

Abstract
Many sequence variants in predisposition genes are of uncertain clinical significance, and classification of these variants into high- or low-risk categories is an important problem in clinical genetics. Classification of such variants can be performed by direct epidemiological observations, including cosegregation with disease in families and degree of family history of the disease, or by indirect measures, including amino acid conservation, severity of amino acid change, and evidence from functional assays. In this study, we have developed an approach to the synthesis of such evidence in a multifactorial likelihood-ratio model. We applied this model to the analysis of three unclassified variants in BRCA1 and three in BRCA2. The evidence strongly suggests that two variants (C1787S in BRCA1 and D2723H in BRCA2) are deleterious, three (R841W in BRCA1 and Y42C and P655R in BRCA2) are neutral, and one (R1699Q in BRCA1) remains of uncertain significance. These results provide a demonstration of the utility of the model.
Introduction
The identification of specific genes involved in a number of common diseases has resulted in the integration of genetic testing into clinical practice. For many of these genes, the sequence variants that are identified include known deleterious (often protein-truncating) mutations, recognized polymorphisms assumed to be neutral in terms of disease risk, and other variants (usually with missense changes) of uncertain clinical relevance. The last category poses problems for genetics counseling, since tested individuals and their families are given a seemingly ambiguous result, unless sufficient evidence is available that a given missense change is deleterious. In the case of the breast cancer susceptibility genes BRCA1 (MIM 113705) and BRCA2 (MIM 600185), these so-called unclassified variants (UCVs) account for approximately half of all unique variants detected (other than common polymorphisms) (see Breast Cancer Information Core [BIC] database Web site) and were identified in 13% of all women tested in one study (Frank et al. 2002). Thus, if one accepts that more rigorous screening and/or other preventive measures are useful in lowering morbidity and mortality in individuals who carry a high-risk deleterious mutation in these genes, a relatively large number of them could be helped by the classification of these variants as neutral or deleterious. Although the present article focuses on BRCA1 and BRCA2, similar issues occur in genetic testing for other common disorders for which major susceptibility genes have been identified.
To address this important clinical problem, various types of evidence may help to classify such variants as deleterious or neutral, with respect to the disease of interest. These include frequency of the variant in cases and controls, co-occurrence of the variant with a known deleterious mutation in one or more tested individuals (under the assumption that either homozygosity for true deleterious mutations is embryonically lethal or homozygotes will at least have a clearly recognizable phenotype), cosegregation of the variant with disease in families, occurrence of disease in relatives of index cases with a given variant, the nature and position of the amino acid substitution, the degree of conservation of amino acids among species, and the results of functional assays. Each of these sources of evidence has particular strengths and limitations in addressing the general problem of causality of sequence variants. These lines of evidence are summarized in table 1.
Table 1.
Types of Evidence Potentially Useful for UCV Classification
| Line of Evidence | Advantage(s) | Disadvantage(s) |
| Frequency in cases and controls | Provides a direct estimate of associated cancer risk | Variants are rare, so such studies would need to be prohibitively large (10,000+) |
| Co-occurrence (in trans) with deleterious mutations | If homozygotes and compound heterozygotes are assumed to be embryonically lethal (or vanishingly rare), we can often classify a variant as neutral on the basis of a single observation | Much less power to show causality; quantification is dependent on the assumed fitness of the homozygous genotype, which is not known with precision |
| Cosegregation with disease in pedigrees | Easily quantifiable and directly related to disease risk; not susceptible to uncertainties in mutation frequencies or population stratification | Requires sampling of additional individuals in the pedigrees (particularly additional cases), which may be difficult to achieve |
| Family history | Usually available for most variants without additional data or sample collection; potentially very powerful | Dependent on family ascertainment scheme; could be biased in stratified populations with heterogeneous ascertainment, so not as robust as cosegregation; power may be low for infrequent variants |
| Species conservation and amino acid–change severity | Can be applied to every possible missense change in the BRCA1 and BRCA2 genes; does not require extensive family history; complete conservation is predictive if enough evolutionary time sequence is available | Only indirectly related to disease risk; the magnitude of odds ratios is not sufficient to classify variants without additional information |
| Functional studies | Can evaluate biologically the variant's effect on the protein's ability to perform some key cellular functions | May only be relevant for variants in certain domains of the protein; the function tested may not be related to cancer causation |
| Loss of heterozygosity | Straightforward to quantify as an adjunct to cosegregation data; robust | Requires tumor material |
| Pathological classification | Potentially powerful for BRCA1 tumors in which the pathological characteristics are quite distinct; quantifiable | Prediction is weak when routine pathology data are used; systematic evaluation requires tumor material; weakly predictive for BRCA2 |
We and others have examined such classification schemes by use of a variety of approaches. For example, in terms of cosegregation of variants within a pedigree, Thompson et al. (2003) provided a method of calculating odds of causality for UCVs by use of complete pedigree data. Petersen et al. (1998) performed a similar study but used a more restricted approach. In terms of conservation of amino acids across species, a number of studies have been done. Miller and Kumar (2001) validated the hypothesis that missense variants at highly conserved/invariant residues would more often be deleterious, whereas highly variable changes would more likely be neutral. With regard to BRCA1, Fleming et al. (2003) and Abkevich et al. (2004) used the conservation of BRCA1 residues in a variety of mammalian and nonmammalian species to make preliminary classifications of 139 (Fleming et al. 2003) and 146 (Abkevich et al. 2004) putative missense mutations. In terms of functional assays, examination of BRCA1 has been limited to two functional domains: the RING finger (residues 24–64) and the BRCT domain (residues 1642–1863). Functional mammalian and yeast-based assays have focused on transcriptional activation by the BRCT domain (Vallon-Christersson et al. 2001). Recently, Mirkovic et al. (2004) used the three-dimensional protein structure to develop a rule-based system for the classification of variants, applying this approach to 57 observed constitutional missense variants in the BRCT domain of BRCA1. For BRCA2, analyses of functional domains have focused on the DNA-binding region between amino acids 2373 and 3256 (Yang et al. 2002) and on the eight 40–amino-acid BRC repeats in exon 11 that are associated with interaction of BRCA2 with the RAD51-recombination and DNA-repair protein (Wong et al. 1997; Chen et al. 1999; Davies et al. 2001).
