

Abstract
Haplotypes—that is, linear arrangements of alleles on the same chromosome that were inherited as a unit—are expected to carry important information in the context of association fine mapping of complex diseases. In consideration of a set of tightly linked markers, there is an enormous number of different marker combinations that can be analyzed. Therefore, a severe multiple-testing problem is introduced. One method to deal with this problem is Bonferroni correction by the number of combinations that are considered. Bonferroni correction is appropriate for independent tests but will result in a loss of power in the presence of linkage disequilibrium in the region. A second method is to perform simulations. It is unfortunate that most methods of haplotype analysis already require simulations to obtain an uncorrected P value for a specific marker combination. Thus, it seems that nested simulations are necessary to obtain P values that are corrected for multiple testing, which, apparently, limits the applicability of this approach because of computer running-time restrictions. Here, an algorithm is described that avoids such nested simulations. We check the validity of our approach under two disease models for haplotype analysis of family data. The true type I error rate of our algorithm corresponds to the nominal significance level. Furthermore, we observe a strong gain in power with our method to obtain the global P value, compared with the Bonferroni procedure to calculate the global P value. The method described here has been implemented in the latest update of our program FAMHAP.
Introduction
Data on densely spaced markers within one gene or haplotype block have become a reality. Although the usefulness of haplotype analysis in such a situation is commonly accepted, there is still no consensus on how the analysis should be performed. Multiple testing is an important problem in this context. With n markers, there are 2n-1 marker combinations for which a haplotype-based test can be performed. Hence, for 20 markers, 1,000,000 tests are possible, and it is clear that hardly any P value will withstand a Bonferroni correction by that number. A common approach to reducing the number of tests is to use a sliding window—that is, to test only combinations with a fixed number of neighboring markers. However, the size of such windows has to be chosen in advance, which leads to an important loss of flexibility when haplotypes of interest are shorter than or extend over the chosen window size. Furthermore, cis-acting effects on the disease of markers that are not neighbors are always missed. Finally, correction is still needed for the number of windows and for the number of markers that are analyzed as single loci.
Here, we describe a method that allows for the analysis of all marker combinations or a set of prespecified marker combinations—for example, all marker combinations with ⩽k markers. Suppose that, for each marker combination, we have one test the underlying statistic of which depends on the haplotype distribution with respect to these markers. We consider the global null hypothesis—that none of the marker combinations shows association with the disease. It is then natural to try to assess the global significance with the statistic Tmax, which is the maximum of the statistics over all marker combinations. Since the distribution of Tmax is generally unknown, P values are obtained through Monte Carlo simulations. The Tmax approach has been used, for instance, by McIntyre et al. (2000) for the single-locus analysis of several markers by use of the transmission/disequilibrium test (TDT) by Spielman et al. (1993). In our situation, however, a maximum statistic will not suffice, since the test statistics for combinations with different numbers of markers are usually not comparable. Therefore, we replace Tmax with Pmin, the smallest raw P value found among the combinations. Since the distribution of Pmin is not known, significance has to be assessed with Monte Carlo simulations. This approach was considered, for instance, by Lazzeroni and Lange (1998) and by Jannot et al. (2003). Although the null distribution of the test statistic for a single hypothesis is known in situations described by Lazzeroni and Lange (1998) and by Jannot et al. (2003), in the context of haplotype analysis, even these null distributions are often unknown. For example, a method to perform a haplotype-based TDT-like test for samples of families with a single affected child has been given by Zhao et al. (2000) and has been extended to general nuclear families by Knapp and Becker (2003). A transmission/nontransmission table for haplotypes is constructed, in which the possible transmission patterns of each family are weighted with a relative likelihood based on estimated haplotype frequencies. The distribution of the corresponding test statistic under the null hypothesis is unknown, and the raw P value has to be obtained through permutation replicates, in which the transmission/nontransmission status of the haplotypes is randomly permuted. Therefore, a nested simulation strategy seems to be necessary to obtain the distribution of Pmin, since, in each of the permutation replicates, the raw P value for each marker combination is needed and has to be obtained through permutation replicates. This will often be computationally unfeasible.
Although the raw P value for each test has to be obtained through permutation replicates, it is possible to assess the overall significance for all marker combinations without nested simulations. Ge et al. (2003) have shown that a single set of permutations is sufficient to obtain raw P values for each marker combination and to estimate the distribution of Pmin. They applied this idea to a multiple-testing problem that occurs with microarray data. We provide an adaption of their idea, taking into account sample size, running time, and computer-storage requirements that differ between microarray and haplotype data. We check the validity and power of our approach with a simulation study under two disease models. Finally, we demonstrate its applicability by reanalyzing a recently published association study.
Methods
Association Testing Using Multiple Tightly Linked Markers
Consider n tightly linked markers, and let A={1,…,n}. Any nonempty subset B of A is called a “marker combination.” Zhao et al. (2000) proposed a TDT-like association test for the hypothesis HB0—that there is no disease association with marker combination B—which can be applied to samples consisting of nuclear families with a single affected child. Their approach was subsequently extended to samples of nuclear families with an arbitrary number of children (Knapp and Becker 2003). In brief, this test proceeds with the following steps.
-
1.
The maximum-likelihood frequency estimates for each haplotype of marker combination B are computed.
-
2.
In the case that the observed single-marker genotypes of a family allow for more than one haplotype explanation, the frequency estimates of step 1 are used to calculate weights (which sum to 1 within a family) for each of these possible haplotype explanations.
-
3.
With ambiguous families replaced by a set of weighted haplotype explanations, the test statistic of any extension of the TDT for a single-marker locus with >2 alleles can be used to calculate the value TB0 for the observed data. Here, we consider the statistic proposed by Spielman and Ewens (1996), which requires calculation of a table of transmitted/nontransmitted haplotypes. Each affected child contributes 2 units to this table. Only families with complete genotype information for all loci of marker combination B contribute to this table.
