by 259% (Welch et al. 2000).
Cancer of the lung has frequently been cited as an example of a malignancy that is solely determined by the environment (Doll and Peto 1981; Blot and Fraumeni 1996). The risks associated with cigarette smoking (Burch 1980; Doll and Peto 1981; Carbone 1992; Doll et al. 1994; Blot and Fraumeni 1996) and certain occupations, such as mining (Seaton 1984), shipbuilding, and petroleum refining (Blot and Fraumeni 1976; Blot et al. 1979; Gottlieb and Steadman 1979) are well established. There is little doubt that the majority of lung cancer cases are attributable to cigarette smoking and other behavioral and environmental risk factors (Beckett 1993). However, numerous studies also suggest the involvement of genetic risk factors.
Investigators have long hypothesized that individuals differ in their susceptibility to environmental insults (Motulsky 1957; Heath 1958; Friberg 1959). More than 40 years ago, Tokuhata and Lilienfeld (1963) provided epidemiologic evidence for familial aggregation of lung cancer after accounting for personal smoking, which suggested the possible interaction of genes, shared environment, and common lifestyle factors in the etiology of lung cancer. Fraumeni et al. (1975) reported an increased risk of lung cancer mortality in siblings of probands with lung cancer, and positive family history has consistently been found to be a risk factor for lung cancer in a number of case-control studies (reviewed in Bailey-Wilson et al. 1998; Sellers and Bailey-Wilson 1998; Etzel et al. 2003).
Genetic modeling studies have also suggested that familial aggregation of lung cancer may be due to inheritance of only a few genetic factors. Segregation analyses of Louisiana families gave evidence for inheritance of a rare major autosomal gene that acts in conjunction with cigarette smoking to produce an earlier age at onset of the cancer (Sellers et al. 1990, 1991, 1992). Under this model, heterozygotes with the susceptibility allele and with average levels of cigarette smoking had relative risks of 14, 11.8, and 6.2 at age 50 years, 60 years, and 70 years, respectively, compared with noncarriers who had average levels of smoking. Additional segregation analyses of these data examining cohort and polygenic effects continued to indicate the effects of an allele on inherited susceptibility to lung cancer and also on risk for a broader group of smoking-related cancers (lung, oral cavity, esophagus, nasopharynx, larynx, pancreas, bladder, kidney, and uterine cervix) after smoking was allowed for (Chen et al. 1991; Bailey-Wilson et al. 1992). Gauderman et al. (1997) applied a Gibbs sampling method to the same lung cancer data set and found evidence for a dominant major locus with significant effects of smoking, but no evidence of gene-environment statistical interaction on the logistic scale.
Similar findings were reported by Yang et al. (1999), who noted evidence for Mendelian codominant inheritance with modifying effects of smoking and chronic bronchitis, using families of nonsmoking cases diagnosed at ages 40–59 years. Daw et al. (in press) performed oligogenic segregation analysis of time to onset of lung cancer on >2,000 families. The results of their work indicate the likely segregation of risk-conferring alleles at three or four genetic loci. The low number of estimated loci strongly suggests that genetic linkage studies could identify genetic factors for familial lung cancer (FLC).
On the basis of the evidence from these studies of the existence of cancer susceptibility genes that may act in conjunction with cigarette smoking to increase risk of lung and other throat cancers, we performed a genomewide-scan linkage study in families selected for aggregation of lung cancer. We considered lung, laryngeal (ICD-9 161.0–161.9), oropharyngeal (ICD-9 146.3–146.9), or hypopharyngeal (ICD 148.0–148.9) cancer, henceforth grouped together under the term “lung and throat (LT) cancer,” to be pleiotropic effects of the same gene. The high case-fatality rate (15% 5-year survival) and low resection rate (25%) makes the study of families with lung cancer particularly challenging, because it is difficult to collect adequate numbers of biospecimens for DNA analysis. A multidisciplinary collaborative effort was necessary to identify and accrue large numbers of families with FLC to test the hypothesis that there are genetic variants that greatly increase the risk of developing lung cancer. Recognizing the complexity of modeling the impact of environmental factors for this complex disease, we made an a priori decision that a simple autosomal dominant affected-only model would constitute our primary analytic strategy. Further analyses in which we also evaluated the evidence for linkage, allowing for the effects that smoking behavior, age, and sex have on the risk for lung cancer, were performed and are described.
Material and Methods
Data Collection
Data were collected by the FLC recruitment sites of the Genetic Epidemiology of Lung Cancer Consortium (GELCC): the University of Cincinnati, University of Colorado, Karmanos Cancer Institute, Saccomanno Research Institute, Louisiana State University Health Sciences Center, Mayo Clinic, and Medical College of Ohio. To date, of the 26,108 lung cancer cases screened at GELCC sites, 13.7% had at least one first-degree relative with lung cancer. Following the initial family history screening process, we collected, from 3,541 probands and/or their family representatives, data regarding additional persons affected with any cancers in the extended family, vital status of affected individuals, availability of archival tissue, and willingness of family members to participate in the study. Full pedigree development and biospecimen collection were performed on 771 families with three or more first-degree relatives affected with lung cancer. Cancers were verified by medical records, pathology reports, cancer registry records, or death certificates for 69% of individuals affected with LT, and by reports of multiple family members for the other 31% of family members affected with LT. Elsewhere, studies have shown that reports from family members give high accuracy rates for lung cancer diagnoses (Sellers et al. 1987; King et al. 2002). Only a small percentage (∼11%) of these families had sufficient biospecimens available to be informative for linkage analysis, and 52 were genotyped. From these 52 families, we accrued 654 blood samples (595 from family members and 59 from spouses), 10 buccal cell samples, and 78 archival blocks containing normal tissue. Archival blocks of lung, throat, or laryngeal tumors were collected from 61 family members, and blocks of other tumor types were collected from an additional 60 family members. Two families are African American, and one family has mixed racial composition (African American, Creole, and white); the remaining families are white.
Data were sent from each FLC collection site to the central phenotype-data management center in Cincinnati, where they were reviewed, verified, and merged, prior to being transmitted to M. D. Anderson Cancer Center for creation of files for linkage analyses. The Cincinnati site developed a comprehensive database of the familial pedigrees with lung cancer, including information on family history, affection status, tissue acquisition, and clinical and epidemiologic data.
Sample Preparation and Genotyping
Blood, buccal cells, and archival biospecimens were used as sources of DNA for genotyping family members of the kindreds with lung cancer. DNA isolated from blood has been genotyped at the Center for Inherited Disease Research (CIDR, a National Institutes of Health–supported core research facility), and DNA from buccal cells and archival tissue and sputum were genotyped at the University of Cincinnati.
DNA from archival tissue for genotyping was obtained from 10 10-μm paraffin sections containing normal tissue. The archival tissue blocks were examined at UT Southwestern Medical Center, and sections of normal tissue were prepared for genotyping at the University of Cincinnati. We required the specimen to have at least 50% normal cells for the global genotyping. DNA was isolated from paraffin sections, and sputum samples were isolated by a modified Wright and Manos (1990) procedure, performed by incubating the tissue with 0.5 μg/l of Proteinase K in 1× PCR buffer with NP-40 and Tween 20 for 1 h at 55°C. This was followed by a 95°C incubation for 10 min to inactivate the Proteinase K. Additionally, we included an extraction of the isolated DNA with 24:1 (v:v) chloroform:isoamyl alcohol. DNA was isolated from the buccal cells and from whole blood using the Puregene Kit (Gentra Systems) in accordance with the manufacturer’s protocols.
