

Abstract
We have screened the nearly complete DNA sequence of the human Y chromosome for microsatellites (short tandem repeats) that meet the criteria of having a repeat-unit size of ⩾3 and a repeat count of ⩾8 and thus are likely to be easy to genotype accurately and to be polymorphic. Candidate loci were tested in silico for novelty and for probable Y specificity, and then they were tested experimentally to identify Y-specific loci and to assess their polymorphism. This yielded 166 useful new Y-chromosomal microsatellites, 139 of which were polymorphic, in a sample of eight diverse Y chromosomes representing eight Y-SNP haplogroups. This large sample of microsatellites, together with 28 previously known markers analyzed here—all sharing a common evolutionary history—allowed us to investigate the factors influencing their variation. For simple microsatellites, the average repeat count accounted for the highest proportion of repeat variance (∼34%). For complex microsatellites, the largest proportion of the variance (again, ∼34%) was explained by the average repeat count of the longest homogeneous array, which normally is variable. In these complex microsatellites, the additional repeats outside the longest homogeneous array significantly increased the variance, but this was lower than the variance of a simple microsatellite with the same total repeat count. As a result of this work, a large number of new, highly polymorphic Y-chromosomal microsatellites are now available for population-genetic, evolutionary, genealogical, and forensic investigations.
Introduction
Microsatellites, or short tandem repeats (STRs), consist of repetitions of a 1–6-bp unit. The number of repetitions (repeat count) can vary between individuals, so microsatellites have proved to be useful markers in several areas of genetics, including gene mapping, forensic investigations, and evolutionary studies. Microsatellites from the nonrecombining portion of the human Y chromosome have an important role in forensic genetics, where they have become the markers of choice, particularly in cases involving sexual assault or in paternity testing when the putative father is not available (Jobling et al. 1997; Kayser et al. 1997); they are also used increasingly in genealogical research (Jobling 2001) and find a major application in evolutionary studies as markers for male lineages (Kayser et al. 2001; Stumpf and Goldstein 2001; Jobling and Tyler-Smith 2003).
Y-chromosomal microsatellites are used in two ways: (1) to distinguish lineages (the number of markers and their variability will determine the degree of discrimination) and (2) to provide information about lineage relationships (the number of markers and the extent to which their properties are understood will influence the reliability of the inferences). Standard forensic databases use either 9 or 11 Y-chromosomal microsatellites (Roewer et al. 2001; Kayser et al. 2002), and evolutionary studies have used up to 16, but some populations contain many individuals who share the same 16-locus haplotype. For example, 14% of the Parsi population in Pakistan share one 16-locus haplotype (Mohyuddin et al. 2001), and ∼4% (32/720) of men from a large part of Asia share the same 15-locus haplotype, attributed to Genghis Khan (Zerjal et al. 2003). Similarly, 13% (26/200) of men from Finland share one 16-locus haplotype (Hedman et al. 2004). Thus, even 15 or 16 Y-chromosomal microsatellites are insufficient for some applications. Conversely, in rare cases, father and son differ because a mutation has occurred (Kayser et al. 2000); forensic or paternity-testing calculations must take this possibility into account (Kayser and Sajantila 2001; Rolf et al. 2001). For reliable evolutionary inferences, large numbers of microsatellites are needed (e.g., >20), and the mutational properties of each locus should be well understood (Stumpf and Goldstein 2001). There is thus a need for both additional loci and a better understanding of their properties.
Prior to this study, only 53 different Y-chromosomal microsatellites (counting all loci that can be analyzed separately) were known, of which 52 were published (Chen et al. 1994; Mathias et al. 1994; Jobling et al. 1996; Kayser et al. 1997; White et al. 1999; Ayub et al. 2000; Iida et al. 2001, 2002; Bosch et al. 2002; Redd et al. 2002; Mohyuddin et al. 2004) and one was deposited in the Genome Database (GDB). This contrasts sharply with the number of autosomal microsatellites, of which several hundred are available for each human chromosome (Dib et al. 1996). Genomewide analysis of the published human DNA sequence has demonstrated that the density of microsatellites ⩾12 bp in length (combining all repeat-unit lengths) is similar on all chromosomes, including the euchromatic portion of the Y chromosome (Subramanian et al. 2003), so more Y-chromosomal loci should be present. Most of the loci investigated by Subramanian and colleagues were short and monomorphic, but longer, polymorphic microsatellites can readily be identified from sequence data by use of the program Tandem Repeats Finder (Benson 1999; Ayub et al. 2000). We have therefore screened the nearly complete Y-chromosomal DNA sequence for microsatellites with a unit size that is ⩾3 and a repeat count that is ⩾8. These criteria were chosen to yield loci that would have a high probability of showing variation and that would be free from the “stuttering” that complicates the scoring of mono- and dinucleotide microsatellites. Loci were then taken through a series of sequential tests for (1) novelty, (2) in silico design of locus-specific primers, (3) experimental validation of Y-specific amplification, (4) polymorphism screening, and (5) DNA sequencing. This screen resulted in the development of 166 novel Y loci, 139 of which were polymorphic in a small but diverse sample of eight Y chromosomes. This large number of loci, together with 28 previously known markers analyzed here, provided an opportunity to test some of the properties of microsatellites: the relationship between microsatellite length or repeat count and variance, the effect of sequence complexity, and the potential role of an origin from retrotransposons, such as Alu and LINE elements. An analysis of these human Y-chromosomal microsatellites in nonhuman primates is currently under way (A. Erler, M. Stoneking, and M. Kayser, unpublished data).