-
4. The P value PB0 is assessed by simulation. In each replicate of this simulation, a sample is constructed in which all marker genotypes of all children are either left unchanged or replaced by the two nontransmitted parental alleles, with equal chance. Since only fully genotyped families are used, this is equivalent to changing the transmission/nontransmission status of each possible haplotype explanation of a family. Let TBi denote the value of the test statistic obtained for the ith replicate. Then, PB0 is the fraction of permutation replicates resulting in a test statistic greater than or equal to the test statistic of the real data; that is,
where t denotes the number of permutation replicates and |{i:TBi⩾TB0}| denotes the number of elements of a set A.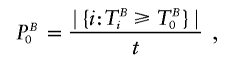
Testing More Than One Hypothesis
Now suppose that not only a single marker combination B should be tested for its association with the disease, but that a set ℬ of marker combinations should be evaluated. Let 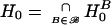 be the global null hypothesis—that none of the marker combinations B∈ℬ is associated with the disease. Our central goal is to construct a test for this global hypothesis. The preceding section described a method to obtain PB0 for each hypothesis HB0. PB0 is called “raw P value” or “unadjusted P value” for hypothesis HB0. It seems intuitively reasonable that the testing procedure for the global hypothesis H0 should depend on
be the global null hypothesis—that none of the marker combinations B∈ℬ is associated with the disease. Our central goal is to construct a test for this global hypothesis. The preceding section described a method to obtain PB0 for each hypothesis HB0. PB0 is called “raw P value” or “unadjusted P value” for hypothesis HB0. It seems intuitively reasonable that the testing procedure for the global hypothesis H0 should depend on 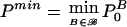 ; that is, the global null hypothesis is rejected in the case that the minimum of the raw P values is sufficiently small. Thus, it is necessary to determine the distribution of Pmin in the case that H0 holds true. Again, this can be achieved by simulation. Since, in each replication of this simulation, it is necessary to obtain the P value for each B (which requires simulations itself), nested simulations seem to be necessary. The obvious drawback of such nested simulations is running time. However, we show here how these nested simulations can be avoided.
; that is, the global null hypothesis is rejected in the case that the minimum of the raw P values is sufficiently small. Thus, it is necessary to determine the distribution of Pmin in the case that H0 holds true. Again, this can be achieved by simulation. Since, in each replication of this simulation, it is necessary to obtain the P value for each B (which requires simulations itself), nested simulations seem to be necessary. The obvious drawback of such nested simulations is running time. However, we show here how these nested simulations can be avoided.
The basic idea is to use the same set of permutation replicates to determine the empirical distribution of Pmin that has been used to determine PB0. For each hypothesis B∈ℬ and for each permutation replicate i=1,…, t, the uncorrected P value of the ith permutation replicate is calculated as
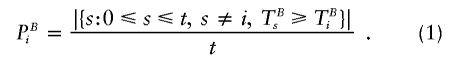 |
(Note that the real data serves as a “permutation replicate” for the calculation of PBi.) For i>0, let 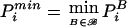 be the minimum of the uncorrected P values for all marker combinations in the ith permutation replicate. Then, the P value for the global hypothesis H0 is calculated as
be the minimum of the uncorrected P values for all marker combinations in the ith permutation replicate. Then, the P value for the global hypothesis H0 is calculated as
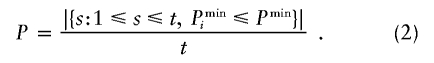 |
Table 1 provides an example that explains the strategy. (Of course, >10 permutation replicates are needed in practice.) For the real data, the smallest raw P value is obtained for marker combination B={1,2}, so that Pmin=.10. We observe a smaller Pmini for the permutation replicates i∈{2,3,4}. Thus, the P value for the global hypothesis H0 is estimated to be .30.
Table 1.
Hypothetical Example for Three Tightly Linked Markers and 10 Permutation Replicates
|
Values for Marker Combination B = |
|||||||||||||||
| {1} |
{2} |
{3} |
{1,2} |
{1,3} |
{2,3} |
{1,2,3} |
|||||||||
| Data | TBi | PBi | TBi | PBi | TBi | PBi | TBi | PBi | TBi | PBi | TBi | PBi | TBi | PBi | Pmin |
| Real data | 3.00 | .20 | 2.12 | .20 | 2.00 | .40 | 4.48 | .10 | 2.44 | .30 | 3.03 | .20 | 6.47 | .20 | .10 |
| Replicate: | |||||||||||||||
| 1 | 2.01 | .40 | .01 | 1.00 | 2.02 | .30 | 1.42 | .50 | .00 | 1.00 | 3.01 | .40 | 4.47 | .40 | .30 |
| 2 | 3.12 | .00 | 4.12 | .00 | 2.12 | .20 | 5.44 | .00 | .44 | .50 | 7.33 | .00 | 9.47 | .00 | .00 |
| 3 | 2.99 | .30 | 3.12 | .10 | 3.22 | .00 | 1.44 | .30 | 4.44 | .10 | 3.02 | .30 | 8.47 | .10 | .00 |
| 4 | 3.02 | .10 | 1.02 | .50 | 2.22 | .10 | 1.43 | .40 | 5.44 | .00 | 6.33 | .10 | 5.47 | .30 | .00 |
| 5 | 1.44 | .50 | 1.12 | .30 | 1.99 | .50 | 1.41 | .60 | .03 | .90 | 3.00 | .50 | .02 | 1.00 | .30 |
| 6 | .99 | .60 | 1.11 | .40 | .74 | 1.00 | 1.40 | .70 | .04 | .80 | 2.99 | .60 | .37 | .90 | .40 |
| 7 | .78 | .70 | .12 | .60 | .75 | .90 | 4.44 | .20 | .34 | .60 | 2.87 | .70 | .47 | .80 | .20 |
| 8 | .13 | 1.00 | .11 | .70 | 1.00 | .80 | 1.34 | .80 | 1.44 | .40 | 2.84 | .80 | 1.47 | .70 | .40 |
| 9 | .55 | .80 | .02 | .90 | 1.88 | .60 | .44 | 1.00 | 3.44 | .20 | 1.33 | .90 | 2.47 | .60 | .20 |
| 10 | .42 | .90 | .10 | .80 | 1.07 | .70 | 1.24 | .90 | .24 | .70 | 1.04 | 1.00 | 3.47 | .50 | .50 |
The idea to calculate the raw P values (PB0) and the empirical distribution of Pmin from a single set of permutation replicates is the basis of a method, recently proposed by Ge et al. (2003), for obtaining so-called minP-adjusted P values (Westfall and Young 1993). Therefore, the following section summarizes the algorithm of Ge et al. (2003), and the subsequent section describes the details and modifications of our adaption of this basic idea for testing the global null hypothesis.