The CIDR global genotyping set consisted of 392 markers (15 families) or 388 markers (37 families). PCR amplifications using the primer set for each of the markers were performed at CIDR and the University of Cincinnati. The standard protocol for PCR performed at CIDR can be found on the CIDR Web site. Conditions for PCR using archived DNA were similar to CIDR’s protocol, but with a modification of an increase in the number of cycles to 35. All samples were amplified in an MJ Research Thermocycler. In brief, the cycles were as follows: 95° for 12 min, for 1 cycle; 94° for 45 s, 55° for 1 min, and 72° for 1 min, for 10 cycles; then 89° for 1 min, 55° for 1 min, and 72° for 1 min, for an additional 25 cycles; followed by a final extension at 72° for 10 min. PCR amplifications were performed using a single fluorescently labeled primer obtained from CIDR. After the reactions, PCR products were resolved on an ABI 3100 automated DNA sequencer and were analyzed with genotype software. Because of the reduced amounts of genomic DNA in the archived samples, none of the amplification products were pooled prior to loading onto the 96 wells of a plate for subsequent analysis.
Merging of Genotype Data Generated at CIDR and the University of Cincinnati for Linkage Analyses
Assignment of alleles generated at CIDR and the University of Cincinnati was accomplished by genotyping several samples in common for each gel (or plate) at both facilities. These common samples included CEPH controls 1331-01 and 1331-02 as well as several lymphocyte DNA samples from members of the families with FLC.
Our first step in evaluating the genetic data was to appropriately bin the allele lengths. To allow us to jointly analyze data across different platforms used at CIDR versus those used at the University of Cincinnati, we first compared the raw allele lengths for 16 subjects who had been genotyped on both platforms. We next generated a linear regression to predict CIDR lengths from the University of Cincinnati data and identified any errors in the data as alleles that failed to satisfy the criterion: 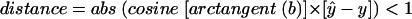 , where
, where  is the predicted value of a point. The prediction of allele lengths from both centers routinely yielded an R2 value >99% for all but two markers (which had R2 values of 97% and 98%). However, the intercepts are routinely different from 0, indicating a shift in allele lengths between labs, and the slope often varied from 1, indicating that without regression adjustment, alleles at the extremes could have been misclassified.
is the predicted value of a point. The prediction of allele lengths from both centers routinely yielded an R2 value >99% for all but two markers (which had R2 values of 97% and 98%). However, the intercepts are routinely different from 0, indicating a shift in allele lengths between labs, and the slope often varied from 1, indicating that without regression adjustment, alleles at the extremes could have been misclassified.
Error Detection
The programs Relative (S.A.G.E. 2002) and PREST (McPeek and Sun 2000) were used to verify relationships among individuals in the data. GAS (GAS 1998), SIBPAIR (Duffy 2002), and PedCheck (O’Connell and Weeks 1998) were used to check for Mendelian inconsistencies. All such errors were corrected by eliminating the genotypes indicated to have been most likely to cause errors.
Linkage Analysis
Families were selected for the study only if they had multiple individuals (at least three) affected with lung cancer. However, for the purpose of the linkage analyses, persons were considered to be affected if they had an LT cancer (resulting in a total of 241 affected persons in these 52 linkage families). Persons affected with other cancers were considered unaffected in these analyses. Marker allele frequencies were calculated separately and linkage analyses were performed separately for the white American and African American families, with the results combined in overall tests of linkage.
Two-point parametric and multipoint parametric linkage tests were performed with the FASTLINK (Cottingham et al. 1993) and SIMWALK2 programs (Sobel and Lange 1996; Sobel et al. 2001), with the use of an autosomal dominant low-penetrance model under the assumption of a susceptibility allele frequency of 0.01, 10% penetrance in gene carriers, and 1% penetrance in non–gene carriers. Because this model assumes a very low penetrance for unaffected individuals, they can have any of the genotypes with the probabilities not influenced by their phenotype, so that this model is virtually identical to an affected-only parametric analysis (Terwilliger and Ott 1994). The nonzero sporadic rate further allows for individuals who have lung cancer due solely to environmental exposures (i.e., cigarette smoking). Several studies have shown that approximately correct models recover most of the available power in a parametric linkage analysis (Hodge et al. 1997; Greenberg et al. 1998; Abreu et al. 1999; Durner et al. 1999; Greenberg and Abreu 2001; Abreu et al. 2002). Since ∼90% of the affected individuals in these pedigrees smoked cigarettes, a weighting of only the affected individuals in this analysis has the effect of allowing for smoking status and ignoring information from unaffected subjects. LOD scores under the assumption of heterogeneity (HLODs) were computed for both two-point and multipoint analyses (Ott 1999).
We also performed several other types of analyses that incorporated the effects of smoking, age, and sex into our linkage study. Two-point parametric linkage analysis was performed using LODLINK, from the Statistical Analysis for Genetic Epidemiology (S.A.G.E.) package. When using LODLINK, we assumed the best-fitting model from segregation analysis (Sellers et al. 1990), an autosomal codominant locus with susceptibility allele frequency of 0.052, and penetrance functions that included age and cigarette smoking as covariates in the model. Since the codominant susceptibility allele in this model is uncommon, predicted segregation of disease in families would appear similar to dominant inheritance.
The current implementation of LODLINK only allows two-point linkage analysis, yet it is well known that multipoint linkage analysis can be more powerful than two-point linkage, especially in situations where matings are not fully informative (as in these families with large numbers of unsampled deceased parents). The complex “gene-environment” model used in LODLINK has not yet been implemented in any multipoint linkage analysis program. Therefore, to incorporate effects from smoking in multipoint analysis, we also used a method described elsewhere (Shete et al. 2002) in which a liability class is defined for each subject. To construct the liability classes, we used the same logistic model that was derived from the segregation analysis by Sellers et al. (1990) and that was used in the LODLINK analyses described above.
Nonparametric analyses also were performed with variance-components methods. Two approaches were used—one that incorporated age at onset and the other that used binary outcomes. Since LT cancer has a variable age-at-onset component, we can incorporate age and other measured environmental risk factors by using time-to-event data within a linkage analysis framework. The rationale behind this approach is based on the assumption that if there is significant age-at-onset variability, then the use of survival analysis techniques will provide additional information in linkage studies. We used mixed-effects Cox models (Amos et al. 2001; Therneau 2003; Pankratz et al., in press). This method made it possible to simultaneously estimate fixed and random effects (of smoking and sex) on censored survival data without specifying the censored trait distribution. It also allowed us to retain the full flexibility of Cox regression while exploiting the broad capabilities of variance-components models. For individuals who developed the disease, the age at onset represented the observed time data. For those who were free of disease at the examination or interview, their age at the time of the exam or interview was used as their observed data. These individuals represented censored observations. To determine the significance of the associations between age at onset and the measured environmental risk factors and additional genetic factors, proportional hazards regression analyses were performed with the S-Plus coxme function that incorporates the multipoint identity by descent (IBD) calculated by SIMWALK2 (Sobel and Lange 1996; Sobel et al. 2001).
The second nonparametric approach used the variance-components approach (binary option), as implemented in the SOLAR linkage package (Amos 1994; Almasy and Blangero 1998; Williams et al. 1999), to scan the genome for regions linked to the binary trait (affected and unaffected), with multipoint IBD calculated by SIMWALK2 (Sobel and Lange 1996; Sobel et al. 2001).
We also have performed subset analyses on the families with the most affected persons and with affected persons in two or more generations. Of the 52 families, 38 met our initial inclusion criterion of having four or more affected persons. A smaller subset of 23 families had five or more affected relatives in two or more generations. This latter group may be most likely to reflect genetic susceptibility due to inheritance of an autosomal dominant factor.