A comprehensive model is needed, in which all these sources of evidence can be used together to create a combined assessment of a particular sequence variant of interest. In this comprehensive model, both quantitative and qualitative evidence would be properly weighted to arrive at a final classification. Ideally, the end result would be the overall odds of causality—that is, the ratio of the likelihood of the observed data under the hypothesis of causality to that under the hypothesis of neutrality. If all of the various types of evidence were quantifiable in the same way, this would be straightforward. However, each type of evidence depends on different models and underlying assumptions, and some are more suitable to quantification and formulation as a likelihood ratio than others. Here, we focus primarily on the relevant data that can be evaluated directly on a genetic/epidemiological basis, as these data are easily quantifiable in terms of likelihood ratios; moreover, they are most directly related to the clinical outcome of interest—that is, the risk of developing cancer for a carrier of the particular sequence variant under consideration.
Methods
For clarity, we assumed that all variants in the gene of interest can be classified into two categories: “mutations” (M) that predispose to a high risk of breast and ovarian cancer and “neutral variants” (V) that cause no risk. Thus, we make the important simplifying assumption that variants do not have an intermediate risk. Almost all protein-truncating variants are known, with high probability, to be mutations. The aim is to determine whether or not other variants are likely to be deleterious mutations. These include amino acid substitutions, in-frame deletions, silent mutations, and some intronic changes. We would like to determine statistically the posterior probability that each variant (V) is a mutation (M), given the available data:
 |
The statistical analysis focuses on the likelihood ratio (Pr[Data|M]/Pr[Data|V]). The choice of an appropriate prior probability (Pr[M]) that a new variant is a mutation is uncertain. However, given that there is a high frequency of such variants and that only a few of the variants can be unequivocally classified as mutations, it is clear that the probability is low. At least 70% of the families with breast or ovarian cancer that exhibit clear linkage to BRCA1 or BRCA2 have been shown to harbor deleterious mutations; a significant fraction of the remaining families (at least those linked to BRCA1) harbor large-scale rearrangements. We believe that the prior probability of a given UCV being deleterious is <10% and may be closer to 1%. This suggests that the appropriate likelihood threshold for declaring a variant to be deleterious should be at least 1,000:1. The appropriate threshold for declaring against causality is not as critical, since this decision does not affect genetics counseling. For the purposes of classification in the BIC, we suggest a likelihood ratio of 100:1 against causality as a useful criterion. Of course, the choice of threshold in each clinical situation will vary according to the particular circumstances.
Specific Contributions of Individual/Family Data Components
Co-occurrence with deleterious mutations
A variety of mouse studies (Gowen et al. 1996; Liu et al. 1996; Hohenstein et al. 2001) have indicated that homozygosity for Brca1 is embryonically lethal. This finding is reinforced by the clear deficit of BRCA1 homozygotes and compound heterozygotes, compared with expected numbers, among a series of individuals with the founder mutations 185delAG and 5382insC (0 observed vs. 6.5 expected) (Frank et al. 2002; Abkevich et al. 2004). For each BRCA1 variant under consideration, we first examined the frequency of the mutation in the Myriad Genetics Laboratories database, which contains complete full-sequence data for both BRCA1 and BRCA2 from >20,000 individuals, as well as rudimentary family and patient history. In the following analysis, we assumed that individuals homozygous for a deleterious mutation in BRCA1 or BRCA2 are extremely rare. If the variant is neutral, the probability of an individual with the variant also carrying (in trans) a deleterious mutation, p1, can be roughly estimated as half the overall frequency of deleterious mutations in the population being studied. If the variant is deleterious, this probability becomes
 |
Thus, if one observes the variant n times, k of which are in conjunction with a deleterious mutation, the appropriate likelihood ratio is given by the following binomial likelihood ratio:
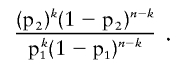 |
For BRCA1, the frequency of deleterious mutations in the Myriad Genetics Laboratories database is 0.088 (1,765 known BRCA1 deleterious mutations in 20,000 tests). Taking into account the evidence that BRCA1 homozygotes and compound heterozygotes are vanishingly rare and quite likely to be embryonically lethal, we assumed p2=0.0001 for these calculations.