Algorithm for Step-Down minP-Adjusted P Values, by Ge et al. (2003)
First, it should be noted that the goal of Ge et al. (2003) is slightly different from our goal to construct a test for the global null hypothesis H0. Their algorithm aims to obtain so-called minP-adjusted P values 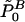 for each hypothesis HB0. If the global null hypothesis H0 holds true, the probability is ⩽α that one or more of these adjusted P values is ⩽α. Therefore, it is evident that the rule “reject H0 in the case that the smallest of the adjusted P values is ⩽α” defines a level-α test for H0. Since the smallest adjusted P value belongs to the same hypothesis for which the unadjusted P value is smallest, for the purpose of testing H0, it would be sufficient to calculate only the adjusted P value for the hypothesis that possesses the smallest unadjusted P value, instead of calculating all adjusted P values
for each hypothesis HB0. If the global null hypothesis H0 holds true, the probability is ⩽α that one or more of these adjusted P values is ⩽α. Therefore, it is evident that the rule “reject H0 in the case that the smallest of the adjusted P values is ⩽α” defines a level-α test for H0. Since the smallest adjusted P value belongs to the same hypothesis for which the unadjusted P value is smallest, for the purpose of testing H0, it would be sufficient to calculate only the adjusted P value for the hypothesis that possesses the smallest unadjusted P value, instead of calculating all adjusted P values 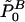 , B∈ℬ. The initial step of the algorithm of Ge et al. (2003, cf. step 0 in box 4 on p. 21) requires obtaining raw P values for each hypothesis. However, this requirement is a direct consequence of their goal to obtain
, B∈ℬ. The initial step of the algorithm of Ge et al. (2003, cf. step 0 in box 4 on p. 21) requires obtaining raw P values for each hypothesis. However, this requirement is a direct consequence of their goal to obtain 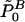 for all B∈ℬ, and it can be dropped in the case that only the adjusted P value for the hypothesis with the smallest unadjusted P value has to be calculated.
for all B∈ℬ, and it can be dropped in the case that only the adjusted P value for the hypothesis with the smallest unadjusted P value has to be calculated.
Second, a minor technical difference between the algorithm of Ge et al. (2003) and our approach is that these authors use
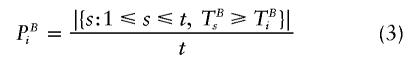 |
instead of (1) for calculation of uncorrected P values of the ith permutation replicate. Recently, there has been debate on how to estimate P values by Monte Carlo methods. North et al. (2002, 2003) prefer the estimate (r+1)/(n+1), where n is the number of permutation replicates and r is the number of these replicates that produce a test statistic greater than or equal to that calculated for the actual data. Broman and Caffo (2003) and Ewens (2003) favor the traditional estimate r/n. Now, Ge et al. (2003) employ this traditional estimate in obtaining the raw P values PB0, whereas their application of equation (3) in obtaining raw P values of the ith permutation replicate makes use of the proposal by North et al. (2002, 2003). We agree with Broman and Caffo (2003) that neither r/n nor (n+1)/(r+1) should be considered incorrect, but we believe that it is reasonable to use the same approach for both purposes—that is, calculation of raw P values for the real data and for the permutation replicates. Therefore, we employ equation (1) instead of equation (3). Note that, in the case that all possible permutation replicates are evaluated in obtaining PBi, equation (3) is appropriate. Indeed, when all replicates can be conducted, there is one replicate that is identical to the real data. Hence, it is reasonable to compare a replicate to itself, since the real data is compared to itself as well. In contrast to the microarray problem considered by Ge et al. (2003), the set of all permutation replicates is always much too large for our problem of haplotype-association testing.
The main merit of the algorithm by Ge et al. (2003) is that it does not require storage of the whole table TBi(i=1,…, t; B∈ℬ) of test statistics per hypothesis B and permutation replicate i, but only requires storage of the test statistics for a single hypothesis at a time. The algorithm is presented in the context of microarray data, for which the number of hypotheses (i.e., the number of genes for which expression is measured) is usually large compared with the number of individuals considered (typically < 20). For haplotype data, the number of marker combinations tested is low for <10 SNPs and can reasonably be limited by consideration of only marker combinations with <4 or 5 markers when more SNPs are typed. Here, a computer memory problem occurs when many permutation replications have to be conducted (e.g., in the case that very small global P values have to be estimated), whereas, for microarray data, even the number of all possible permutations of the real data (i.e., permutations of the disease status of the individuals) is small, because the total number of individuals is very small. Therefore, we have implemented an approach to solve the computer memory problem that is more appropriate for haplotype data.
