

To the Editor:
We would like to comment on the Schork and Greenwood (2004) article dealing with the inherent “bias” toward the null hypothesis in the context of nonparametric linkage analysis. The authors point out that, in certain situations, a loss of evidence for linkage can result from the practice of assigning expected allele-sharing values to affected relative pairs that are uninformative for their identity-by-descent (IBD) status. They explained this by setting up a likelihood function and studying its properties by simulation, clearly illustrating the negative impact of using expected IBD values for uninformative pairs. However, we would like to point out that their likelihood does not reflect how the majority of nonparametric linkage analysis programs compute statistics in practice. Indeed, the “problem” has been known and well discussed for years. Some of the concerns we discuss here have also been raised by Cordell (2004).
Schork and Greenwood (2004) set up the likelihood formulation as follows. Let ni be the number of sib pairs sharing i alleles IBD (i=0, 1, or 2). If all families had unambiguous IBD sharing, then the LOD score evaluated at the sharing vector (p0, p1, p2) is calculated as
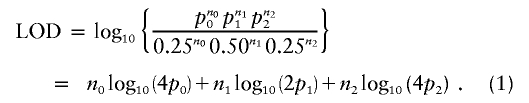 |
In their model, Schork and Greenwood (2004) said that fully uninformative sibling pairs contribute 0.25, 0.50, and 0.25, respectively, to the counts n0, n1, and n2 used in equation (1). If so, then the presence of uninformative sib pairs can lower the LOD score. However, in most software implementations, expected allele-sharing values are not used to compute nonparametric LOD scores. For example, consider the maximum LOD score (MLS) statistic proposed by Risch (1990). Let wi be the probability of the observed marker phenotypes of the pair, given that they share i alleles IBD (i=0, 1, or 2). Then, the likelihood of the observed marker data for the pair is given by
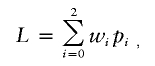 |
where pi is the posterior probability that the pair shares i alleles IBD, given that both members of the pair are affected. Suppose, in addition, that we know that n2,1 pairs share either 2 or 1 alleles, n2,0 pairs share either 2 or 0 alleles, n1,0 pairs share either 1 or 0 alleles, and nun is the number of pairs that are fully uninformative. According to Risch (1990), the LOD score can be written as
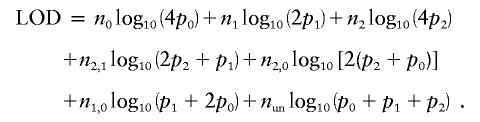 |
Maximizing this likelihood gives consistent and asymptotically unbiased estimates of the IBD-sharing probabilities. Cordell (2004) confirms this by simulation.
To verify that most implementations of nonparametric linkage statistics are not altered by uninformative families, we used FastSLINK (Ott 1989; Weeks et al. 1990; Cottingham et al. 1993) to simulate 200 fully genotyped affected–sib-pair families under disease model 1 of Schork and Greenwood (2004). The disease locus was completely linked to a two-allele marker with equally frequent alleles. We then used a variety of programs to compute linkage statistics on two data sets: (1) all 200 families and (2) the 147 families that remained after removal of the fully uninformative families. As shown in table 1, the majority of the linkage statistics, as implemented in widely used software, are exactly the same for the two data sets.
Table 1.
Comparison of Linkage Statistics Analyses Using All 200 Families and Using Only the 147 Informative Families
|
Result for |
|||
| Statistic and Software | All 200 Families | 147 Informative Families | Reference |
| Mean test Z value: | |||
| SIBPAL | 14.07 | 17.56 | Haseman and Elston 1972 |
| MLS LOD score (2 df): | |||
| SPLINK | 36.34 | 36.34 | Holmans 1993 |
| MLS LOD score (1 df): | |||
| GeneHunter | 22.20 | 22.20 | Kruglyak and Lander 1995 |
| ASPEX sib_phase | 22.20 | 22.20 | Hinds and Risch 1996 |
| NPL Sall Z score: | |||
| GeneHunter | 6.70 | 7.82 | Kruglyak et al. 1996 |
| Allegro | 7.82 | 7.82 | Gudbjartsson et al. 2000 |
| Merlin | 7.82 | 7.82 | Abecasis et al. 2002 |
| GeneHunter-Plus Sall LOD score: | |||
| GeneHunter-Plus | 22.20 | 22.20 | Kong and Cox 1997 |
| Allegro | 22.20 | 22.20 | Gudbjartsson et al. 2000 |
| Merlin | 22.20 | 22.20 | Abecasis et al. 2002 |
There are two statistics in table 1 that are less significant when all 200 families are used than when the uninformative families are removed. These two statistics are the GeneHunter NPL Sall Z score and the SIBPAL mean test Z value. In both of these cases, the reduction in evidence for linkage is caused by the use of the “perfect data approximation” to compute the variance of the statistics. The “perfect data approximation” performs well if most of the families are informative for IBD sharing, but, as the proportion of uninformative families increases, it becomes increasingly conservative, leading to a loss of power (Kruglyak et al. 1996). In fact, the loss of power due to “bias” that Schork and Greenwood (2004) identify is, mathematically, exactly the samething as the loss of power due to the “perfect data approximation.”