Methods
Database Screen for Tandem Repeats
We obtained 23 Mb of the four genomic contigs (NT_011896, NT_011878, NT_0113, and NT_011903) that represent the known sequence of the euchromatic nonrecombining region of the human Y chromosome (NRY) (GenBank Web site [initial sequence download, March 2001; recheck, April 2002]). These sequences were used as input for the program Tandem Repeats Finder (Benson 1999; Tandem Repeats Finder Web site). A set of 475 STRs was chosen from the output files by visual examination, according to the following criteria: (1) unit size of 3–6 bp and (2) a perfect match of an array of eight or more copies. These sequences were compared with the National Center for Biotechnology Information (NCBI) Homo sapiens genomic sequence database by use of BLAST (BLAST Web site) to identify loci that matched other sequences on the Y chromosome or elsewhere in the genome. Primers were designed within the 200 bp of DNA flanking each microsatellite by use of the program Primer3 (Rozen and Skaletsky 2000; Primer3 Web site). For microsatellites exhibiting high sequence similarity to other loci, at least one primer with two or more mismatches at the 3′ end was designed. In total, 149 microsatellites were excluded from further experimental work either because no primer could be designed that was exempt from formation of hairpins and primer dimers or because no locus-specific primer could be found. In addition, 45 known microsatellites were identified in our data set, which reduced the total number of new Y-specific candidate microsatellites to 281. To allow the subsequent combination of microsatellites for multiplex PCR, we designed primers preferentially for the same annealing temperature (∼60°C) and chose a range of amplicon lengths (∼100–400 bp).
DNA Samples
DNA samples of three male and two female human individuals were used for locus evaluation. DNA samples from eight males belonging to the binary-marker haplogroups A, B, C, E, I, J, K*, and R were used for polymorphism testing (nomenclature according to the Y Chromosome Consortium [2002]). To cover the maximum amount of Y-chromosome genetic diversity, individuals were ascertained from different Y-SNP haplogroups. These males originated from the following geographic regions: A: Africa (San); B: Africa (Biaka Pygmy); C, E, and K*: Asia (Pakistan); and I, J, and R: Europe (United Kingdom, Portugal, United Kingdom, respectively).
PCR Optimization
PCR was optimized, and microsatellites were genotyped and sequenced in five different laboratories. Although there were minor differences in protocols between laboratories, we give here the protocol followed in the Leipzig laboratory. Details of the slight variations used in other laboratories are available from the authors on request.