Algorithm for Testing the Global Null Hypothesis H0
The algorithm for testing the global null hypothesis—that none of the marker combinations B∈ℬ is associated with the disease—has been implemented in the program FAMHAP (Becker and Knapp 2004), which originally was developed to obtain maximum-likelihood estimates of haplotype frequencies from samples consisting of arbitrary nuclear families. Two options are provided for describing the set ℬ of marker combinations B: (1) with option ”maxmarker = k,” only marker combinations B consisting of ⩽k (k⩽n) loci are considered; (2) with option “window = yes,” only marker combinations of neighboring markers are considered. With a defined ℬ, the program proceeds with the following steps.
-
1.
Estimate frequencies for the full n-locus haplotypes from the data. For each marker combination B∈ℬ, obtain frequencies for each haplotype of B by summing all corresponding n-locus haplotype frequencies. Calculate and store the table of transmitted/nontransmitted haplotypes (cf. step 3 in association test) for each B∈ℬ and for each family with complete marker genotypes at all marker loci of B.
-
2.
For all B, compute the test statistic TB0.
-
3.
For each family, randomly transpose (cf. step 4 in association test) the table of transmitted/nontransmitted haplotypes (cf. step 1) for all B∈ℬ. Recompute and store TBi for these modified data.
-
4.
Repeat step 3 t times.
-
5.
Calculate PBi by use of equations (1) and
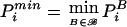 .
. -
6.
Calculate the P value for the global hypothesis H0 by use of equation (2).
The memory requirement to store TBi for each permutation replicate i=1,…,t and for each marker combination B∈ℬ is of the order ∣ℬ∣× t. With 1,000 marker combinations and 100,000 permutation replicates, 108 test statistics have to be stored, and it is obvious that the number of permutation replicates cannot be increased much further. However, a huge number of permutation replicates is needed only for estimating small global P values or for performing the test of the global null hypothesis at small type I error rate (North et al. 2002). For this, the program provides the option “alpha = <value>,” which enforces the condition that only the <value> × t permutation replicates with the highest test statistics are stored for each marker combination. This information is sufficient to decide if the P value corresponding to the global null hypothesis is ⩽value. Indeed, PB⋆i>α in the case that TB⋆i does not belong to the <value> × t permutation replicates with the highest test statistic for marker combination B⋆.It follows that 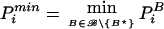 (i.e., the concrete value of PB⋆i is irrelevant for calculating Pmini), or Pmini>α (i.e., even the concrete value of Pmini is irrelevant for deciding whether the global P value is ⩽α). With option “alpha = <value>,” the storage requirement is reduced to <value> × ∣ℬ∣ × t. Typically, <value> × ∣ℬ∣ is <1 in situations in which the number of permutation replicates is too large to allow storage of the whole table of test statistics.
(i.e., the concrete value of PB⋆i is irrelevant for calculating Pmini), or Pmini>α (i.e., even the concrete value of Pmini is irrelevant for deciding whether the global P value is ⩽α). With option “alpha = <value>,” the storage requirement is reduced to <value> × ∣ℬ∣ × t. Typically, <value> × ∣ℬ∣ is <1 in situations in which the number of permutation replicates is too large to allow storage of the whole table of test statistics.
Because of the discreteness of the distribution of Pmin, generally some of the permutation replicates will give Pmini=Pmin. In accordance with equation (2), all of these permutation replicates have to be counted for determination of the global P value for H0. As an example, suppose that, in table 1, permutation replicate 2 was the real data and the real data were one of the permutation replicates, with Pmin=.0. Since Pmini=.0 for permutation replicates 3 and 4 as well, the global P value is .2, as calculated by equation (2). Now, it is reasonable to enforce an order for data sets with identical smallest raw P value by consideration of the second smallest raw P value, which will be denoted Pmin2 for the real data and Pmin2i for permutation replicate i. Then, the P value for the global null hypothesis can be calculated by use of
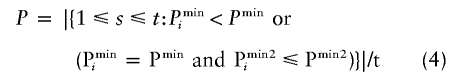 |
instead of equation (2). For the example of table 1 (and under the assumption that permutation replicate 2 was the real data), raw P values are .0 for two further marker combinations (i.e., Pmin2=.0), whereas the second smallest raw P values for permutation replicates 3 and 4 are .1. Calculated by use of equation (4), the global P is .0 instead of .2. Of course, it is possible that the second smallest P values of two permutation replicates are identical as well and that it would be necessary to consider the third smallest P values, and so on. However, the number of such instances (which is printed out by our program) will be small in practice, and our implementation takes into account only the improvement that can be obtained from the consideration of the second smallest raw P value.
Note that there is a potential conflict in using equation (4) instead of equation (2) with the option “alpha = <value>.” If the second smallest raw P value for the real data is > value, this value will not have been stored, and the global P value has to be determined by use of equation (2). However, if Pmin2⩽ value, the improved determination of the global P value can be applied even in combination with option “alpha = <value>.” In addition, the discreteness of the distribution of Pmin is most pronounced in the case that the number of permutation replicates is relatively small, whereas, for a large number of permutation replicates, the difference between equation (2) and equation (4) will be less important. On the other hand, the option “alpha = <value>” is not required if the number of permutation replicates is small.
Simulations
We performed a simulation study for samples of 233 trios and samples of 175 nuclear families with two affected children. We tried to model the situation of many markers within a small region with just five SNP markers. We simulated three core SNPs, which can be viewed as tagging SNPs of a haplotype block, and one SNP on both sides of the block, each of which was only in moderate linkage disequilibrium (LD) with the core block. These SNPs model the borders of the analyzed region. The core region of the three SNPs comprised five haplotypes. We simulated a diallelic disease locus under a dominant and a recessive model, with a prespecified relative risk of 2 and an attributable fraction of 0.20. We assumed a recombination fraction of zero between the disease locus and the core block. The first of the five core haplotypes had the same frequency as the disease allele and was assumed to be in in complete LD with the disease allele. The other haplotypes at the core block were chosen from a uniform distribution. We additionally simulated a neighboring SNP on the left and on the right of the core block, depending on the core haplotype. The extent of the LD between each core haplotype and the neighboring markers can be found in table 2. Under all scenarios, we used 3,000 simulated data sets and computed the global P value with 4,000 permutation replicates for each simulated data set. We also checked the size of our test by simulations under the null hypothesis. For this purpose, we enforced linkage equilibrium between the disease locus and the markers. Empirical significance levels and power were computed as the portion of simulated data sets for which the global P value or the Bonferroni-corrected smallest raw P value, respectively, was ⩽.05.