For BRCA2, the corresponding estimate for the frequency of deleterious mutations is 0.059. The fitness issue here is slightly more problematic, since BRCA2 compound heterozygotes have been found among individuals with the rare recessive disease Fanconi anemia type D1 (Howlett et al. 2002; Wagner et al. 2004). However, it is reasonable to assume that compound heterozygotes for deleterious mutations in BRCA2 are extremely rare in adults, since the Fanconi anemia phenotype usually leads to death in early childhood. Taking into account the additional uncertainty associated with BRCA2 homozygosity, we assumed p2=0.001 for these calculations. One complication that arises in these data is the distinction between mutations occurring in cis and those in trans. Although the parental origin of the mutations is rarely known, mutations occurring in cis can often be recognized by recurrent observation of the same mutation/variant combination, and we have ignored these instances in our calculations.
The frequency of variants in groups of individuals, classified by likelihood of being a mutation carrier (i.e., family history)
A substantial amount of family history information is available for BRCA1 and BRCA2. The most important source, given the scope and completeness of the genotyping, is the data obtained from sequencing by Myriad Genetics Laboratories. The rationale here is that mutation prevalence is known to be strongly dependent on certain key factors (disease status of the proband, age at diagnosis, and number and age of relatives with breast or ovarian cancer), so these characteristics should also predict the prevalence of a new disease-causing variant, whereas the prevalence of a neutral variant should be independent of family history. As a “proof of principle,” we have examined the confirmed deleterious missense mutation BRCA1 C61G, for which there are 57 occurrences in the Myriad Genetics Laboratories database with family history information available. We compared the family histories of these 57 index cases with those of all known deleterious mutations in the database by use of a multinomial likelihood-ratio model, resulting in odds in favor of causality of >1,000,000:1, showing the potential utility of this approach, at least for relatively frequent variants.
Cosegregation data
To assess causality from the cosegregation data, we used the statistical model described by Thompson et al. (2003). For these calculations, we assumed an allele frequency of the variant of 0.0001 and used the BRCA penetrance estimates that were based on the recent meta-analysis of 22 population-based studies (Antoniou et al. 2003), with pooling across age groups, if necessary, depending on the level of detail of the family history information. Although family-based estimates might be more appropriate, we preferred to use these estimates, since the criteria for testing differ markedly among testing centers and the use of the population data would, if anything, be conservative. We do not, at present, allow for the possibility that a variant observed in the proband is a de novo mutation, although this could easily be incorporated into the model. Because, in many cases, complete pedigree data were unavailable, we relied on crude family history information and constructed complete pedigrees by creating individuals of unknown phenotype and genotype to connect the individuals in the pedigree. Note that, since analysis of cosegregation is conditional on the phenotypes in the family, the data on cosegregation can be considered independent of the data on family history (FH). The data from the co-occurrence of the variant with deleterious mutations are independent of the other information as well, so that these three likelihood ratios can be evaluated independently and multiplied:
 |
Incorporation of the Data on Sequence Conservation, Nature of Substitution, and Functional Characteristics
These data are more difficult to evaluate statistically than the data described above, since there is no direct link between these data and cancer risk. Our approach was to start with an initial model that was based on the limited number of already-classified missense variants for which data are available, and then, using the individual-specific data described above, we iteratively refined the estimated parameters as variants were classified into either deleterious or neutral categories. We describe below, in more detail, some initial models for this process.
Severity of the amino acid substitution
The idea here is to use a score for the type of substitution and to derive the likelihood ratio on the basis of the distribution of this score in known neutral variants and known deleterious mutations. One approach is to use the chemical-difference matrix proposed by Grantham (1974) to produce a score (Grantham matrix score [GMS]) for the observed substitution in the variant that is being investigated (GMSUV). We then determined the probability density function of the two distributions of scores, f(GMS;θM) and f(GMS;θV), where the form and parameterization, θ, of f() depends on the distribution of the data. The likelihood ratio for these data is then given by
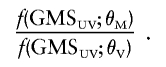 |
As a preliminary strategy for incorporating these data, we calculated the mean and SD of the GMS in known deleterious BRCA1 missense mutations (excluding those that are known to be splice mutations), as well as that for known missense changes that are clearly neutral (e.g., common polymorphisms). For true deleterious missense mutations, the mean and SD were 133 and 65, respectively, whereas, for neutral variants, the corresponding values were 65 and 39. Given the apparent relationship between the mean and SD, we assumed that the distribution of f(GMS; θ) was lognormal, although, at present, there are insufficient numbers of known deleterious and neutral variants available to test the fit to this (or any other) distribution. This approach assumes that the mechanism of action in cancer causation is the change in the protein associated with the missense UCV. For variants near the intron/exon boundary, however, this assumption may not be valid, and the variant may be associated with disease through alternative splicing. To avoid this problem, such variants could be evaluated for their potential effect on splicing by use of a predictive algorithm, such as that used in the Berkeley Drosophila Genome Project (see Berkeley Drosophila Genome Project Web site). If possible, these variants were assessed through evaluation of alternative splicing by use of mRNA from blood samples of patients carrying the variant.