Candidate loci from computational investigation were investigated for male specificity by establishing PCR assays. All loci underwent PCR-based optimization procedures by use of DNA samples from three male and two female individuals. Two parameters were tested: (1) annealing temperature and (2), if necessary, MgCl2 concentration. Two different PCR systems were used: system A and system B. For system A, PCR was performed by analyzing 10–20 ng genomic DNA in a total volume of 25 μl. Final concentrations were as follows: 1× SuperTaq buffer and 0.35 U SuperTaq (HT Biotechnology), MgCl2 at 1.5 mM + (n × 0.7 mM; n= 1, 2, 3, 4), 800 μM dNTPs (200 μM each), 200 nM of each primer, and 9 nM TaqStart monoclonal antibody (BD Biosciences Clontech). Before preparation of the PCR master mix, the SuperTaq polymerase was mixed with TaqStart antibody. Cycling conditions (on an MJ PTC-200/225) were as follows: initial denaturation at 94°C for 3 min; touchdown with 8 cycles of 60 s at 94°C, 60 s at Ta (annealing temperature) + 4°C (minus 0.5°C/cycle), and 60 s at 60°C; 32 cycles of 60 s at 94°C, 60 s at Ta, and 60 s at 60°C; and a final extension of 10 min at 60°C. In the case of an annealing temperature >60°C, the elongation steps and the final extension step were performed at the annealing temperature. For PCR system B, 1× PCR buffer I (containing 1.5 mM MgCl2), and 0.5 U Amplitaq Gold (Roche Molecular Systems), 200 μM dNTPs (50 μM each), and 1 μM of each primer were used. Cycling conditions (on an MJ PTC-200/225) were as follows: initial denaturation at 95°C for 14 min; 32 cycles of 45 s at 94°C, 45 s at Ta, and 45 s at 72°C; and a final extension of 5 min at 72°C. For both systems, the initial annealing temperature was 60°C. When nonspecific PCR products were observed, the annealing temperature was increased in steps, and when no PCR product was observed, it was decreased. PCR products were separated and visualized via conventional agarose gel electrophoresis and ethidium bromide staining (2% SeaKem LE agarose, 1× TBE buffer, 400 ng/ml ethidium bromide). Normally, PCR optimization was performed until a single (or multiple) male-specific PCR fragment(s) was (were) obtained, with no amplification of female DNA. However, in some cases, PCR products were obtained from female DNA, but those loci were used only if there was a clear length difference between the male and female products. No male-specific protocol could be developed for 67 loci, which were excluded from further analysis, and 48 loci could not be optimized at all. In total, male-specific amplification assays could be developed for 166 new Y-chromosomal microsatellites.
Microsatellite Genotyping
To screen for polymorphisms in the new Y-chromosomal microsatellites, eight male DNA samples were genotyped by use of fluorescence-based fragment-length analysis. In addition, a female DNA sample was used as a control. One primer from each pair was fluorescently labeled (TAMRA, FAM, JOE, or HEX), and the optimized PCR conditions (see above and the online-only tab-delimited data set) were utilized. Amplification products were separated by electrophoresis through a 5% Long Ranger polyacrylamide gel (FMC Bioproducts) by use of an ABI 377 sequencer (Perkin-Elmer Applied Biosystems). Allele sizes were determined in relation to internal size standards (ROX400 and ROX500) by use of GeneScan 2.1 software (Perkin-Elmer Applied Biosystems). Three different amplification products, labeled with different fluorescent dyes, were analyzed simultaneously. The loading amount was determined by a semiquantitative comparison to a standard concentration on an agarose gel (see above for conditions). In addition to the newly described markers, 22 of the published Y-chromosomal microsatellites were also analyzed in the same eight DNA samples, by use of published protocols: DYS19, DYS388, DYS389I, DYS389II, DYS390, DYS391, DYS392, DYS393 (Kayser et al. 1997, 2002), DYS385a, DYS385b (Kittler et al. 2003), DYS425 (Thomas et al. 1999), DYS434, DYS435, DYS436, DYS437, DYS348, DYS439 (Y-GATA-A4) (Ayub et al. 2000), Y-GATA-7.1 (DYS460), Y-GATA-7.2 (DYS461), Y-GATA-A10, Y-GATA-H4, and Y-GATAC4 (DYS635) (White et al. 1999). An additional six of the previously known markers were typed by use of newly designed primers and protocols: DYS426 (Y3C8), DYS454 (Y4S29), DYS443 (Y4C12), DYS444 (Y4C37), DYS449 (Y4C169), and BV005731 (Y4C83). Overall, 194 different Y-chromosomal microsatellites, made up of 166 new and 28 previously known markers, were typed and analyzed in this study.
Microsatellite DNA Sequencing
Two alleles of different lengths were sequenced for each locus. This allowed the variable array(s) to be identified and fragment sizes to be calibrated as repeat counts. Unlabeled PCR products were purified with the Wizard purification kit (Promega) or the QIAquick PCR purification kit (Qiagen). Cycle sequencing was performed by use of the ABI BigDye Terminator Cycle Sequencing Ready Reaction kit (PE Applied Biosystems). Sequencing products were purified by use of an isopropanol-based protocol and were separated via a 5% Long Ranger denaturing polyacrylamide gel (FMC Bioproducts) on an ABI 377 sequencer (PE Applied Biosystems). The results were analyzed by use of Sequencing Analysis Software 3.4 (PE Applied Biosystems) and SeqManII, version 5.00 (DNASTAR). Loci with multiple male-specific amplification products were excluded from sequence analysis. Of the 166 new Y-chromosomal microsatellites, we obtained complete de novo sequence information for 120 markers. For the remaining markers, the PCR primers used for sequencing were located too close to the start of the repeat for complete repeat information to be read, or (in rare cases) sequence analysis was unsuccessful for unknown reasons. From the 28 previously known Y microsatellites used here, we obtained complete de novo sequence information for 19 markers. Thus, a total of 139 Y-chromosomal microsatellites were successfully sequenced in this study.