The negative effects of the “perfect data approximation” can be illustrated by a simple example. Consider the sib-pair IBD-sharing statistic
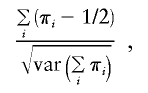 |
where πi is the estimated proportion of alleles shared IBD for the ith affected sib pair. Suppose we have two data sets: (1) 50 fully informative affected–sib-pair families and (2) 50 fully informative and 50 uninformative families. Suppose πi in our fully informative families takes on the values 0, 1/2, and 1, with probabilities1/8, 1/2, and 3/8, respectively, whereas πi is 1/2 in our uninformative families. The numerator of the statistic is identical for both data sets. However, different approaches to computing the variance in the denominator can lead to different statistic values for the two data sets. Under the “perfect data approximation,” the value of the statistic is 2.50 for the first data set and 1.77 for the second data set—an undesirable reduction in the evidence for linkage. Use of the correct variance (given that the number of uninformative families remains constant) leads to statistic values of 2.50 for both data sets. Another option is to use the empirical variance, which reflects the alternative hypothesis rather than the null hypothesis and can be quite powerful; the empirical variance gives an expected IBD-sharing statistic of 2.50 for both example data sets. A score test using empirical variances was one of the best statistics in a recent evaluation of methods for QTL mapping using selected sibling pairs (T.Cuenco et al. 2003).
To avoid the negative consequences of using the “perfect data approximation,” Kong and Cox (1997) proposed a nonparametric statistic that performs much better in the presence of uninformative families. This statistic has been implemented in GeneHunter-Plus (Kong and Cox 1997), Allegro (Gudbjartsson et al. 2000), and Merlin (Abecasis et al. 2002) and, as illustrated by our simple simulation experiment in table 1, is insensitive to the presence of fully uninformative families. Similarly, in the context of the Haseman-Elston (HE) test (Haseman and Elston 1972), in which trait values are regressed on IBD sharing, the problem of using estimated IBD sharinghas long been recognized. For example, Kruglyak and Lander (1995) developed a missing-value regression approach to compute a modified HE test that has much better behavior in the presence of uninformative families than the original test.
Whereas it is always useful to remind the scientific community that proper statistical analyses of linkage data requires deep insight into the potential weaknesses of the chosen methodology and software implementation, we feel that Schork and Greenwood’s concerns are overstated. Indeed, as we have shown, not only has this potential problem been known since at least the mid-1990s, but, in addition, the majority of implementations of linkage statistics in commonly used software do not suffer from this “bias” toward the null hypothesis in the presence of uninformative families. Furthermore, the use of highly informative markers in a multipoint analysis will result in very few families being fully uninformative for IBD sharing.
Acknowledgments
This work was supported by the University of Pittsburgh and National Institutes of Health grants 5D43TW006180-02 and 5R01MH064205-06. Some of the results of this paper were obtained using the S.A.G.E. package of genetic epidemiology software, which is supported by U.S. Public Health Service Resource grant RR03655 from the National Center for Research Resources.
References
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR (2002) Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 30:97–101 10.1038/ng786 [DOI] [PubMed] [Google Scholar]
- Cordell HJ (2004) Bias toward the null hypothesis in model-free linkage analysis is highly dependent on the test statistic used. Am J Hum Genet 74:1294–1302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottingham RW, Idury RM, Schäffer AA (1993) Faster sequential genetic linkage computations. Am J Hum Genet 53:252–263 [PMC free article] [PubMed] [Google Scholar]
- Gudbjartsson DF, Jonasson K, Frigge ML, Kong A (2000) Allegro, a new computer program for multipoint linkage analysis. Nat Genet 25:12–13 10.1038/75514 [DOI] [PubMed] [Google Scholar]
- Haseman JK, Elston RC (1972) The investigation of linkage between a quantitative trait and a marker locus. Behav Genet 2:3–19 [DOI] [PubMed] [Google Scholar]
- Hinds D, Risch N (1996) The ASPEX package: affected sib-pair exclusion mapping. Available at: http://aspex.sourceforge.net/. Accessed August 2, 2004
- Holmans P (1993) Asymptotic properties of affected–sib-pair linkage analysis. Am J Hum Genet 52:362–374 [PMC free article] [PubMed] [Google Scholar]
- Kong A, Cox NJ (1997) Allele-sharing models: LOD scores and accurate linkage tests. Am J Hum Genet 61:1179–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES (1996) Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet 58:1347–1363 [PMC free article] [PubMed] [Google Scholar]
- Kruglyak L, Lander ES (1995) Complete multipoint sib-pair analysis of qualitative and quantitative traits. Am J Hum Genet 57:439–454 [PMC free article] [PubMed] [Google Scholar]
- Ott J (1989) Computer-simulation methods in human linkage analysis. Proc Natl Acad Sci USA 86:4175–4178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risch N (1990) Linkage strategies for genetically complex traits. III. The effect of marker polymorphism on analysis of affected relative pairs. Am J Hum Genet 46:242–253 [PMC free article] [PubMed] [Google Scholar]
- Schork NJ, Greenwood TA (2004) Inherent bias toward the null hypothesis in conventional multipoint nonparametric linkage analysis. Am J Hum Genet 74:306–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- T.Cuenco K, Szatkiewicz JP, Feingold E (2003) Recent advances in human quantitative-trait–locus mapping: comparison of methods for selected sibling pairs. Am J Hum Genet 73:863–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks DE, Ott J, Lathrop GM (1990) SLINK: a general simulation program for linkage analysis. Am J Hum Genet 47:A204 [Google Scholar]