Conservation of the variant amino acid across species
Although mutations at fully conserved amino acids are plausibly likely to be deleterious, it is not known whether such mutations are invariably associated with an increased cancer risk. Using sequence data from the genes orthologous to human BRCA1 and BRCA2 in six and four additional species, respectively, Abkevich et al. (2004) derived a mathematical model for BRCA sequence variation in which they postulated two types of amino acid substitutions: one under functional constraint and therefore slowly substituting (SS), and the other under no selective pressure and therefore fast substituting (FS). Thus, a UCV that results in an amino acid substitution at an SS position might be expected to be deleterious, whereas a UCV that occurs at an FS position is more likely to be neutral. On the basis of the observed multiple sequence alignments and a mathematical model, the relative fraction of the two types of changes can be estimated for each possible number of different residues seen in the multiple sequence alignment, and the relative odds of a variant being of either type can be calculated under the model. For example, under this model, a UCV in BRCA1 that changes a completely conserved amino acid is 10.4 times (125:12) more likely to be of the SS variety (and, hence, more likely to be deleterious). If this classification were completely concordant with the risk classification, these would also be the odds in favor of causality. For BRCA2, a similar procedure can be used, although the limited number of species for which sequence data are available reduces the discriminatory power. As more BRCA sequence data become available, these models will undoubtedly be improved.
Functional data
These data are perhaps the most difficult to put into a likelihood-based framework. This is because there are a number of functional assays, each of which potentially tests a different function of the protein. To incorporate these data into the model, it will be necessary to have a larger set of variants with both (1) clear classification (according to the specified thresholds) of the deleterious and neutral categories and (2) functional data from a variety of different assays. For this reason, we have used functional data as qualitative supporting evidence, without directly incorporating these data into the likelihood-based evaluation.
On the basis of the data for which we have good initial models relevant to cancer risk, we can easily combine the relevant odds of causality. Those variants that are classified with high probability (i.e., with odds for or against causality reaching predefined thresholds) can then be used to evaluate and refine statistical models relating to functional or sequence-conservation data. As more variants are classified, these models will become more discriminating and, hence, more useful in the classification of variants for which there is insufficient family history and cosegregation data to achieve a clinically useful level of evidence for or against causality. This process is detailed in the flowchart in figure 1.
Figure 1.

Flowchart of the procedure for classification of sequence variants of unknown clinical significance. ER = estrogen receptor status; LR = likelihood ratio.
Results
To illustrate the model, we have selected three UCVs in BRCA1 and three in BRCA2 for analysis with the approaches described above. The likelihood ratios for each of the components in the analysis, as well as the combined odds for each of the six variants analyzed, are discussed below and are summarized in table 2.
Table 2.
Odds in Favor of Each Variant Being Deleterious for the Six Variants Discussed in the Text, for Each Source of Information and Overall
|
Odds in Favor of Causality for |
||||||
|
BRCA1 |
BRCA2 |
|||||
| Data Source | C1787S | R1699Q | R841W | Y42C | P655R | D2723H |
| Co-occurrence | 1.2 | 1.4 | .028 | 8.9 × 10−11 | .007 | 2.0 |
| Cosegregation | 1,694 | 2.84 | 4 × 10−9 | 6.7 × 10−7 | .48 | 13,731 |
| GMS | 1.5 | .48 | 1.31 | 3.49 | 1.35 | .98 |
| Conservation | 10.4 | 10.4 | .006 | .194a | .004a | 5.0 |
| Overall odds | 31,692 | 20 | 8.7 × 10−13 | 4 × 10−17 | .00002 | 134,563 |
Deleted residue counted as a substitution.
BRCA1
C1787S
This variant has been observed four times, but it has not been detected in any individual who also carried a clear deleterious mutation. Two available families show evidence of cosegregation with disease-yielding combined odds in favor of causality from the cosegregation data of 1,694:1. Incorporation of the data on co-occurrence yields overall odds in favor of causality of 2,032:1. Thus, on the basis of the family data alone, this variant could be classified as a disease-associated mutation. The cysteine residue is completely conserved, including in Xenopus and in the pufferfish Tetraodon. The substitution to serine is associated with a GMS of 112, compared with the average GMS for known polymorphisms of 60, the expected GMS value of 78 for a random missense change, and a GMS of 133 for 16 previously characterized deleterious missense mutations. The genomic data give odds of 15.5:1 in favor of the variant being deleterious, consistent with the pedigree data. This sequence variant has not yet been characterized functionally, but its effect on the three-dimensional protein structure has been modeled, and it is predicted to impact protein function (Mirkovic et al. 2004). It should be mentioned that the C1787S variant is always seen (presumably in cis) with an additional variant, G1788D.