Alu/LINE Associations
The association of microsatellites with Alu and LINE elements was investigated by use of RepeatMasker (RepeatMasker Web Server) to identify repeated elements in the 200 bp of flanking DNA on each side of each microsatellite. An association was counted when the microsatellite lay at the 3′ end of the element at the location of the polyA tail. Cases in which a microsatellite was located either internally or at the 5′ end of an element, as well as instances in which a gap existed between the polyA tail and the microsatellite, were not considered to be associations.
Microsatellite Locus Delimitation, Repeat Count, and Complexity
There are several ways to classify microsatellite sequences. One classification method considers all repetitive units within a given PCR amplicon as part of the same multiple microsatellite locus. However, the location of the PCR primers does not necessarily provide the most biologically relevant delimitation of a repetitive sequence. We therefore decided to use more-specific criteria reflecting biological relevance. Identifying the beginning and end of a simple microsatellite is trivial, but it can be complicated for a complex microsatellite. A variation in sequence along a repeat array can represent a change to another part of the same microsatellite (making it a complex locus) or to the end of the microsatellite. Rules must be chosen to decide between these possibilities. We note that, according to the definition used here, an amplicon may contain more than one microsatellite locus. We adopted the following five criteria for defining the ends of a locus. First, the analysis began with the array of ⩾8 homogeneous units identified by Tandem Repeats Finder. Second, when a variant unit was encountered, it was classified as (1) an insertion/deletion (only insertions/deletions of one base were allowed); (2) a base substitution creating a new unit sequence within the same locus; or (3) the end of the locus. These classifications were attempted in order (1, 2, 3), to reflect their parsimony and biological relevance: for example, (GATA)n (GAT)1 (GATA)m was preferred over the alternative description, (GATA)n (GATG)1 (ATAG)m, since a single-nucleotide (“A”) deletion provides a more parsimonious description of the structure than two changes in unit sequence. Third, if the variant structure could not be explained by a single-base insertion/deletion, the phase of the repeat was maintained and the next step was to decide whether the locus continued with a different repeat unit or whether it ended. For it to continue with a second unit sequence, there must be two or more copies of the new unit. If there was only one, the locus could either (1) continue with a return to the first repeat unit, (2) continue with a third repeat unit, or (3) end. For example, (GATA)n (GACA)2, (GATA)n (GACA)1 (GATA)m, and (GATA)n (GACA)1 (GAAA)m (where n and m are >2) were all considered to be single loci, but (GATA)n (GACA)1 (GAAA)1 (GATA)m was considered to end after (GATA)n, with (GATA)m as a separate locus. Fourth, apart from single-base insertions/deletions, changes in unit length represent different loci; dinucleotide repeats, for example, were not considered to be part of the trinucleotide (or other) repeats of interest in this study, even if they were directly adjacent. Finally, isolated arrays of only two tandem repeats were not counted. The repeat count is then the number of units within a locus; therefore, in the first example in this paragraph, the repeat count is (n + 1 + m).
Microsatellites have conventionally been classified as “simple” if they contain an uninterrupted run of units sharing the same sequence (homogeneous array) and as “complex” if there is an interruption or change in the sequence. However, complexity is a quantitative characteristic, so we developed an analog of the Nei gene-diversity formula (Nei 1987) to quantify it:
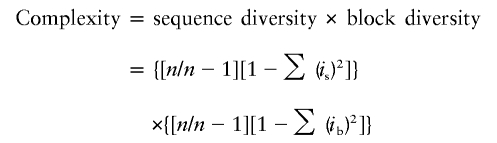 |
where n stands for the number of units, is stands for the frequency of the ith sequence, and ib stands for the frequency of the ith block. This represents the probability that two randomly chosen units from the same locus have different sequences, multiplied by the probability that two randomly chosen units from the same locus lie in different blocks. Complexity varies from 0 for a simple microsatellite such as (AAT)13 to 1 for a nonrepeated sequence. In practice, complexity values >0.6 are rare; the average value for the complex loci listed in the online-only tab-delimited data set was 0.20, and the highest value observed was 0.69.
Variance Analysis
Repeat variance was calculated if genotypes from six or more (usually eight) of the eight males were available. Multicopy loci and loci with more than one polymorphic repeat (observed in our sequencing data) were excluded from variance analyses.