Table 2.
Haplotype Distribution
|
LD between Core Haplotypeand Neighboring SNP |
||
| Core Haplotype | Lefta | Rightb |
| 111 | .30 | .30 |
| 112 | .30 | .30 |
| 121 | .30 | .30 |
| 211 | .60 | .60 |
| 222 | .65 | .65 |
Probability that the SNP to the left of the core block carries allele 1, given the core haplotype.
Probability that the SNP to the right of the core block carries allele 1, given the core haplotype.
Results
We have integrated our method into our program FAMHAP, which was developed for haplotype frequency estimation in nuclear families. In particular, haplotype frequencies and lists of haplotype explanations that are necessary for the computation of the underlying test statistic can be computed internally. Thus, time-consuming communication between different software packages can be avoided. The simulation study was performed on a Pentium III PC with 512 megabytes of main memory. The evaluation of a single data set with 4,000 permutation replicates took 10 s, on average, to obtain the raw P values for all 31 marker combinations and the corresponding global P value. A total of 3,000 simulated data sets had to be evaluated for each of 56 different situations, since we conducted simulations under the null hypothesis and the alternative, under two disease models, for two different data structures and for seven different configurations of “maxmarker” and “window.” In total, our simulation study ran for ∼56×3,000×10s, that is, ∼19.5 d. With nested simulations, 4,000×4,000 permutation replicates would have been necessary to obtain the global P for a single simulated data set. Thus, our simulation study with nested simulations would have taken 4,000×19.5 d, which is ∼214 years and would not have been feasible.
In table 3, the empirical significance levels for the different data structures and disease models are shown. (The disease model is relevant only for the haplotype frequencies.) In addition, we varied the number of marker combinations tested, by allowing only marker combinations with ⩽k = 1, 2, 3, and 5 markers (“maxmarker = k”), or by allowing only combinations of neighboring markers (“window = yes”). Under all models and marker combinations, the true type I error rate for the test that rejects H0 in the case that the smallest P value is ⩽.05 is markedly above .05. As expected, this effect becomes stronger as the number of tested combinations increases. These high true type I error rates show that even with a considerable amount of LD, multiple testing is still an important problem. On the other hand, Bonferroni correction leads to true type I error rates that are too small, because it ignores the LD of the region. Indeed, when many marker combinations are tested, the true level for the Bonferroni correction is ∼0.02 rather than 0.05. With our global P values, however, under none of the scenarios did we observe a significant deviation from the nominal level α=0.05. Thus, our method is a valid testing procedure that adequately accounts both for the number of tests and for the LD of a region. The nominal level is also maintained with samples consisting of nuclear families with two children, which confirms that we obtain a valid test for association from this family structure. Table 4 shows the power of testing for the uncorrected P values of the marker combinations of the core region, whereas table 5 shows the results of the power studies with Bonferroni correction and our global P values. In accordance with the simulation setting, the power of the uncorrected tests in table 4 is smallest for the single markers, increases when 2-marker haplotypes are considered, and is best for the 3-marker combination, irrespective of the data structure or disease model. Table 5 shows that the power improvement of the haplotype analysis is maintained even after the multiple testing is taken into account. In general, the power under our global P values is considerably higher than under Bonferroni-corrected P values and lies about halfway between the power obtained by Bonferroni-corrected and uncorrected P values of the best combination, which represents the upper limit of what can be reached (for instance, if one knew the best combination in advance from an independent source). Both for Bonferroni-corrected and for global P values, the power increases with the number of considered markers and reaches its optimum when only combinations of ⩽3 neighboring markers are considered (“maxmarker = 3”; “window = yes”). This is consistent with our simulation setup, which modeled a 3-marker disease haplotype of neighboring markers. However, whereas the power of the Bonferroni correction drops when too many combinations are tested (“maxmarker = 5”; “window = no”), tests of too many combinations have only a slight impact on the power when our global P values are computed. This shows that our method is able to capture the dependence of the tests and that it is not negatively affected by the consideration of effects of a high order or cis-acting effects if there are no such effects. Besides the comparison of the Bonferroni-corrected and our global P values, we consistently observe a higher power for the nuclear families with two affected children than for the trio sample, although the sample size for both data structures is equal with respect to genotyping effort. The better power of data from nuclear families with two affected children may partially be explained by the fact that families with multiple affected siblings are genetically more loaded and therefore lead to an increased power (Risch and Teng 1998; Fingerlin et al. 2004).
Table 3.