R1699Q
This mutation has been observed seven times in the Myriad Genetics Laboratories database, but it has never been detected in an individual with a deleterious mutation. This provides odds of 1.4:1 in favor of it being a deleterious mutation. Three small families with multiple individuals who were tested for this variant were available for analysis, leading to an overall cosegregation-based odds ratio of 2.8:1 in favor of causality for this variant. The combined odds from these two sources are 4:1 in favor of causality, and, therefore, this variant cannot be classified on the basis of this evidence alone. As with C1787S, the arginine residue is completely conserved. However, the change from arginine to glutamine yields a GMS of 43, lower than many of the known polymorphic substitutions. The combined odds ratio from the genomic data is 4.99:1, again slightly in favor of causality. In mammalian cells, this sequence variant showed clear loss of transcriptional activation capability (Vallon-Christersson et al. 2001). It should be noted that another alteration in this same codon, R1699W, is considered by Myriad Genetics Laboratories to be a deleterious mutation, on the basis of both functional (Koonin et al. 1996; Vallon-Christersson et al. 2001) and cosegregation data.
R841W
This variant has been observed in the Myriad Genetics Laboratories database 57 times, with 1 of those observations occurring in an individual who also carried a known deleterious mutation. Analysis of cosegregation in six pedigrees with multiple individuals tested showed quite convincing evidence against this variant being a high-risk allele (250,000,000:1 against causality). Thus, this variant can be unequivocally assigned to the neutral/nondisease-associated sequence variant category.
In contrast to the previous two BRCA1 UCVs discussed above, this residue shows considerable variation among the various orthologues, with three alternative amino acids present and no conservation other than in the Pan troglodytes sequence. The amino acid associated with this variant, tryptophan, is not found in any of the five other species with sequence data available. The sequence and substitution data give odds of ∼130:1 against causality, which supports the genetic data. Barker et al. (1996) have suggested that this variant may be associated with a modest increased risk of breast cancer. Since our approach considers only the hypotheses that the variant is high penetrance or is neutral, we cannot exclude the possibility that R841W is associated with a more moderate risk.
BRCA2
Y42C
This mutation has been observed 144 times, 8 of which were in patients who also carried a known BRCA2 deleterious mutation in trans with Y42C. We analyzed 17 pedigrees with this UCV and the overall odds against causality from these data were ∼1,500,000:1. Thus, on the basis of the pedigree cosegregation data alone, the odds are overwhelming against causality, and the co-occurrence data provides, if anything, even stronger evidence against causality.
For this variant, the tyrosine residue is conserved in chicken but is deleted in the Tetraodon sequence. The change from a tyrosine to a cysteine is one of the most severe changes, as measured by the GMS (194). Thus, the evidence based on sequence conservation and severity of the amino acid substitution is equivocal (combined odds, 1.3:1 against causality). However, as noted above, the co-occurrence and cosegregation data are overwhelmingly against Y42C being a deleterious BRCA2 allele.
P655R
This variant has been detected 63 times, twice with a known deleterious mutation. Ten pedigrees were analyzed for this variant and, taken together, exhibited weak evidence against causality (2:1). The combined evidence from the pedigree and co-occurrence data is 298:1 against causality, which would exceed our suggested threshold for classifying this as a neutral variant. This residue is conserved in rat and dog but is deleted in chicken and Tetraodon. The proline-to-arginine change is associated with a GMS of 103, a score that is between the average value for neutral changes and the value for BRCA1 deleterious mutations.
D2723H
This variant has been observed in the Myriad Genetics Laboratories database 24 times and has never appeared with a proven deleterious mutation. The variant yields odds in favor of causality under the BRCA2 co-occurrence model of 2.0:1. All 10 pedigrees with multiple individuals tested for this variant showed complete cosegregation with breast and ovarian cancer, yielding overall odds of 13,731:1 in favor of causality. Thus, the pedigree data provide odds of ∼57,000:1 in favor of causality—more than sufficient to classify the variant as deleterious by use of the suggested threshold of 1,000:1. The aspartate residue is completely conserved as far out as Tetraodon, although the GMS for this substitution is only 81. A BRCA2 protein carrying this variant showed disrupted DNA-repair capacity after exposure to gamma irradiation and mitomycin-c, similar to the deleterious truncating mutation 6174delT. Moreover, in 293T human embryonal kidney cancer cells, the BRCA2 protein with D2723H showed aberrant cellular localization, compared with the wild-type protein (K. Wu, S. Hinson, A. Ohashi, S. Tavtigian, A. Deffenbaugh, D. Goldgar, and F. Couch, unpublished data). Thus, in our view, the BRCA2 D2723H variant can be classified unequivocally as a deleterious BRCA2 allele.