Results
Table 1 summarizes characteristics of the 52 pedigrees. There were 223 persons affected with lung cancer; 13 persons affected with laryngeal (ICD-9 161.0–161.9), oropharyngeal (ICD-9 146.3–146.9), or hypopharyngeal (ICD 148.0–148.9) cancer; and 5 persons affected with both primary lung cancer and one of the other three primary cancers. The median number of affected persons per family was four. There were 36 families with affected persons in two or more generations. Six pedigrees contained affected family members in three generations, and one other pedigree contained affected individuals in four generations. Many families also contained affected persons with onset of lung cancer at an early age. The median age at onset for lung, laryngeal, and pharyngeal cancer was 60 years, less than the median value of 70 years for age at diagnosis in the general white population (Ries et al. 2000). Minimum age at onset within the family may be a better indicator of the potential for existence of a susceptibility gene, since it is not influenced by older sporadic cases. In our families, the minimum age at onset within the family ranged from 29 years to 68 years with a median value of 50.5 years.
Table 1.
Characteristics of the 52 Familial Pedigrees with Lung Cancer
|
No. of Affected Individualsa |
|||||
| No. of AffectedIndividualsper Pedigree | No. of Pedigrees | Total | DirectlyGenotyped | With InferrableGenotypes | No. of Unaffected Individuals Directly Genotyped |
| 3 | 14 | 42 | 23 | 8 | 105 |
| 4 | 12 | 48 | 18 | 8 | 100 |
| 5 | 13 | 65 | 27 | 8 | 181 |
| 6 | 7 | 42 | 12 | 10 | 106 |
| 7 | 4 | 28 | 10 | 5 | 68 |
| 8 | 2 | 16 | 7 | 0 | 11 |
| Total | 52 | 241 | 97 | 39 | 571 |
Affected individuals had lung, laryngeal, oropharyngeal, or hypopharyngeal cancer.
For the 241 affected individuals in the 52 families with FLC (table 1), 86 blood samples were available for global genotyping. Of 149 individuals in the 52 families who were deceased at the time of pedigree development, we were able to globally genotype 50 (34%). DNA from archival tissue specimens was used to globally genotype 11 affected family members. We reconstructed the genotypes of 39 persons, using genotypes of spouses and offspring, including genotypes from archival tissue of five spouses. Genotyping was also performed for markers on chromosomes 6 and 12 for five deceased affected persons whose archival specimens yielded limited DNA. Without the ability to globally genotype archival specimens, we would not have genotypes from 15% of the affected family members needed for linkage analysis.
Figure 1 shows the maximum (recombination fractions of ⩽40%) two-point homogeneity LOD scores across the genome, under the simple dominant low-penetrance model without inclusion of age and smoking exposure. Positive HLOD scores that approached or exceeded 1.0 were calculated for markers on chromosomes 1, 4, 6, 9, 12, 20, and 21. Two-point analysis gave an HLOD of 0.94 at D6S2436 in all families and an HLOD of 1.5 in the 38 families with four affected relatives, whereas for the 23 highest risk families, the two-point HLOD score was 2.1. Figure 2 shows that inclusion of age and smoking in the LODLINK models gives maximum two-point HLOD scores of 1.48 on chromosome 6 at C6S1848 in all families, but, for marker D6S2436, the LODLINK results including smoking and sex effects yielded lower HLOD scores than the simple model did. On chromosome 12, LODLINK yielded two-point HLODs of 1.54 at marker D12S372, 1.1 at D12S375, and 0.4 at D12S2070. The HLOD scores that were obtained using the model that incorporated the covariates were higher in some regions than the maximum two-point HLODs that were obtained under the simple dominant low-penetrance affected-only model with no covariates.
Figure 1.

Maximum two-point homogeneity LOD scores under the simple dominant low-penetrance model without inclusion of age and smoking exposure. Individual chromosomes are indicated at the top of the graph.
Figure 2.

Two-point maximum HLOD scores on chromosome 6, under the Sellers et al. (1990) model (Env) that includes age and cigarette-smoking exposure and is used in LODLINK, compared with two-point HLOD scores under the simple dominant low-penetrance model without inclusion of age and smoking exposures (No Env). Inclusion of age and smoking exposure increases the evidence in favor of linkage on 6q.
Multipoint parametric linkage under the simple dominant low-penetrance affected-only model yielded a maximum HLOD of 2.79 at 155 cM (marker D6S2436) on chromosome 6q in the 52 families (fig. 2), with 67% of families estimated to be linked. Multipoint analysis of the 38 families with four affected relatives gives an HLOD of 3.47 at this same location, with 78% of families estimated to be linked, whereas for the 23 highest risk families, the multipoint HLOD score is 4.26, with 94% estimated to be linked (fig. 3 and table 2). Conversely, linkage analysis of the 14 families with three affected relatives yielded negative LOD scores. A predivided samples test for heterogeneity (Ott 1999) that compared the evidence for linkage in multigenerational families with five affected relatives (23 families) to the other familes was significant (P=.007). The 1-HLOD support interval in the 23 multigenerational families extends from C6S1848 at 146 cM to 164 cM near marker D6S1035.
Figure 3.

Plot of chromosome 6 parametric multipoint HLOD scores (affected-only dominant model, no environmental covariates) calculated by SIMWALK2, in all 52 families (HLOD-All), in the 38 families with four or more affected individuals (HLOD-38), and in the 23 multigenerational families with five or more affected individuals (HLOD-23).
Table 2.
Multipoint HLOD Scores for Susceptibility to LT Cancer
|
Multipoint HLODa in |
|||||
| Chromosome | Locus | Position | All 52 Families | 38 Familiesb | 23 Familiesc |
| 6q | D6S2436 | 155 | 2.79 | 3.47 | 4.26 |
| 12q | D12S2070 | 125 | .60 | .89 | .63 |
| 20 | Near D20S470 | 37 | .98 | 1.20 | 1.18 |
| 14 | D14S306 | 44 | 1.06 | 1.09 | .84 |
Simple dominant model using SIMWALK2.
Families with four or more affected individuals.
Multigenerational families with five or more affected individuals.
The estimate of the proportion of linked families from heterogeneity linkage analysis is known to be imprecise and may be inaccurate in studying complex traits, although the test for linkage in the presence of heterogeneity is robust and powerful (see, e.g., Greenberg and Abreu 2001; Whittemore and Halpern 2001; Hodge et al. 2002; Vieland and Logue 2002). However, the relative increase in the estimated proportion of linked families in subsets of families with an increasing number of affected individuals suggests a decrease in heterogeneity in the more informative families. In the subset of 23 highly informative families, 14 (61%) have LOD scores >0.0 at D6S2436, 11 (48%) have LOD scores >0.2, and 8 (35%) have LOD scores >0.4. These data demonstrate that more than half of these 23 pedigrees contribute toward the positive LOD score on 6q.
Further multipoint analysis that includes smoking behavior and that uses a liability class for each subject yielded somewhat lower HLOD scores. The maximal HLOD score from analysis of all 52 families was 2.63, with the HLOD score of 3.04 for the white families. For the 38 families with ⩾4 cases, the HLOD score was 1.96, and for the 23 multigenerational pedigrees, the HLOD score was 2.90.
Multipoint linkage analysis under the simple autosomal dominant decreased-penetrance model yielded HLODs close to 1.0 on chromosomes 12, 14, and 20 (table 2). Among the 19 families that showed positive LOD scores on chromosome 6, we also found positive multipoint LOD scores on chromosome 12, with a maximum HLOD score of 1.6 at D12S2070, and 72% of families were estimated to show linkage.
Nonparametric multipoint analyses of LT cancer with the use of the variance-components approach (binary option) and the mixed-effects Cox models (with multipoint IBD sharing calculated by SIMWALK2) also gave support for linkage of LT cancer in the 6q region at C6S1848 at 146 cM (LOD=2.46 and 2.01, respectively) in the 23 highest risk families with sex and smoking status (yes/no) as covariates (fig. 4). On chromosome 12, these nonparametric methods also gave evidence of linkage for LT cancer (LOD=0.36 [coxme] and 0.63 [binary]) in the 23 highest risk families with sex and smoking status (yes/no) as covariates.
Figure 4.