Empirical Significance Level at α=0.05
|
Empirical Significance Level under Model |
|||||||
| Recessive |
Dominant |
||||||
| Data Structure,Maxmarker,aand Windowb | No. of Combinations | Bestc | Bonferronid | Global | Bestc | Bonferronid | Global |
| Trios: | |||||||
| 1: | |||||||
| … | 5 | .192 | .039 | .051 | .193 | .024 | .051 |
| 2: | |||||||
| Yes | 9 | .240 | .031 | .049 | .239 | .032 | .054 |
| No | 15 | .272 | .026 | .046 | .279 | .024 | .052 |
| 3: | |||||||
| Yes | 12 | .249 | .025 | .052 | .252 | .024 | .051 |
| No | 25 | .301 | .023 | .047 | .322 | .020 | .049 |
| 5: | |||||||
| Yes | 15 | .260 | .024 | .049 | .268 | .017 | .050 |
| No | 31 | .322 | .022 | .047 | .331 | .017 | .053 |
| Nuclear families: | |||||||
| 1: | |||||||
| … | 5 | .161 | .034 | .046 | .173 | .038 | .048 |
| 2: | |||||||
| Yes | 9 | .212 | .027 | .047 | .217 | .032 | .048 |
| No | 15 | .270 | .025 | .048 | .272 | .025 | .048 |
| 3: | |||||||
| Yes | 12 | .239 | .023 | .044 | .240 | .027 | .051 |
| No | 25 | .325 | .022 | .047 | .306 | .021 | .049 |
| 5: | |||||||
| Yes | 15 | .258 | .022 | .046 | .261 | .024 | .049 |
| No | 31 | .326 | .019 | .047 | .319 | .018 | .050 |
Maximum number of markers in combination B.
When “window = yes,” only combinations of neighboring markers are tested.
Uncorrected smallest P value.
Bonferroni-corrected smallest P value.
Table 4.
Empirical Power of Testing a Single Marker Combination (α=0.05)
|
Power under Model |
||
| Data Structure andSNP Markers Tested | Recessive | Dominant |
| Trios: | ||
| 2 | .399 | .213 |
| 3 | .407 | .211 |
| 4 | .420 | .225 |
| 2,3 | .571 | .350 |
| 2,4 | .569 | .355 |
| 3,4 | .586 | .341 |
| 2,3,4 | .735 | .719 |
| Nuclear families: | ||
| 2 | .618 | .385 |
| 3 | .609 | .386 |
| 4 | .612 | .378 |
| 2,3 | .818 | .635 |
| 2,4 | .825 | .622 |
| 3,4 | .805 | .640 |
| 2,3,4 | .934 | .942 |
Table 5.
Empirical Power of Testing a Set of Marker Combinations (α=0.05) for the Global Hypothesis
|
Empirical Power under Model |
|||||
| Recessive |
Dominant |
||||
| Data Structure,Maxmarker,and Window | No. of Combinations | Bonferronia | Global | Bonferronia | Global |
| Trios: | |||||
| 1: | |||||
| … | 5 | .409 | .441 | .230 | .250 |
| 2: | |||||
| Yes | 9 | .469 | .556 | .277 | .360 |
| No | 15 | .432 | .553 | .264 | .359 |
| 3: | |||||
| Yes | 12 | .526 | .601 | .436 | .524 |
| No | 25 | .439 | .580 | .339 | .479 |
| 5: | |||||
| Yes | 25 | .499 | .599 | .422 | .521 |
| No | 31 | .437 | .582 | .352 | .499 |
| Nuclear Families | |||||
| 1: | |||||
| … | 5 | .664 | .729 | .429 | .497 |
| 2: | |||||
| Yes | 9 | .753 | .819 | .556 | .640 |
| No | 15 | .743 | .823 | .559 | .666 |
| 3: | |||||
| Yes | 12 | .799 | .862 | .778 | .837 |
| No | 25 | .747 | .843 | .721 | .808 |
| 5: | |||||
| Yes | 25 | .779 | .854 | .773 | .840 |
| No | 31 | .728 | .835 | .714 | .810 |
The power of the procedure, which is based on Bonferroni-corrected smallest raw P value.
Application to a Real Data Set
After an initial finding by Straub et al. (2002), Schwab et al. (2003) have recently reported supportive evidence for association of schizophrenia with multilocus haplotypes in the 6p22.3 gene, dysbindin. They typed markers rs3213207 (SNP 1), rs1011313 (SNP 2), rs2619528 (SNP 3), rs760761 (SNP 4), rs2619522 (SNP 5), and rs1018381 (SNP 6) in a sib-pair sample (78 families) and in an independently ascertained trio sample (125 families). Schwab et al. (2003) reported the results of association analyses performed with the methods of Zhao et al. (2000) and Knapp and Becker (2003) for all marker combinations of ⩽5 markers and for the sib-pair sample, the trio sample, and the combined sample. The greatest evidence for association was found in the combined sample (without six Israeli families) for the combination of SNPs 2 and 4. However, with the software that was used at that time, it was not possible to conduct >200,000 permutation replicates. None of these replicates resulted in a test statistic greater than or equal to the statistic of the real data for this marker combination. Schwab et al. (2003) concluded that the raw P{2,4}0 was almost certainly smaller than 2×10-5 for this 2-marker combination and applied a Bonferroni correction. With our new implementation, we were able to conduct 108 permutation replicates, and we obtained a raw P value of P{2,4}0=2×10-6 for this marker combination. For very small P, the coefficient of variation (i.e., SD divided by the mean) of a raw P value estimated by t permutation replicates is 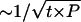 . Therefore, t=108 permutation replicates are adequate to ensure that the coefficient of variation of the raw P value is ∼10% in the case that the true raw P value for a specific marker combination is ∼10−6. Our program (with option “alpha = 0.001”) required 52 h (with a Pentium III PC) to obtain the results summarized in table 6.
. Therefore, t=108 permutation replicates are adequate to ensure that the coefficient of variation of the raw P value is ∼10% in the case that the true raw P value for a specific marker combination is ∼10−6. Our program (with option “alpha = 0.001”) required 52 h (with a Pentium III PC) to obtain the results summarized in table 6.
Table 6.
P Values Based on 108 Permutation Replicates for the Dysbindin Combined-Family Sample (without Israeli Families)
|
P |
|||
| Maxmarker | No. of Combinations | Bonferronia | Global |
| 1 | 6 | .000324 | .000212 |
| 2 | 21 | .000044 | .000023 |
| 3 | 41 | .000086 | .000030 |
| 4 | 56 | .000118 | .000032 |
| 5 | 62 | .000130 | .000033 |
| 6 | 63 | .000132 | .000033 |
Bonferroni-corrected smallest raw P value.