Statistical Tests
Multiple linear regression analyses, Mann-Whitney U tests, and Wilcoxon signed-rank tests were performed by use of the software package SPSS, version 11 (SPSS). The direction of mutational changes was inferred by use of MacClade (Maddison and Maddison 1992; MacClade Web site).
Results
Identification of New Y-Chromosomal Microsatellites
Screening of 23 Mb of Y-chromosomal sequence by use of the program Tandem Repeats Finder revealed 475 loci that met the criteria of a unit size of ⩾3 bp and a perfect array with a repeat count of ⩾8. Of these 475 loci, 45 corresponded to previously known loci and 149 did not allow suitable primers to be designed, providing 281 novel candidate loci for further evaluation (table 1). Primers were synthesized for these loci and were tested experimentally in DNA samples of three males and two females, with the result that 166 new microsatellite loci showed male-specific amplification (table 1). Details of these loci have been deposited in the Genome Database (see Genome Database Web site); they are also given in the online-only tab-delimited data set. There were 51 trinucleotide repeat loci, 100 tetranucleotide repeat loci, 14 pentanucleotide repeat loci, and 1 hexanucleotide repeat locus (table 2). The 166 new loci identified here increase the number of available microsatellites for the human Y chromosome to 219, with the distribution of repeat types shown in figure 1. In the 23 Mb of Y-chromosomal DNA sequence available, there are thus almost 10 useful microsatellites per Mb. The 219 microsatellites are distributed throughout the entire euchromatic region of the Y chromosome (fig. 2), except near the centromere (which is not represented in the sequence database), although there is a reduction in their density in the recently transposed X-Y homologous region on Yp.
Table 1.
Summary of Y-Chromosomal Microsatellite Discovery Stages
|
No. of Loci |
||
| Stage of Analysis | Excluded | Remaining |
| Tandem Repeats Finder output | … | 475 |
| Novel loci | 45 | 430 |
| Suitable primer pair in silico | 149 | 281 |
| Male-specific amplification | 115 | 166 |
| Polymorphic in 8 diverse Y chromosomes | 27 | 139 |
Table 2.
Number of Y-Chromosomal Microsatellites Classified According to Unit Size[Note]
|
No. of Microsatellites Classified as |
||||||
| Data Set | Dinucleotide | Trinucleotide | Tetranucleotide | Pentanucleotide | Hexanucleotide | Total |
| Previously known | 4 | 5 | 35 | 7 | 2 | 53 |
| New nonpolymorphic | NA | 18 | 8 | 1 | 0 | 27 |
| New polymorphic | NA | 33 | 92 | 13 | 1 | 139 |
| Total (old + new) | 4 | 56 | 135 | 21 | 3 | 219 |
Note.— NA = not analyzed.
Figure 1.

Unit-size distribution of 53 previously known and 166 new human Y-chromosomal microsatellites.
Figure 2.

Localization of the 219 currently known microsatellites on the euchromatic part of the human Y chromosome
Copy Number of the New Y-Chromosomal Microsatellites
Multicopy Y-chromosomal microsatellites are common and are accounted for by their location in the palindromes P1–P8 (which themselves carry some sequences related to each other) that make up 25% of the euchromatin (Skaletsky et al. 2003). Among the 166 loci, 38 were expected to produce multiple Y-chromosomal fragments, because the primer pairs showed 100% sequence identity to more than one location in the published Y-chromosome sequence. Of these 38 loci, 25 did indeed show multiple Y-specific bands in the PCR analysis, but 13 showed only a single product in all individuals, perhaps reflecting difficulties in sequence assembly, gene-conversion events leading to homogeneity between copies (Rozen et al. 2003), or insufficient evolutionary time for the alleles to diverge in repeat number. It is more surprising that three loci (DYS507, DYS514, and DYS518), each with a single location in the sequence database, produced two PCR fragments experimentally in one, two, or three of the eight chromosomes, respectively. These could not be accounted for by mispriming from known related sites, but they may arise from mutations that create additional sites or duplication/gene-conversion polymorphisms on the Y chromosome, as has been observed at known single-copy Y-chromosomal microsatellites (e.g., Bosch and Jobling 2003; Santos et al. 1996; for a summary, see Kayser et al. 2000). The number of multicopy Y-chromosomal microsatellites identified here is a minimum, since multiple copies with identical microsatellite length (due to insufficient evolutionary time for the alleles to mutate) would escape detection. All loci with detectable multiple copies were excluded from further statistical analyses because correct allele-locus assignment is impossible.