Plot of chromosome 6 nonparametric multipoint linkage analysis using the variance-components approach (binary option) and the mixed-effects Cox models with multipoint IBD sharing calculated by SIMWALK2, for the 23 multigenerational families with five or more affected individuals.
The results of our multipoint analyses indicated much stronger evidence for linkage from multipoint analysis, compared with the two-point analysis. To evaluate the impact that sparseness in availability of subjects in the extended pedigrees has on LOD score calculations, we used a simulation approach implemented with SLINK (Ott 1989; Weeks et al. 1990). We simulated data for the 23 multigenerational families, assuming penetrance of 40% in carriers and 4% in noncarriers, and then analyzed the data, assuming penetrance of 10% in carriers and 1% in noncarriers. We then performed linkage analysis of the disease susceptibility and 1 or 2 markers (the order was D6S2436–0.02 cM–disease–10 cM–D6S1035, and we used D6S2436 alone for two-point analyses). We also simulated data under the assumption that we could only obtain samples from those individuals who actually gave samples (sparse case), versus the situation in which all individuals give samples (dense case). In sparse pedigrees, the increase in LOD scores from two-point to three-point analyses was 34%, whereas, for dense pedigrees, it was only 19%. We anticipate that simulations using C6S1848 might further indicate a gain in LOD scores from multipoint analysis, but four-point analysis using SLINK was prohibitively time consuming. These studies suggest that, for sparse pedigrees, multipoint analysis is relatively more informative than it is for dense pedigrees with no missing genotypes of affected persons.
To explore the effect that smoking has on risk for lung cancer among carriers of the susceptibility haplotype, we first obtained from SIMWALK2 the most likely haplotypes from 21 of the 23 multigenerational pedigrees (in 2 pedigrees, we were unable to clearly identify a susceptibility haplotype). Then, we scored all individuals as either carriers (n=223, of which 87 were affected, and 90.4% of the affected individuals smoked) or noncarriers (n=344, of which 19 were affected, and 83% of affected individuals smoked). We then performed a Cox proportional hazards survival analysis using SAS 8.0 to assess the effects of smoking and sex separately among carriers and noncarriers of the susceptibility haplotype, treating time to onset of LT cancer as an endpoint and sex and cigarette pack-years as predictors. The results of this analysis showed a mild effect of increasing smoking on risk for lung cancer among carriers (hazards ratio [HR] per pack-year = 1.004, P=.13), whereas, among noncarriers, there was a significant effect of smoking on risk for lung cancer (HR per pack-year = 1.018, P=.0023). When smoking was treated as a categorical yes/no variable, we found that smoking increased risk for both carriers (HR = 3.31, P=.002) and noncarriers (HR = 2.95, P=.10). These sets of observations suggest that smoking at any level increases risk in carriers of inherited susceptibility from a locus on chromosome 6q, whereas increasing smoking increases risk for noncarriers.
Discussion
These results provide clear evidence for a major susceptibility locus on chromosome 6q influencing LT cancer risk—particularly in the multigenerational densely affected families—with characteristics consistent with an autosomal dominant or codominant major locus. As expected, the multipoint analyses that test for linkage in the presence of heterogeneity (multipoint HLODs) gave the most power in our analyses. In addition to the compelling evidence for linkage to chromosome 6q, we also found suggestive evidence for linkages to some other regions (Lander and Kruglyak 1995). There is also some indication of a possible epistatic interaction between the putative loci on chromosomes 6 and 12, but more data are required to confirm this interaction.
The 1-HLOD support interval on 6q obtained from the multipoint analysis of the multigenerational families extends from C6S1848 at 146 cM (147.95 Mb) to 164 cM (159.94 Mb) near D6S1035. We have also provided the location of the markers on 6q in million base pairs (Mb), since the chromosomal regions of allelic loss are reported in the literature in Mb (see fig. 5 legend). The interval supporting linkage overlaps a genomic region on 6q that exhibits allelic loss in non–small-cell lung carcinoma (Petersen et al. 1997; Virmani et al. 1998; Luk et al. 2001; Goeze et al. 2002). These studies used cytogenetic techniques of comparative genomic hybridization (CGH) and/or fluorescent in situ hybridization to detect large regions of chromosomal imbalance on 6q. Berrieman et al. (2004) detected allelic loss between 6q25-qter for 50% of the samples examined, and Goeze et al. (2002) detected deletion between 6q14–24 in at least 60% of the 59 primary lung tumors examined. The study by Petersen et al. (1997) examined 50 non–small-cell lung carcinomas by CGH, and 46% exhibited chromosomal imbalance in the interval supporting linkage to 6q. Luk et al. (2001) examined 23 lung tumors and detected 6q loss in 30% of the tumors. There are no published studies that examined lung tumors to define a minimum region of deletion on 6q by loss-of-heterozygosity (LOH) analysis. However, two studies did detect LOH with several markers on 6q (located between 152 Mb and 167 Mb) in 50% of the lung tumor/lung cancer cell lines examined (Merlo et al. 1994; Virmani et al. 1998). These analyses of sporadic non–small-cell lung carcinomas clearly demonstrate frequent allelic loss on regions of 6q that overlap our linkage interval.
Figure 5.

Minimum regions of chromosomal deletions of various tumor types, located within the 6q linkage region. The HLOD-score plot for the 23 families (fig. 3) is reproduced with the scale of the X-axis in physical distances (Mb) instead of cM. Horizontal lines denote regions of minimum chromosomal deletions for various tumor types. The number above each line denotes the reference in which the information about chromosomal deletions was obtained (see list of references below). Some of the studies detected two or more distinct regions of deletion and, thus, the same reference number may appear above more than one horizontal line. The numbers 1–4 on deletion lines represent minimal regions of deletion for breast cancer; 5 and 6, for ovarian cancer; 7, for mesothelioma; 8 and 9, for pancreatic cancer; 10, for SCC of oral cavity; 11, for melanoma; and 12, for Hodgkin lymphoma. Markers are indicated by the symbol “♦” and are placed from left to right: D6S474 (112.92 Mb), D6S1040 (130.97 Mb), D6S1009 (137.28 Mb), C6S1848 (147.95 Mb), D6S2436 (154.70 Mb), D6S1035 (159.94 Mb), D6S1277 (164.21 Mb), and D6S1027 (168.98 Mb). The references for each horizontal line are: (1) Noviello et al. 1996; (2) Utada et al. 2000; (3) Zeller et al. 2003; (4) Cesari et al. 2003; (5) Hansen et al. 2002; (6) Cesari et al. 2003; (7) Jensen et al. 2003; (8) Abe et al. 1999; (9) Barghorn et al. 2001; (10) Tong et al. 2004; (11) Millikin et al. 1991; and (12) Re et al. 2003.
Numerous other tumor types also have allelic loss on regions of 6q which overlap the interval supporting linkage, including breast tumor (Noviello et al. 1996; Utada et al. 2000; Cesari et al. 2003; Zeller et al. 2003), ovarian tumor (Hansen et al. 2002; Cesari et al. 2003), mesothelioma (Jensen et al. 2003), pancreatic tumor (Abe et al. 1999; Barghorn et al. 2001), squamous cell carcinoma (SCC) of the oral cavity (Tong et al. 2004), melanoma (Millikin et al. 1991), and Hodgkin lymphoma (Re et al. 2003). These studies utilized LOH analyses to determine regions of minimum chromosomal deletion. Figure 5 illustrates the overlap of these minimum regions of deletion within our interval supporting linkage on 6q. The horizontal lines denote regions of deletion, and the numbers above each line denote the reference from which the data were obtained (see fig. 5 legend). Since some studies identified two or more distinct minimum regions of deletion, a number may appear above more than one horizontal line. The majority of the detected regions of allelic loss on 6q are located in our linkage region (fig. 5), although deletions are observed in two other locations. For example, allelic loss has been detected in prostate tumors, non-Hodgkin lymphomas, and acute lymphoblastic leukemias in a region proximal (90–105 Mb) to our linkage region (Merup et al. 1998; Srikantan et al. 1999; Zhang et al. 2000; Hyytinen et al. 2002; Verhagen et al. 2002; Konishi et al. 2003). Tumor-suppressor genes may also reside on 6q in a region telomeric to our linkage region (Tibiletti et al. 2000).