Note that the full table of test statistics for all marker combinations and all permutation replicates consists of 63×108 entries, which would require 24 gigabytes of memory. The algorithm of Ge et al. (2003) reduces this requirement to 380 megabytes by storing only test statistics for the permutation replicates of a single marker combination at a time. The disadvantage of their approach for this example is related to running time. Ge et al. (2003) discussed two ways to guarantee that the same ordered set of permutation replicates is used for each marker combination. The first way is to reset the random number generator at the same fixed value for each hypothesis. Since the dysbindin combined sample consists of 203 families and each permutation replicate requires permutation of transmitted/nontransmitted haplotypes in each family, 203×108 calls of the random number generator are necessary for generating 108 permutation replicates. The time required for these calls is ∼1.5 h with our hardware. In the case that these calls have to be repeated for each marker combination, the additional running time is 62×1.5 h=93 h. The second way discussed by Ge et al. (2003) is to recode and store each permutation. However, 203 bits (∼26 bytes) of storage are required to store a single permutation, resulting in a requirement of 2.4 gigabytes to store all 108 permutations. In addition, we expect that the coding and recoding of permutations would substantially increase the running time.
The results presented in table 6 for the dysbindin data suggest an advantage of haplotype analysis as compared with single-marker analysis, even after correction for the increased number of tests, since the global P value for “maxmarker = 1” is higher by a factor >6 than the global P value obtained by consideration of all marker combinations (i.e., “maxmarker = 6”). Also, in accordance with the results of our simulation study, it can be seen from table 6 that the global P value does not increase much when too many marker combinations are tested. Since the smallest raw P value corresponds to B={2,4}, the option “maxmarker = 2” results in the smallest global P value. However, consideration of all marker combinations increases the global P value by a factor of only <1.5, although the number of single hypotheses increases by a factor of 3. Comparison of the P value obtained by Bonferroni correction with the global P value obtained by our method reveals that the ratio of these values increases from ∼1.5 (for the set of single marker combinations) up to 4 (for the set of all marker combinations).
Discussion
We implemented a method that adequately accounts for the multiple-testing problem that occurs in the context of haplotype analysis. With the help of Ge et al.'s (2003) idea to reduce a nested permutation algorithm to a single permutation algorithm, computer running time is reduced drastically, and a power study becomes feasible. Our implementation is optimized for the application to haplotype analysis. Computer storage can be reduced by consideration, for each marker combination, of only the replicates with a raw P value ⩽α. With this feature, the analysis of the dysbindin data could be performed with the necessary number of permutation replicates. Furthermore, we obtained a considerable gain in speed. Since it was not necessary to proceed with one hypothesis at a time, we did not have to do repeated callsof the random number generator for each marker combination.
In the simulation study, we have shown that our method adequately accounts both for the LD of a set of tightly linked markers and for the induced multiple-testing problem. In contrast to Bonferroni correction of the smallest P value, our method avoids being overly conservative. Consistently, power increases substantially when global P values are considered instead of Bonferroni-corrected P values. Our method allows for the simultaneous consideration of different marker combinations and comprises, in particular, the analysis of single markers. In this way, our method can be a step toward a unified strategy to judge the significance of the association of a phenotype with a genomic region as a whole. The need for this is highlighted by the strongly increased true significance levels for uncorrected P values shown in table 3.
However, some flexibility remains with respect to the set of marker combinations that will be considered. With few markers, it is possible to consider all combinations, and our simulations show that, in general, not much power is lost, even when the marker combination with the strongest association does not consist of all markers. On the other hand, we can hope to detect high-order interactions or cis-acting effects if they are present. For a densely spaced marker system of 20–50 markers, it is computationally impossible to consider all combinations, even with our approach. In such a situation, it is useful to consider only marker combinations with ⩽3 markers but also to allow combinations of markers that are not neighbors. In this way, the number of tests is restricted to a level that is computationally feasible, but all single-, two-, and three-locus haplotype effects can be detected, including cis-acting effects and haplotype effects that are blurred by markers that arose in the region after the mutation event(s).
Our simulations for family data show that haplotype analysis can be more powerful than single-locus analysis when the background LD is taken into account adequately, as by our method, and also after correction for the multiple testing. Studies for case-control data under a coalescent model with a simple Bonferroni correction did not detect a gain in terms of power for haplotype analysis (Kaplan and Morris 2001).
Of course, our simulation study covers only a limited range of possible disease scenarios, but we do not think that it favors haplotype analysis inadequately. On the contrary, we modeled only a single disease haplotype, but the benefit of haplotype analysis versus single-locus analysis is stronger with multiple disease alleles (Morris and Kaplan 2002). Thus, on average, the power gain is likely to be even stronger than the gain found in our study. In addition, we modeled markers that were in moderate LD, and we would expect our method to perform even better under strong LD, both in comparison with single-marker analysis and in comparison with Bonferroni-corrected P values.
Besides these issues, our simulation study suggests that haplotype analysis is particularly favorable for nuclear families with multiple affected siblings. At least as far as single-marker analysis is concerned, case-control data has higher power than TDT analysis of trio data, under many disease scenarios, since the latter pays a price for being robust against population stratification. However, we observed that nuclear families with two affected children were much more powerful than trios. Hence, a comparison of nuclear families with multiple affected siblings and case-control data, on the basis of an equal genotyping effort, would be highly interesting. In general, nuclear families allow for very precise haplotype reconstruction (Becker and Knapp 2002; Schaid 2002), such that the haplotypic information can be fully exploited. Furthermore, as mentioned above, families with multiple affected siblings are genetically more loaded and therefore lead to an increased power (Risch and Teng 1998; Fingerlin et al. 2004). Thus, the comparison can be refined to a comparison with case-control data in which the cases are index cases from affected siblings. We are currently working on an implementation for case-control data, which will yield the possibility to compare the performance of case-control and nuclear-family data. (Note that, for case-control data also, the underlying distribution is often unknown, since asymptotic theory is difficult to apply when the number of different haplotypes is high.) Besides that, the idea of Ge et al. (2003) is quite general and can be applied to obtain global P values for different kinds of test statistics, including tests for specific genetic models and models of interaction. For family data, the principle has been implemented in the latest update of our program FAMHAP (Becker and Knapp 2004).