Microsatellite Origins from Retroposon Elements
A large proportion of the microsatellites in the human genome are thought to have originated from the polyA tails of retroposons, principally the Alu and LINE-1 elements. For example, one study found that 72% (275/381) of the tri- to hexanucleotide repeats with a total length of >16 bp, from 2.8 Mb of genomic DNA, were closely associated with the 3′ end of a retroposon (Nadir et al. 1996). In an in silico survey, which included some loci that could not be analyzed experimentally, we found that 114/257 (44%) of Y-chromosomal microsatellites were associated with the 3′ end of Alu repeats and 26/257 (10%) were associated with LINE-1–element 3′ ends. This suggests that, overall, at least 140/257 (54%) of Y-chromosomal microsatellites originated from retroposons. The significantly lower proportion of retroposon-associated microsatellites on the Y chromosome than on autosomes may be due to the different criteria used to identify the microsatellites, the greater length of the Y loci, or the presence of fewer retroposons on the Y chromosome (Callinan et al. 2003). The number of simple microsatellite loci associated with retroposons (81/118 [69%]) was significantly greater than that of complex loci (50/139 [36%]; χ2 test: P<.0001). The difference in the proportion of simple versus complex microsatellites associated with Alu elements was not significant, but a significantly greater number of simple loci were associated with LINE-1 elements (22/118 [19%]) than were complex microsatellites (4/139 [3%]; P<.0001) (fig. 3). The largest number of LINE-1 associations was with trinucleotide repeats (14/22 simple loci). There is thus a remarkable tendency for LINE-1 elements that give rise to microsatellites meeting our criteria to form simple trinucleotides: 14/26 LINE-1–associated microsatellites were simple trinucleotides, compared with 33/231 non-LINE-1–associated microsatellites (χ2 test: P<.0001).
Figure 3.

Relative frequencies of retroposon-associated Y-chromosomal microsatellites with different complexities.
Variability of the New Y-Chromosomal Microsatellites
Of the 166 new Y-chromosomal microsatellites, 139 were found to be polymorphic among eight Y chromosomes that belong to different haplogroups defined by binary markers. The distribution of these among microsatellite unit-size classes is shown in table 2. The average total number of repeats among 19 nonpolymorphic loci was 9.7 (8.6 for the longest homogeneous array), compared with an average total length of 14.4 repeat units (11.8 for the longest homogeneous array) among 102 polymorphic loci, a significant difference (Mann-Whitney U test: Z=-4.658 and P<.001 for the total array; Z=-5.367 and P<.001 for the longest homogeneous array [the analysis considers only loci for which repeat-count information was available from de novo sequence analysis]). Low repeat count is therefore associated with a lack of variation in this sample, as might be expected.
Microsatellite variability can be summarized by a number of measures, including diversity and variance. We show both of these in the online-only tab-delimited data set, but we consider variance to be the more informative because it takes into account not only whether or not a difference exists between alleles but also the magnitude of any difference. Variance is therefore the measure used in the analyses below. Trinucleotide repeats tend to have low or high variances, whereas microsatellites with larger units are more likely to have intermediate variances (fig. 4). The increased representation of trinucleotide repeats in the low-variance class may reflect a dependence of variability on absolute length (see below), such that loci with a trinucleotide repeat count of 8 have often not accumulated mutations in the set of chromosomes investigated. Their increased representation in the high-variance classes may result from the high mutation rate characteristic of some trinucleotide loci, such as those associated with trinucleotide-expansion diseases.
Figure 4.

Variance characteristics of Y-chromosomal microsatellites with different repeat-unit lengths.