Discussion
Although we have focused on the BRCA1 and BRCA2 genes, many of the methods described here are quite general and can be used for any hereditary disease in which the genes responsible are characterized by many sequence variants for which it is difficult to assess a clear association with disease. As genetic testing for common, multifactorial diseases moves into clinical practice, the problems associated with the interpretation of sequence variants of unknown significance will result in psychological stress for patients and families and an increased burden on genetics counselors. In addition to the obvious clinical utility of developing and implementing a rigorous classification procedure for UCVs, the process could raise interesting questions about the biological basis of the disease predisposition conferred by the gene being studied. If, for example, a particular sequence variant shows conclusive evidence of causality on the basis of epidemiological data but functions normally in a specific assay, this would lead us to infer that the function being tested is perhaps not relevant to the disease process.
In addition to the main factors discussed extensively above, a number of other pieces of data could aid the classification of unknown sequence variants. These might be somewhat dependent on the disease and the gene being studied. For example, for BRCA1, we could take advantage of the fact that there is strong evidence that the pathology of BRCA1 tumors differs from that of tumors in noncarriers of the same age (Breast Cancer Linkage Consortium 1997; Lakhani et al. 1998). Provided that one assumes that the pathological characteristics of tumors are not dependent on other familial factors, the odds based on pathological characteristics can be multiplied across all tumors carrying that specific germline UCV.
Another piece of information that could potentially be incorporated into such models, at least for many tumor-suppressor genes, is loss of heterozygosity (LOH) in tumors carrying the putative causal variant. For example, in BRCA1, ∼85% of tumors exhibit LOH at BRCA1, compared with ∼30% of breast cancers in noncarriers. Moreover, the LOH invariably involves the wild-type chromosome (Cornelis et al. 1995). Similar arguments apply to BRCA2 and to several other cancer-predisposition genes. Methods for incorporating LOH data into linkage analysis have been developed (Rebbeck et al. 1994), and this approach could be used to extend the cosegregation analysis.
For almost all the lines of evidence we have considered, it is clearly easier to obtain high odds in favor of neutrality than it is to show causality. This is similar to the situation in linkage analysis in which a single recombinant event is sufficient to exclude tight linkage but a much larger number of events is required to provide significant evidence in favor of linkage. It should be emphasized that our classification evaluation is based on the relative likelihood of the observed data under two specific hypotheses: that of complete neutrality of the variant (i.e., it confers no increased risk of disease) and that of what we have termed as “causality” (i.e., the risk of disease conferred by the variant under consideration is comparable to the risk conferred by known mutations). Whether this is appropriate for all disease genes (and for which ones) is an important consideration in the application of this approach to a particular problem. If, for example, a particular missense mutation were associated with an intermediate risk, it might be classified in the deleterious or the neutral category, depending on the type of data available. Clearly, in this situation, more sophisticated models will be required. One of the expectations for such intermediate-risk variants is that they will prove difficult to classify, in spite of a substantial amount of data. This is a result of the potential for conflicting data from the various sources, which would make it difficult to achieve the specified thresholds for classification as either neutral or deleterious. If sufficient pedigrees are available for such variants and if these pedigrees have a reasonable number of individuals typed for the variant, it may be possible to estimate directly (through pedigree or case-control studies) the risk associated with the variant, although the derived estimate is likely to have wide confidence limits.
The classification of variants should ideally be based on clinical observations, since these are directly related to cancer risk and, hence, are the most relevant and also the most straightforward to quantify. On the basis of clinical information and our assumed models and thresholds, we were able to classify five of the six variants we studied as either deleterious or neutral. The additional value of the genomic data in these cases was less clear, but, in general, the genomic data supported the clinical data. It is interesting to note that, in each case, the score derived from species conservation pointed in the same direction as the clinical data, although the odds were much weaker. The GMSs, however, were inconsistent, giving odds in the opposite direction in four of the five classifiable cases and calling into question the utility of this measure in the classification process.
In summary, we believe that this multidisciplinary approach to evaluation of sequence variants of unknown significance provides a system of checks and balances and avoids overreliance on one source of information. This should result in more-reliable classification of such variants, which in turn will improve the clinical utility of genetic tests now being offered to patients and their families. The work presented here represents only the first step in an ongoing process. Additional work remains to be done, including the examination of the robustness of the method against violations of basic assumptions, the incorporation of this uncertainty into the model, the validation of each of the individual components through the accumulation of large amounts of additional data, and the development of other approaches to the integration of the various components into a comprehensive model.