Acknowledgments
We are very grateful to Professors Sibylle G. Schwab and Dieter B. Wildenauer for providing the dysbindin family data. Our work was supported by grant Kn 378/1 (Project D1 of FOR 423) from the Deutsche Forschungsgemeinschaft.
Electronic-Database Information
The URL for data presented herein is as follows:
- FAMHAP, http://www.uni-bonn.de/~umt70e/becker.html (for haplotype frequency estimation)
References
- Becker T, Knapp M (2002) Efficiency of haplotype frequency estimation when nuclear family information is included. Hum Hered 54:45–53 10.1159/000066692 [DOI] [PubMed] [Google Scholar]
- ——— (2004) Maximum-likelihood estimation of haplotype frequencies in nuclear families. Genet Epidemiol 27:21–32 10.1002/gepi.10323 [DOI] [PubMed] [Google Scholar]
- Broman KW, Caffo BS (2003) Simulation-based P values: response to North et al. Am J Hum Genet 72:496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewens WJ (2003) On estimating P values by Monte Carlo methods. Am J Hum Genet 72:496–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fingerlin TE, Boehnke M, Abecasis GR (2004) Increasing the power and efficiency of disease-marker case-control association studies through use of allele-sharing information. Am J Hum Genet 74:432–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Dudoit S, Speed TP (2003) Resampling-based multiple testing for microarray data analysis. Test 12:1–77 [Google Scholar]
- Jannot AS, Essioux L, Reese MG, Clerget-Darpoux F (2003) Improved use of SNP information to detect the role of genes. Genet Epidemiol 25:158–167 10.1002/gepi.10256 [DOI] [PubMed] [Google Scholar]
- Kaplan N, Morris R (2001) Issues concerning association studies for fine mapping a suspectibility gene for a complex disease. Genet Epidemiol 20:432–457 10.1002/gepi.1012 [DOI] [PubMed] [Google Scholar]
- Knapp M, Becker T (2003) Family-based association analysis with tightly linked markers. Hum Hered 56:2–9 10.1159/000073727 [DOI] [PubMed] [Google Scholar]
- Lazzeroni LC, Lange K (1998) A conditional interference framework for extending the transmission/disequilibrium test. Hum Hered 48:67–81 10.1159/000022784 [DOI] [PubMed] [Google Scholar]
- McIntyre LM, Martin ER, Simonsen KL, Kaplan NL (2000) Circumventing multiple testing: a multilocus Monte Carlo approach for association. Genet Epidemiol 19:18–29 [DOI] [PubMed] [Google Scholar]
- Morris R, Kaplan N (2002) On the advantage of haplotype analysis in the presence of multiple disease susceptibility alleles. Genet Epidemiol 23:221–233 10.1002/gepi.10200 [DOI] [PubMed] [Google Scholar]
- North BV, Curtis D, Sham PC (2002) A note on the calculation of empirical P values from Monte Carlo procedures. Am J Hum Genet 71:439–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— (2003) A note on the calculation of empirical P values from Monte Carlo procedures. Am J Hum Genet 72:498–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risch N, Teng J (1998) The relative power of family-based and case-control designs for linkage disequilibrium studies of complex human diseases. I. DNA pooling. Genome Res 8:1273–1288 [DOI] [PubMed] [Google Scholar]
- Schaid DJ (2002) Relative efficiency of ambiguous vs. directly measured haplotype frequencies. Genet Epidemiol 23:426–443 10.1002/gepi.10184 [DOI] [PubMed] [Google Scholar]
- Schwab SG, Knapp M, Mondabon S, Hallmayer J, Borrmann-Hassenbach M, Albus M, Lerer B, Rietschel M, Trixler M, Maier W, Wildenauer DB (2003) Support for association of schizophrenia with genetic variation in the 6p22.3 gene, dysbindin, in sib-pair families with linkage and in an additonal sample of triad families. Am J Hum Genet 72:185–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielman RS, Ewens WJ (1996) The TDT and other family-based tests for linkage disequilibrium and association. Am J Hum Genet 59:983–989 [PMC free article] [PubMed] [Google Scholar]
- Spielman RS, McGinnis RE, Ewens WJ (1993) Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am J Hum Genet 52:506–516 [PMC free article] [PubMed] [Google Scholar]
- Straub RE, Jiang Y, MacLean CJ, Ma Y, Webb BT, Myakishev MV, Harris-Kerr C, Wormley B, Sadek H, Kadambi B, Cesare AJ, Gibberman A, Wang X, O’Neill FA, Walsh D, Kendler KS (2002) Genetic variation in the 6p22.3 gene, DTNBP1, the human ortholog of the mouse dysbindin gene, is associated with schizophrenia. Am J Hum Genet 71:337–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westfall PH, Young SS (1993) Resampling-based multiple testing: examples and methods for P-value adjustment. John Wiley and Sons, New York [Google Scholar]
- Zhao H, Zhang S, Merikangas KR, Trixler M, Wildenauer DB, Sun F, Kidd KK (2000) Transmission/disequilibrium tests using multiple tightly linked markers. Am J Hum Genet 67:936–946 [DOI] [PMC free article] [PubMed] [Google Scholar]