There is considerable overlap between these regions of minimum deletion shown in figure 5, including the following: (1) overlap of regions denoted by horizontal lines 1, 2, 4, 6, and 8, with the minimum region of overlap around markers D6S305 and D6S1599; (2) overlap of lines 12, 11, 5, 3, and 1, with the minimum region of overlap between D6S978 and D6S1637; (3) overlap of lines 11, 9, 7, 3, and 1, with the minimum region of overlap between D6S1648 and D6S1055; and (4) overlap of lines 11, 10, 8, 7, and 1, with the minimum region of overlap between D6S270 and D6S308. Also, some of the overlap occurs between minimum regions of deletion detected in different tumor types (e.g., horizontal lines 1 and 3 depict regions of deletion detected from breast tumors, line 5 from ovarian tumors, line 11 from melanomas, and line 12 from Hodgkin lymphomas). These data are consistent with the existence of one or more tumor-suppressor genes in the linkage region that we have identified for LT cancer in this study. There are several interesting candidate genes in this region, including four putative tumor-suppressor genes: SASH1 (148.8 Mb), LATS1 (150 Mb), IGF2R (160.45 Mb), and PARK2 (161.7 Mb). Also, other genes in this region are involved in some aspects of the regulation of cellular proliferation or the prevention of DNA damage. The sequencing of exons in these candidate genes is underway in families that show evidence of linkage of lung, laryngeal, oropharyngeal, or hypopharyngeal cancer to 6q markers.
We have detected compelling evidence of linkage on 6q using a simple dominant low-penetrance affected-only model. This linkage model was chosen as our primary analytical approach because of uncertainty about the strength of the relationship between smoking behavior and lung cancer risk in the high-risk families we are studying. As indicated in our prior studies of Li-Fraumeni syndrome (Hwang et al. 2003), smoking could have a much less important role in the context of genetic susceptibility than in the general population, and parametric models that rely heavily on smoking behavior might lead to a deflation of LOD scores for detection of linkage. Furthermore, possible deflation of LOD scores could occur if there is a common genetic susceptibility between smoking behavior and LT cancers. In addition, since ∼90% of the affected family members in our studies smoked, a weighting of only the affected individuals in this low-penetrance model has the effect of allowing for smoking status while ignoring information from unaffected subjects. The nonzero sporatic rate (1%) allows for individuals who are not gene carriers to be affected as a result solely of their smoking exposure, and the relatively low penetrance (10%) for gene carriers allows nonsmoking unaffected persons to have a high probability of being a gene carrier. Finally, simple models such as this one are known to recover a large portion of the linkage information available for many types of complex traits, as long as the mode of inheritance (dominance) at the trait locus is correctly specified (Hodge et al. 1997; Greenberg et al. 1998; Abreu et al. 1999, 2002; Durner et al. 1999; Greenberg and Abreu 2001).
The detection of significant linkage on 6q by the simple model, without explicitly including smoking history as a covariate in the model, does not imply that smoking exposure is an unimportant risk factor for lung cancer in family members who are carriers of the genetic risk factor but, rather, it implies that the genetic factor on 6q is strong enough to be detected by the low-penetrance affected-only linkage model with our pedigrees. Smoking exposure is also an important risk factor, since 90% of the affected individuals in the 52 pedigrees were smokers, and 89% of individuals in the 23 multigenerational families with 5 or more affected members smoked. The Cox modeling that we performed suggested that the inferred carriers of a 6q mutation may be sensitive to any level of smoking, rather than suggesting an increasing gradient of risk with increasing smoking behavior, as is usually observed in the general population. In the segregation analysis approach used by Sellers et al. (1990), the difference in the effect of smoking behavior on risk could not be effectively modeled because of the difficulties in fitting a covariate by unmeasured genotype interactions.
Because it is possible that better modeling of the joint effect of genetic factors and smoking exposure might improve power to detect linkage, we also included smoking behavior in two-point and multipoint parametric analyses and in multipoint nonparametric linkage analyses. Our two-point parametric analyses with the use of LODLINK gave stronger evidence of linkage to some parts of the 6q region than did the simple model in two-point analyses (fig. 2). However, the results from the multipoint analysis showed decreased (but still strong) evidence for linkage when smoking behavior was included in the analysis. It is possible that the model previously derived from segregation analysis of a population-based case series of lung cancer is not appropriate for the highly selected families we are studying in this linkage study, as discussed above. If the Sellers et al. (1990) model inaccurately models the differential effect of smoking behavior on carriers and noncarriers, then the use of it could yield lower evidence for linkage, particularly in the multipoint analysis, which is known to be more sensitive to model misspecification than the two-point linkage analysis (Risch and Giuffra 1992). Work by both Dizier et al. (1993) and Durner et al. (1999) has shown that the use of models that are the result of population-based segregation analysis in the linkage analysis of a very complex trait can reduce or conceal evidence of linkage.
Nonparametric multipoint analysis incorporating age and smoking status as covariates and using the variance-components and mixed-effects Cox models also support the same genomic region found by the parametric models. An advantage of the nonparametric linkage analysis is nonspecification of a model. Thus, when similar evidence for linkage is obtained with nonparametric analysis, then the evidence of linkage from the parametric model is substantiated. It is also known that multipoint parametric analysis can be more powerful than the nonparametric approach, especially under dominant inheritance (Durner et al. 1999; Greenberg and Abreu 2001). In future studies, and particularly once the gene predisposing to lung cancer in these families is identified, an evaluation of the effect that smoking has on lung cancer risk will be of great value in understanding carcinogenesis for carriers.
In the study of complex traits such as LT cancer, genetic heterogeneity is expected and has been observed for other cancers with hereditary components, such as breast cancer, colorectal cancer, malignant melanoma, and prostate cancer. Our results are consistent with this expectation. In other complex diseases, subdivision by clinical characteristics, such as age at onset and pattern of inheritance in the pedigrees, has often resulted in reduction of heterogeneity. As we subdivided our pedigrees into those that were more consistent with a strong genetic susceptibility (38 families with four or more affected relatives) and further subdivided them into those that were most consistent with an autosomal dominant susceptibility locus (23 families with five or more affected individuals in two or more generations), the evidence for linkage at 6q increased substantially, and the estimate of the proportion of linked families increased, approaching 1.0 for the subset of 23 multigenerational pedigrees. In fact, in this subset, the homogeneity LOD score and the HLOD were virtually identical, suggesting that these clinical familial characteristics had substantially reduced the heterogeneity.
Elsewhere, Hodge et al. (1997), Greenberg and Abreu (2001), and Abreu et al. (2002) have shown that there is some inflation of the type I error rate (false-positive rate) when one uses multiple penetrance models and when one computes HLOD scores as opposed to LOD scores. These authors have shown that the critical LOD threshold of 3.0 should be increased by 0.3 for maximizing over penetrance only (Hodge et al. 1997), by 0.47 when two-point HLODs are used (Abreu et al. 2002), and by 0.7 when multipoint HLODs are used (Greenberg and Abreu 2001). Thus, these authors suggest that adding 1 LOD unit to the significance threshold of LOD=3.0 (corresponding to a P value of .0001), which results in a corresponding critical threshold of HLOD=4.0, is a somewhat conservative adjustment for an analysis in which multipoint HLODs are calculated and multiple penetrance models are used. Our observed multipoint HLOD of 4.26 in the multigenerational families would satisfy these criterion.