Acknowledgments
Members of the BIC Steering Committee are (in alphabetical order) Merete Bjørnslett (Department of Cancer Genetics, Norwegian Radium Hospital, Oslo), Larry Brody (National Human Genome Research Institute, Bethesda), Georgia Chenevix-Trench (Queensland Institute of Medical Research, Brisbane, Australia), Fergus J. Couch (Mayo Clinic College of Medicine, Rochester, MN), Amie M. Deffenbaugh (Myriad Genetics Laboratories, Salt Lake City), Peter Devilee (Department of Human Genetics, Leiden University, Leiden, Netherlands), Douglas Easton (Cancer Research UK, Genetic Epidemiology Unit, Strangeways Research Laboratory, Cambridge, United Kingdom), Charis Eng (Clinical Cancer Genetics Program, Division of Human Genetics, Ohio State University, Columbus), William Foulkes (McGill University, Montreal), David Goldgar (Unit of Genetic Epidemiology, International Agency for Research on Cancer, Lyon, France), Kathi Malone (Fred Hutchinson Cancer Research Center, Seattle), Alvaro N. A. Monteiro (H. Lee Moffitt Cancer Center, Tampa, FL), Kate Nathanson (Department of Medicine, Medical Genetics, University of Pennsylvania School of Medicine, Philadelphia), Susan Neuhausen (Epidemiology Division, Department of Medicine, University of California at Irvine, Irvine), Sharon Plon (Department of Pediatrics, Baylor College of Medicine, Houston), Csilla Szabo (Lyon, France), Sean Tavtigian (Unit of Genetic Cancer Susceptibility, International Agency for Research on Cancer, Lyon, France), and Tom Walsh (Department of Genetics, University of Washington, Seattle). This work was supported by National Institutes of Health awards CA92309 (to A.N.A.M.), CA81203 (to D.E.G.), and CA82267 (to F.J.C.). F.J.C. also acknowledges support from the Breast Cancer Research Foundation and the U.S. Army Medical Research and Materiel Command (DAMD17-1-00-0328). D.F.E. is a Principal Research Fellow of Cancer Research UK. D.F.E. and D.E.G. received support for this work from the Canadian Institutes of Health Research through the INHERIT BRCAs research program. The authors gratefully acknowledge the technical assistance of Helene Renard, Colette Bonnardel, and Yvette Granjard.
Electronic-Database Information
The URLs for data presented herein are as follows:
- Berkeley Drosophila Genome Project, http://www.fruitfly.org/seq_tools/splice.html (for splice-site analysis)
- Breast Cancer Information Core (BIC) database, http://research.nhgri.nih.gov/bic/
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for BRCA1 and BRCA2)
References
- Abkevich V, Zharkikh A, Deffenbaugh A, Frank D, Chen Y, Shattuck D, Skolnick M, Gutin A, Tavtigian S (2004) Analysis of missense variation in human BRCA1 in the context of interspecific sequence variation. J Med Genet 41:492–507 10.1136/jmg.2003.015867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoniou A, Pharoah PD, Narod S, Risch HA, Eyfjord JE, Hopper JL, Loman N, et al (2003) Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 72:1117–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DF, Almeida ER, Casey G, Fain PR, Liao SY, Masunaka I, Noble B, Kurosaki T, Anton-Culver H (1996) BRCA1 R841W: a strong candidate for a common mutation with moderate phenotype. Genet Epidemiol 13:595–604 [DOI] [PubMed] [Google Scholar]
- Breast Cancer Linkage Consortium (1997) Pathology of familial breast cancer: differences between breast cancers in carriers of BRCA1 or BRCA2 mutations and sporadic cases. Lancet 349:1505–1510 10.1016/S0140-6736(96)10109-4 [DOI] [PubMed] [Google Scholar]
- Chen CF, Chen PL, Zhong Q, Sharp ZD, Lee WH (1999) Expression of BRC repeats in breast cancer cells disrupts the BRCA2-Rad51 complex and leads to radiation hypersensitivity and loss of G(2)/M checkpoint control. J Biol Chem 274:32931–32935 10.1074/jbc.274.46.32931 [DOI] [PubMed] [Google Scholar]
- Cornelis RS, Neuhausen SL, Johansson O, Arason A, Kelsell D, Ponder BA, Tonin Hamann U, et al (1995) High allele loss rates at 17q12-q21 in breast and ovarian tumours from BRCA1-linked families. The Breast Cancer Linkage Consortium. Genes Chromosomes Cancer 13:203–210 [DOI] [PubMed] [Google Scholar]
- Davies AA, Masson JY, McIlwraith MJ, Stasiak AZ, Stasiak A (2001) Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Mol Cell 7:273–282 10.1016/S1097-2765(01)00175-7 [DOI] [PubMed] [Google Scholar]