Relationship between Microsatellite Sequence and Variability
Simple microsatellites
A major aim of this study was to use a large sample of loci to investigate the factors that influence microsatellite variability. We first consider the completely simple microsatellites (i.e., those loci that consist of one type of repeated unit in an uninterrupted [homogeneous] array within the PCR amplicon); there were 78 such simple microsatellites among the 166 new Y-chromosomal loci described here (see the online-only tab-delimited data set), of which 7 loci are multicopy and thus not suitable for variance analysis. An additional 12 such loci were present in the 28 previously described Y-chromosomal microsatellites included in this study (see the online-only tab-delimited data set). For 65 of these 83 single-copy simple microsatellites, DNA sequence analysis provided direct information on repeat count. To test which sequence factor(s) significantly influence(s) the repeat variance of simple microsatellites, we performed a stepwise multiple linear regression analysis (stepwise criteria: probability of F to enter ⩽0.05; probability of F to remove ⩾0.10) that used the following parameters: average repeat count (i.e., average number of consecutive [homogeneous] repeats), repeat-unit size (i.e., tri-, tetra-, or pentanucleotide repeat), and retrotransposon association (i.e., whether or not a repeat originated from a retrotransposon [Alu, LINE-1] element). When all parameters were considered, a single model including only the average repeat count was obtained; this model showed a highly significant positive correlation of average repeat count to repeat variance (R=0.595; R2=0.344 [adjusted for the number of variables in the model]; F=34.592; P<.001). No significant partial correlation of any of the other parameters with variance was apparent. Thus, among all the parameters tested, average repeat count explains most of the repeat variance (34.4%) of simple microsatellites (fig. 5A). In addition to these completely simple microsatellites that contain a single homogeneous repetitive array within the PCR amplicon, we identified another 50 simple loci that lay within PCR amplicons containing more than one microsatellite locus (by use of our criteria for microsatellite locus delimitation; see the “Methods” section). Of those, 12 loci met the same criteria of length, unit size, and availability of sequence information and were thus applied to multiple regression analysis obtaining similar conclusions: that is, average repeat count is the only sequence parameter that significantly explains repeat variance in simple microsatellites (R=0.695; adjusted R2=0.432; F=9.356; P=.012). This supports not only our conclusion about the importance of average repeat count but also the way we identified repetitive loci in this study.
Figure 5.

A, Relationship between average repeat count and repeat variance for 65 completely simple Y-chromosomal microsatellites (R=0.595; P<.001). B, Relationship between average repeat count of the longest homogeneous array (which normally is variable) and repeat variance for 104 complex Y-chromosomal microsatellites (R=0.591; P<.001).
Complex microsatellites
Complex microsatellites consist of more than one type of repeated unit and/or contain interruptions. Their analysis, in contrast to that of simple microsatellites, allows us to investigate the effect of insertions/deletions and/or different types of repeated units on microsatellite mutability and thus on repeat variance. Of the 166 new Y-chromosomal microsatellites identified in this study, 88 were complex (see the online-only tab-delimited data set), including 17 that are multicopy and thus not suitable for variance analysis. In addition, 16 of the 28 previously known microsatellites analyzed here were also complex (see the online-only tab-delimited data set). For 72 of these 87 single-copy complex loci, DNA sequence analysis provided the repeat-count information needed for statistical analysis. Within these 72 loci, we identified a total of 104 complex microsatellites on the basis of our criteria for microsatellite locus delimitation (see the “Methods” section), which were thus amenable to variance analysis.
To test which sequence factors significantly influence the repeat variance of complex microsatellites, we again performed stepwise multiple linear regression analyses and tested the following parameters: repeat count of the longest homogeneous array, which is normally variable; total repeat count; count of repeats in addition to the longest variable array; unit size; retroposon association; repeat complexity (see the “Methods” section); and total repeat-block count. When all parameters were considered, a single model, which included only the repeat count of the longest homogeneous array (averaged across individuals), was found to predict repeat variance best (R=0.591; adjusted R2=0.343; F=54.837; P<.001). No significant partial correlation of any of the other parameters with the variance was identified (R<0.16; P>.111). Thus, of all the parameters tested, the average repeat count of the longest homogeneous array explained most of the variance (34.3%) of complex microsatellites (fig. 5B). Total repeat count was also significantly correlated with variance when tested separately (R=0.506; adjusted R2=0.249) but less so than the average repeat count of the longest homogeneous array.
The large number of complex microsatellite loci in this study enabled us to test whether the presence of additional repeats (outside the longest homogeneous repeat) influences microsatellite variability. To do this, we compared the average variance at complex microsatellites with that of simple microsatellites with the same average number of repeats. For simple loci, we used the total repeat count, whereas for complex loci the repeat count of the longest homogeneous array was used. Of 14 repeat-count categories for which data were available from both simple and complex loci, we observed that, in 9 categories, the average variance was higher in the complex microsatellites, in 3 categories it was equal, and in 2 categories it was higher in the simple microsatellites (fig. 6A). A Wilcoxon signed-rank test revealed that complex and simple microsatellites differ significantly (P=.05) in their variance. Thus, there is a significantly greater variance at complex microsatellites compared with simple microsatellites with the same average homogeneous repeat count. The higher variance observed at the complex microsatellites is presumably caused by the presence of the additional repeats (outside the longest variable homogeneous array) in the complex microsatellites.
Figure 6.

A, Comparison between simple and complex microsatellites, grouped according to average total repeat count, and complex microsatellites with the same average repeat count in the longest homogeneous array. B, Comparison between simple microsatellites, grouped according to average total repeat count, and complex microsatellites with the same average total repeat count. For repeat count <7, the average repeat variance was 0 for both simple and complex microsatellites.