We intend to follow up the results of this study by fine mapping the most significant regions. We also will attempt to replicate these linkage findings in an independent set of families with FLC. As a result of limited biospecimen availability, effective performance of a linkage study for a rapidly fatal disease such as lung cancer is difficult. However, the ability to globally genotype archival specimens greatly improved the power of our linkage study. In other cancers, discovery of susceptibility genes has led to greater understanding of the biological processes that cause these diseases and ultimately will lead to better methods for prevention and treatment. Likewise, we believe that discovery of lung cancer susceptibility genes will also be important in improving our understanding of this devastating disease.
Acknowledgments
We thank Tori Harris, Leticia B. Borrouso, Gary Lagasse, Shazia Iqbal, Ping Yang, Ph.D., Tracee Shevlin, Cynthia Nixa, Michelle McCullough, Alicia Salkowski, Glenda Sneed, Gracie Ehlert, Teara Carr, Derall Willis, Cassie Kirby, Lynn Mark, Kathryn Burton, Troy Rappold, and Julie Sorensen for assistance with data collection and Richard Levy, M.D. (Oncology/Hematology Care, Inc.), for referral of family probands. We thank Dana Behnemann and Erica Lockwood for assistance with statistical analyses. We thank Medford Klein, M.D., for his assistance. We are grateful to the families who participated in this research. We also thank the Alliance for Lung Cancer Advocacy, Support, and Education for assistance with this project. This work was supported in part by U. S. Public Health Service National Cancer Institute research grants U01 CA76293, R01 HL 71197, R01 CA637000, R01 CA60691, R01 CA87895, SEER N01 CN65064, UT Southwestern SPORE P50 CA070907, Johns Hopkins SPORE P50 CA058184, and Colorado SPORE P50 058187; U. S. Public Health Service National Institute of Environmental Health Sciences research grant P30-ES06096; and U. S. Department of Energy research grant DE-FG02-90ER60939. Some of the results of this study were obtained by the use of the program package S.A.G.E. 3.1, which is supported by U. S. Public Health Resource grant RR03655 from the National Center for Research Resources. Genotyping services for DNA extracted from blood samples were provided by CIDR. CIDR is fully funded through a federal contract from the National Institutes of Health to The Johns Hopkins University, contract N01-HG-65403.
Electronic-Database Information
The URL for data presented herein is as follows:
- Center for Inherited Disease Research (CIDR), http://www.cidr.jhmi.edu
References
- Abe T, Makino N, Furukawa T, Ouyang H, Kimura M, Yatsuoka T, Yokoyama T, Inoue H, Fukushige S, Hoshi M, Hayashi Y, Sunamura M, Kobari M, Matsuno S, Horii A (1999) Identification of three commonly deleted regions on chromosome arm 6q in human pancreatic cancer. Genes Chromosomes Cancer 25:60–64 [DOI] [PubMed] [Google Scholar]
- Abreu PC, Greenberg DA, Hodge SE (1999) Direct power comparisons between simple LOD scores and NPL scores for linkage analysis in complex diseases. Am J Hum Genet 65:847–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abreu PC, Hodge SE, Greenberg DA (2002) Quantification of type I error probabilities for heterogeneity LOD scores. Genet Epidemiol 22:156–169 10.1002/gepi.0155 [DOI] [PubMed] [Google Scholar]
- Almasy L, Blangero J (1998) Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet 62:1198–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amos CI (1994) Robust variance-components approach for assessing genetic linkage in pedigrees. Am J Hum Genet 54:535–543 [PMC free article] [PubMed] [Google Scholar]
- Amos CI, Shete S, Gu X (2001) Variance components analysis for genetic linkage of time to onset for disease. Genet Epidemiol 21:S768–S773 [DOI] [PubMed] [Google Scholar]
- Bailey-Wilson JE, Elston RC, Sellers TA, Rothschild H (1992) Segregation analysis of lung cancer using class A regressive models. Am J Hum Genet 51:A145 [Google Scholar]
- Bailey-Wilson JE, Wiest JS, Anderson MW, Saccomanno G (1998) Genetics of lung cancer. In: Kane MA, Bunn PA (eds) Biology of lung cancer: volume 122 of the lung biology in health and disease series. Marcel Dekker, New York, pp 53–98 [Google Scholar]
- Barghorn A, Speel EJM, Farspour B, Saremaslani P, Schmid S, Perren A, Roth J, Heitz PU, Komminoth P (2001) Putative tumor suppressor loci at 6q22 and 6q23–q24 are involved in the malignant progression of sporadic endocrine pancreatic tumors. Am J Pathol 158:1903–1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckett WS (1993) Epidemiology and etiology of lung cancer. Clin Chest Med 14:1–15 [PubMed] [Google Scholar]
- Berrieman HK, Ashman JNE, Cowen ME, Greenman J, Lind MJ, Cawkwell L (2004) Chromosomal analysis of non-small-cell lung cancer by multicolour fluorescent in situ hybridisation. Br J Cancer 90:900–905 10.1038/sj.bjc.6601569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blot WJ, Fraumeni JF (1976) Geographic patterns of lung cancer: industrial correlations. Am J Epidemiol 103:539–550 [DOI] [PubMed] [Google Scholar]
- ——— (1996) Cancers of the lung and pleura. In: Schottenfeld D, Fraumeni JF (eds) Cancer epidemiology and prevention. 2nd ed. Oxford University Press, New York, pp 637–665 [Google Scholar]
- Blot WJ, Harrington JM, Toledo A, Hoover R, Heath CW Jr, Fraumeni JF Jr (1979) Lung cancer after employment in shipyards during World War II. N Engl J Med 299:620–624 [DOI] [PubMed] [Google Scholar]
- Burch PR (1980) Smoking and lung cancer: tests of a causal hypothesis. J Chron Dis 33:221–238 10.1016/0021-9681(80)90067-3 [DOI] [PubMed] [Google Scholar]
- Carbone D (1992) Smoking and cancer. Am J Med 93:13S–17S 10.1016/0002-9343(92)90621-H [DOI] [PubMed] [Google Scholar]
- Cesari R, Martin ES, Calin GA, Pentimalli F, Bichi R, McAdams H, Trapasso F, Drusco A, Shimizu M, Masciullo V, d’Andrilli G, Scambia G, Picchio MC, Alder H, Godwin AK, Croce CM (2003) Parkin, a gene implicated in autosomal recessive juvenile parkinsonism, is a candidate tumor suppressor gene on chromosome 6q25–q27. Proc Natl Acad Sci USA 100:5956–5961 10.1073/pnas.0931262100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PL, Sellers TA, Bailey-Wilson JE, Rothschild H, Elston RC (1991) Segregation analysis of smoking-associated malignancies: evidence for mendelian inheritance. Am J Hum Genet Supp 49:15 [DOI] [PubMed] [Google Scholar]
- Cottingham RW Jr, Idury RM, Schaffer AA (1993) Faster sequential genetic linkage computations. Am J Hum Genet 53:252–263 [PMC free article] [PubMed] [Google Scholar]
- Daw EW, Ma J, Amos CI, Spitz MR. Monte Carlo Markov chain oligogenic segregation analysis of lung cancer. Eur J Hum Genet (in press) [Google Scholar]
- Dizier MH, Bonaiti-Pellie C, Clerget-Darpoux F (1993) Conclusions of segregation analysis for family data generated under two-locus models. Am J Hum Genet 53:1338–1346 [PMC free article] [PubMed] [Google Scholar]