- Fleming MA, Potter JD, Ramirez CJ, Ostrander GK, Ostrander EA (2003) Understanding missense mutations in the BRCA1 gene: an evolutionary approach. Proc Natl Acad Sci USA 100:1151–1156 10.1073/pnas.0237285100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank TS, Deffenbaugh AM, Reid JE, Hulick M, Ward BE, Lingenfelter B, Gumpper KL, Scholl T, Tavtigian SV, Pruss DR, Critchfield GC (2002) Clinical characteristics of individuals with germline mutations in BRCA1 and BRCA2: analysis of 10,000 individuals. J Clin Oncol 20:1480–1490 10.1200/JCO.20.6.1480 [DOI] [PubMed] [Google Scholar]
- Gowen LC, Johnson BL, Latour AM, Sulik KK, Koller BH (1996) BRCA1 deficiency results in early embryonic lethality characterized by neuroepithelial abnormalities. Nat Genet 12:191–194 10.1038/ng0296-191 [DOI] [PubMed] [Google Scholar]
- Grantham R (1974) Amino acid difference formula to help explain protein evolution. Science 185:862–864 [DOI] [PubMed] [Google Scholar]
- Hohenstein P, Kielman MF, Breukel C, Bennett LM, Wiseman R, Krimpenfort P, Cornelisse C, van Ommen GJ, Devilee P, Fodde R (2001) A targeted mouse BRCA1 mutation removing the last BRCT repeat results in apoptosis and embryonic lethality at the headfold stage. Oncogene 20:2544–2550 10.1038/sj.onc.1204363 [DOI] [PubMed] [Google Scholar]
- Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De Die-Smulders C, Persky N, Grompe M, Joenje H, Pals G, Ikeda H, Fox EA, D’Andrea AD (2002) Biallelic inactivation of BRCA2 in Fanconi anemia. Science 297:606–609 10.1126/science.1073834 [DOI] [PubMed] [Google Scholar]
- Koonin EV, Altschul SF, Bork P (1996) BRCA1 protein products…functional motifs…. Nat Genet 13:266–268 10.1038/ng0796-266 [DOI] [PubMed] [Google Scholar]
- Lakhani S, Jacquemier J, Sloane JP, Gusterson BA, Anderson TJ, van de Vijver MJ, Farid LM, et al (1998) Multifactorial analysis of differences between sporadic breast cancers and cancers involving BRCA1 and BRCA2 mutations. J Natl Cancer Inst 90:1138–1145 10.1093/jnci/90.15.1138 [DOI] [PubMed] [Google Scholar]
- Liu CY, Flesken-Nikitin A, Li S, Zeng Y, Lee WH (1996) Inactivation of the mouse BRCA1 gene leads to failure in the morphogenesis of the egg cylinder in early postimplantation development. Genes Dev 10:1835–1843 [DOI] [PubMed] [Google Scholar]
- Miller MP, Kumar S (2001) Understanding human disease mutations through the use of interspecific genetic variation. Hum Mol Genet 10:2319–2328 10.1093/hmg/10.21.2319 [DOI] [PubMed] [Google Scholar]
- Mirkovic N, Marti-Renom M, Weber B, Sali A, Monteiro A (2004) Structure-based assessment of missense mutations in human BRCA1: implications for breast and ovarian cancer predisposition. Cancer Res 64:3790–3797 [DOI] [PubMed] [Google Scholar]
- Petersen GM, Parmigiani G, Thomas D (1998) Missense mutations in disease genes: a Bayesian approach to evaluate causality. Am J Hum Genet 62:1516–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebbeck TR, Lustbader ED, Buetow KH (1994) Somatic allele loss in genetic linkage analysis of cancer. Genet Epidemiol 11:419–429 [DOI] [PubMed] [Google Scholar]
- Thompson D, Easton DF, Goldgar DE (2003) A full-likelihood method for the evaluation of causality of sequence variants from family data. Am J Hum Genet 73:652–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallon-Christersson J, Cayanan C, Haraldsson K, Loman N, Bergthorsson JT, Brondum-Nielsen K, Gerdes AM, Moller P, Kristoffersson U, Olsson H, Borg A, Monteiro AN (2001) Functional analysis of BRCA1 C-terminal missense mutations identified in breast and ovarian cancer families. Hum Mol Genet 10:353–360 10.1093/hmg/10.4.353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner JE, Tolar J, Levran O, Scholl T, Deffenbaugh A, Satagopan J, Ben-Porat L, Mah K, Batish SD, Kutler DI, MacMillan ML, Hanenberg H, Auerbach AD (2004) Germline mutations in BRCA2: shared genetic susceptibility to breast cancer, early onset leukemia and Fanconi anemia. Blood 103:3226–3229 10.1182/blood-2003-09-3138 [DOI] [PubMed] [Google Scholar]
- Wong AK, Pero R, Ormonde PA, Tavtigian SV, Bartel PL (1997) RAD51 interacts with the evolutionarily conserved BRC motifs in the human breast cancer susceptibility gene BRCA2. J Biol Chem 272:31941–31944 10.1074/jbc.272.51.31941 [DOI] [PubMed] [Google Scholar]
- Yang H, Jeffrey PD, Miller J, Kinnucan E, Sun Y, Thoma NH, Zheng N, Chen PL, Lee WH, Pavletich NP (2002) BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science 297:1837–1848 10.1126/science.297.5588.1837 [DOI] [PubMed] [Google Scholar]