- Doll R, Peto R (1981) The causes of cancer: quantitative estimates of avoidable risks of cancer in the United States today. Oxford University Press, Oxford [PubMed] [Google Scholar]
- Doll R, Peto R, Wheatley K, Gray R, Sutherland I (1994) Mortality in relation to smoking: 40 years’ observations on male British doctors. BMJ 309:901–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy DL (2002) SIBPAIR release 0.99.9, Herston, Queensland, Australia [Google Scholar]
- Durner M, Vieland VJ, Greenberg DA (1999) Further evidence for the increased power of LOD scores compared with nonparametric methods. Am J Hum Genet 64:281–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etzel CJ, Amos CI, Spitz MR (2003) Risk for smoking-related cancer among relatives of lung cancer patients. Cancer Res 63:8531–8555 [PubMed] [Google Scholar]
- Fraumeni JF, Wertelecki W, Blattner WA, Jensen RD, Leventhal BG (1975) Varied manifestations of a familial lymphoproliferative disorder. Am J Med 59:145–151 10.1016/0002-9343(75)90333-2 [DOI] [PubMed] [Google Scholar]
- Friberg L (1959) Smoking habits of monozygotic and dizygotic twins. Br Med J 1:1090–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAS (1998) Genetic Analysis System release, Oxford. Available at http://users.ox.ac.uk/~ayoung/gas.html (accessed July 20, 2004)
- Gauderman WJ, Morrison JL, Carpenter CL, Thomas DC (1997) Analysis of gene-smoking interaction in lung cancer. Genet Epidemiol 14:199–214 [DOI] [PubMed] [Google Scholar]
- Goeze A, Schlüns K, Wolf G, Thäsler Z, Petersen S, Petersen I (2002) Chromosomal imbalances of primary and metastatic lung adenocarcinomas. J Pathol 196:8–16 10.1002/path.1009 [DOI] [PubMed] [Google Scholar]
- Gottlieb MNS, Steadman R (1979) Lung cancer in shipbuilding and related industries in Louisiana. South Med J 72:1099–1101 [DOI] [PubMed] [Google Scholar]
- Greenberg DA, Abreu PC (2001) Determining trait locus position from multipoint analysis: accuracy and power of three different statistics. Genet Epidemiol 21:299–314 10.1002/gepi.1036 [DOI] [PubMed] [Google Scholar]
- Greenberg DA, Abreu PC, Hodge SE (1998) The power to detect linkage in complex disease by means of simple LOD-score analyses. Am J Hum Genet 63:870–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen LL, Jensen LL, Dimitrakakis C, Michalas S, Gilbert F, Barber HRK, Overgaard J, Arzimanoglou II (2002) Allelic imbalance in selected chromosomal regions in ovarian cancer. Cancer Genet Cytogenet 139:1–8 10.1016/S0165-4608(02)00620-9 [DOI] [PubMed] [Google Scholar]
- Heath CW (1958) Differences between smokers and non-smokers. Arch Intern Med 101:377–388 [DOI] [PubMed] [Google Scholar]
- Hodge SE, Abreu PC, Greenberg DA (1997) Magnitude of type I error when single-locus linkage analysis is maximized over models: a simulation study. Am J Hum Genet 60:217–227 [PMC free article] [PubMed] [Google Scholar]
- Hodge SE, Vieland VJ, Greenberg DA (2002) HLODs remain powerful tools for detection of linkage in the presence of genetic heterogeneity. Am J Hum Genet 70:556–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SJ, Cheng LS, Lozano G, Amos CI, Gu X, Strong LC (2003) Lung cancer risk in germline p53 mutation carriers: association between an inherited cancer predisposition, cigarette smoking, and cancer risk. Hum Genet 113:238–243 [DOI] [PubMed] [Google Scholar]
- Hyytinen E-R, Saadut R, Chen C, Paull L, Koivisto PA, Vessella RL, Frierson Jr HF, Dong J-T (2002) Defining the region(s) of deletion at 6q16–q22 in human prostate cancer. Genes Chromosomes Cancer 34:306–312 10.1002/gcc.10065 [DOI] [PubMed] [Google Scholar]
- Jemal A, Tiwari RC, Murray T, Ghafoor A, Samuels A, Ward E, Feuer EJ, Thun M (2004) Cancer statistics, 2004. CA Cancer J Clin 54:8–29 [DOI] [PubMed] [Google Scholar]
- Jensen RH, Tiirikainen M, You L, Ginzinger D, He B, Uematsu K, Xu Z, Treseler P, McCormick F, Jablons DM (2003) Genomic alterations in human mesothelioma including high resolution mapping of common regions of DNA loss in chromosome arm 6q. Anticancer Res 23:2281–2289 [PubMed] [Google Scholar]
- King TM, Tong L, Pack RJ, Spencer C, Amos CI (2002) Accuracy of family history of cancer as reported by men with prostate cancer. Urology 59:546–550 10.1016/S0090-4295(01)01598-9 [DOI] [PubMed] [Google Scholar]
- Konishi N, Nakamura M, Kishi M, Ishida E, Shimada K, Matsuyoshi S, Nagai H, Mitsuru E (2003) Genetic mapping of allelic loss on chromosome 6q within heterogeneous prostate carcinoma. Cancer Sci 94:764–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander E, Kruglyak L (1995) Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11:241–247 [DOI] [PubMed] [Google Scholar]
- Luk C, Tsao MS, Bayani J, Sheperd F, Squire JA (2001) Molecular cytogenetic analysis of non-small cell lung carcinoma by spectral karyotyping and comparative genomic hybridization. Cancer Genet Cytogenet 125:87–99 10.1016/S0165-4608(00)00363-0 [DOI] [PubMed] [Google Scholar]
- McPeek MS, Sun L (2000) Statistical tests for detection of misspecified relationships by use of genome-screen data. Am J Hum Genet 66:1076–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlo A, Gabrielson E, Mabry M, Vollmer R, Baylin SB, Sidransky D (1994) Homozygous deletion on chromosome 9p and loss of heterozygosity on 9p, 6p, and 6q in primary human small cell lung cancer. Cancer Res 54:2322–2326 [PubMed] [Google Scholar]
- Merup M, Moreno TC, Heyman M, Ronnberg K, Grander D, Detlofsson R, Rasool O, Liu Y, Soderhall S, Juliusson G, Gahrton G, Einhorn S (1998) 6q deletions in acute lymphoblastic leukemia and non-Hodgkin’s lymphomas. Blood 91:3397–3400 [PubMed] [Google Scholar]
- Millikin D, Meese E, Vogelstein B, Witkowski C, Trent J (1991) Loss of heterozygosity for loci on the long arm of chromosome 6 in human malignant melanoma. Cancer Res 51:5449–5453 [PubMed] [Google Scholar]
- Motulsky AG (1957) Drug reactions enzymes, and biochemical genetics. JAMA 165:835–837 [DOI] [PubMed] [Google Scholar]
- Noviello C, Courjal F, Theillet C (1996) Loss of heterozygosity on the long arm of chromosome 6 in breast cancer: possibly four regions of deletion. Clin Cancer Res 2:1601–1606 [PubMed] [Google Scholar]
- O’Connell JR, Weeks DE (1998) PedCheck: a program for identifying marker typing incompatibilities in linkage analysis. Am J Hum Genet 63:259–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott J (1989) Computer-simulation methods in human linkage analysis. Proc Natl Acad Sci USA 86:4175–4178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott L (1999) Analysis of human linkage. 3rd ed. Johns Hopkins University Press, Baltimore [Google Scholar]
- Pankratz VS, de Andrade M, Therneau T. The random effects Cox proportional hazards model: variance components for time to onset model. Genet Epidemiol (in press) [DOI] [PubMed] [Google Scholar]
- Petersen I, Bujard M, Petersen S, Wolf G, Goeze A, Schwendel A, Langreck H, Gellert K, Reichel M, Just K, du Manoir S, Cremer T, Dietel M, Ried T (1997) Patterns of chromosomal imbalances in adenocarcinoma and squamous cell carcinoma of the lung. Cancer Res 57:2331–2335 [PubMed] [Google Scholar]
- Re D, Starostik P, Massoudi N, Staratschek-Jox A, Dries V, Thomas RK, Diehl V, Wolf J (2003) Allelic losses on chromosome 6q25 in Hodgkin and Reed Sternberg cells. Cancer Res 63:2606–2609 [PubMed] [Google Scholar]
- Ries L, Eisner M, Kosary C, Hankey B, Miller B, et al (2000) SEER Cancer Statistics Review, 1973–1997. National Cancer Institute, NIH Pub. Mo 00-2789, Bethesda, MD [Google Scholar]
- Risch N, Giuffra L (1992) Model misspecification and multipoint linkage analysis. Hum Hered 42:77–92 [DOI] [PubMed] [Google Scholar]
- S.A.G.E. (2002) Statistical Analysis for Genetic Epidemiology [computer program package]. Statistical Solutions Ltd, Cork, Ireland [Google Scholar]
- Seaton A (1984) Occupational pulmonary neoplasms. In: Morgan WKC, Seaton A (eds) Occupational lung diseases. WB Saunders, Philadelphia, pp 657–675 [Google Scholar]
- Sellers TA, Bailey-Wilson JE (1998) Familial predisposition to lung cancer. In: Roth JA, Cox JD, Hong WK (eds) Lung cancer. Blackwell, Malden, MA, pp 57–71 [Google Scholar]
- Sellers TA, Bailey-Wilson JE, Elston RC, Rothschild H (1991) Evidence for Mendelian factors in early onset lung cancer. In: Origins of human cancer: a comprehensive review. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp 775–780 [Google Scholar]
- Sellers TA, Bailey-Wilson JE, Elston RC, Wilson AF, Ooi WL, Rothschild H (1990) Evidence for Mendelian inheritance in the pathogenesis of lung cancer. J Nat Cancer Inst 82:1272–1279 [DOI] [PubMed] [Google Scholar]
- Sellers TA, Bailey-Wilson JE, Potter JD, Rich SS, Rothschild H, Elston RC (1992) The effect of cohort differences in smoking prevalence on models of lung cancer susceptibility. Genet Epidemiol 9:261–271 [DOI] [PubMed] [Google Scholar]
- Sellers TA, Ooi WL, Elston RC, Chen VW, Bailey-Wilson JE, Rothschild R (1987) Increased familial risk for non-lung cancer among relatives of lung cancer patients. Am J Epidemiol 126:237–246 [DOI] [PubMed] [Google Scholar]
- Shete S, Amos CI, Hwang S-J, Strong LC (2002) Individual specific liability groups in genetic linkage analysis with application to Li-Fraumeni syndrome. Am J Hum Genet 70:813–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobel E, Lange K (1996) Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker sharing statistics. Am J Hum Genet 58:1323–1337 [PMC free article] [PubMed] [Google Scholar]
- Sobel E, Sengul H, Weeks DE (2001) Multipoint estimation of identity-by-descent probabilities at arbitrary positions among marker loci on general pedigrees. Hum Hered 52:121–131 10.1159/000053366 [DOI] [PubMed] [Google Scholar]
- Srikantan V, Sesterhenn IA, Davis L, Hankins GR, Avallone FA, Livezey JR, Connelly R, Mostofi FK, McLeod DG, Moul JW, Chandrasekharappa SC, Srivastava S (1999) Allelic loss on chromosome 6q in primary prostate cancer. Int J Cancer 84:331–335 [DOI] [PubMed] [Google Scholar]
- Terwilliger JD, Ott J (1994) Handbook of genetic linkage. Johns Hopkins University Press, Baltimore and London [Google Scholar]
- Therneau T (2003) Mixed-effect Cox models: sparse matrices, and their use in correlated frailty. Tech rep, Department of Health Sciences Research, Section of Biostatistics, Mayo Clinic, Rochester, MN [Google Scholar]
- Tibiletti MG, Sessa F, Bernasconi B, Cerutti R, Broggi B, Furlan D, Acquanti F, Bianchi M, Russo A, Capella C, Taramelli R (2000) A large 6q deletion is a common cytogenetic alteration in fibroadenomas, pre-malignant lesions and carcinomas of the breast. Clin Cancer Res 6:1422–1431 [PubMed] [Google Scholar]
- Tokuhata GK, Lilienfeld AM (1963) Familial aggregation of lung cancer in humans. J Nat Cancer Inst 30:289–312 [PubMed] [Google Scholar]
- Tong BC, Dhir K, Ha PK, Westra WH, Alter BP, Sidransky D, Koch WM, Califano JA (2004) Use of single nucleotide polymorphism arrays to identify a novel region of loss on chromosome 6q in squamous cell carcinomas of the oral cavity. Head Neck 26:345–352 10.1002/hed.10391 [DOI] [PubMed] [Google Scholar]
- Utada Y, Haga S, Kajiwara T, Kasumi F, Sakamoto G, Nakamura Y, Emi M (2000) Mapping of target regions of allelic loss in primary breast cancers to 1-cM intervals on genomic contigs at 6q21 and 6q25.3. Jpn J Cancer Res 91:293–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhagen PCMS, Hermans KGL, Brok MO, van Weerden WM, Tilanus MGJ, de Weger RA, Boon TA, Trapman J (2002) Deletion of chromosomal region 6q14–16 in prostate cancer. Int J Cancer 102:142–147 10.1002/ijc.10677 [DOI] [PubMed] [Google Scholar]
- Vieland VJ, Logue M (2002) HLODs, trait models and ascertainment: implications of admixture for parameter estimation and linkage detection. Hum Hered 53:23–35 10.1159/000048601 [DOI] [PubMed] [Google Scholar]
- Virmani AK, Fong KM, Kodagoda D, McIntire D, Hung J, Tonk V, Minna JD, Gazdar AF (1998) Allelotyping demonstrates common and distinct patterns of chromosomal loss in human lung cancer types. Genes Chromosomes Cancer 21:308–319 [DOI] [PubMed] [Google Scholar]
- Weeks DE, Ott J, Lathrop GM (1990) SLINK: a general simulation program for linkage analysis. Am J Hum Genet 47:A204 [Google Scholar]
- Welch HG, Schwartz M, Woloshin S (2000) Are increasing 5-year survival rates evidence of success against cancer? JAMA 283:2975–2978 [DOI] [PubMed] [Google Scholar]
- Whittemore AS, Halpern J (2001) Problems in the definition, interpretation, and evaluation of genetic heterogeneity. Am J Hum Genet 68:457–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JT, Van Eerdewegh P, Almasy L, Blangero J (1999) Joint multipoint linkage analysis of multivariate qualitative and quantitative traits. I. Likelihood formulation and simulation results. Am J Hum Genet 65:1134–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright DK, Manos MM (1990) PCR protocols. In: Innis MA, Gelfard DH, Sninsky JJ, White TJ (eds) Sample preparation from paraffin-embedded tissues. Academic Press, San Diego, pp 153–158 [Google Scholar]
- Yang P, Schwartz AG, McAllister AE, Swanson GM, Aston CE (1999) Lung cancer risk in families of nonsmoking probands: heterogeneity by age at diagnosis. Genet Epidemiol 17:253–273 [DOI] [PubMed] [Google Scholar]
- Zeller C, Hinzmann B, Seitz S, Prokoph H, Burkhard-Goettges E, Fisher J, Jandrig B, Schwarz L-E, Rosenthal A, Scherneck S (2003) SASH1: a candidate tumor suppressor gene on chromosome 6q24.3 is downregulated in breast cancer. Oncogene 22:2972–2983 10.1038/sj.onc.1206474 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Matthlessen P, Harder S, Siebert R, Castoldi G, Calasanz J, Wong KF, Rosenwald A, Ott G, Atkin NB, Schlegelberger B (2000) A 3-cM commonly deleted region in 6q21 in leukemias and lymphomas delineated by florescence in situ hybridization. Genes Chromosomes Cancer 27:52–58 [DOI] [PubMed] [Google Scholar]