To determine if the number of these additional repeats influences microsatellite variability, we compared the 50 loci with the smallest numbers of additional repeats (2–4 repeats; average variance 0.615) with the remaining 54 loci with the highest numbers of additional repeats (5–32 repeats; average variance 1.454). The difference in the variance between these two groups of loci is statistically significant (Mann-Whitney U test: Z=-2.07; P=.039). Also, the number of additional repeats (outside the longest variable homogeneous array) at complex loci is positively correlated with the repeat variance (R=0.180), and this approaches statistical significance (F=3.419; P=.067). Together, these results suggest that the number of additional repeats (outside the longest variable homogeneous array) does influence the variance of the longest variable homogeneous array at complex microsatellites.
To examine whether (1) increased repeat count within the longest variable homogeneous array or (2) increased repeat count outside this array has the larger effect on the microsatellite variance at complex loci, we compared the average variance at complex and simple loci with the same total repeat count (averaged across individuals) (fig. 6B). For the 13 total repeat-count categories with data for both simple and complex loci, there were 10 categories in which the complex loci had less variance, 2 in which they were identical, and 1 in which the complex locus had more variance. Thus, complex microsatellites are significantly less variable than simple microsatellites with the same number of total repeats (Wilcoxon signed-rank test: P=.01). Consistent with this, complexity itself has a significantly negative effect on the variance: the correlation coefficient of complexity and the repeat variance was R=-0.195; P=.048.
Phylogeny from Y-Chromosomal Microsatellites
Highly variable microsatellites are expected to be poor markers for reconstructing deep phylogenetic relationships between lineages because recurrent mutations introduce “noise” that obscures the true relationships. We were nevertheless interested to investigate the extent to which the use of a large number of Y-chromosomal microsatellites would overcome this noise. We therefore constructed phylogenies for the eight chromosomes by use of 100 loci (the maximum number accommodated by the Network program) and median-joining/reduced-median networks or a minimum-spanning tree. These all differed from one another, and from the binary-marker phylogeny (results not shown). These findings confirm that even 100 microsatellites do not produce a robust resolution of deep lineages within a molecular phylogeny.
The binary-marker phylogeny, however, allows us to investigate some additional mutational properties of the microsatellites. We used the binary-marker phylogeny to infer the history of mutational changes at the 123 single-copy polymorphic loci for which there were complete data. We inferred that there must have been 132 mutations that resulted in an increase in allele length, compared with 102 decreases, a difference that just achieves statistical significance (χ2=3.85; 1 df; P=.04986). This observation is consistent with the excess of expansions over contractions in allele length observed in other studies (Amos et al. 1996; Kayser et al. 2000).
Discussion
We have performed a comprehensive survey of Y-chromosomal microsatellites. It is likely that our survey includes most of the loci meeting our criteria that exist. A few will have been missed because they lie in gaps in the sequence data; others were excluded because they happened to have a particularly short allele on the chromosome that was sequenced. Some that were discarded during the analysis because we could not design suitable primers or generate specific PCR products could probably be used with improved techniques. Some that were not polymorphic in our sample of eight chromosomes may well show variation in a larger sample. One test of the proportion that we have identified is the fraction of previously known loci that was detected. Of the 48 known loci that met our criteria (i.e., excluding dinucleotides and X-Y homologous loci), we identified 45 (94%). Thus, it is likely that we have identified and characterized the majority of the useful microsatellites on the Y chromosome. Our work therefore provides a fundamental resource for future work in this area.
We foresee several uses for these loci, on the basis of an informed choice of the most suitable loci for each application. In forensics, high variability is usually the most important characteristic, and many new highly variable loci are now available (see the online-only tab-delimited data set). Multicopy loci, such as DYS385 or DYS640, are often the most variable because the different copies vary independently, but they are not ideal for some purposes, such as determining the number of individuals contributing to a mixed stain. A multiplex consisting of highly variable single-copy loci would be easier to interpret. Such a resource, which could amplify up to 20 loci simultaneously (Butler et al. 2002), can now be established. In population genetics, both the variability and the understanding of the mutational mechanism are important. Although high variability is often required, structurally simple loci, which mutate in a simple way so that fragment size is a reliable guide to allele structure, are the best choice, and the new loci provide many additional examples. We therefore list the simple new loci with the highest variances in table 3. If less-variable loci are required—for example, for comparing distantly related lineages (Forster et al. 2000)—they are also available (see the online-only tab-delimited data set).
Table 3.
Most-Variable Simple Single-Copy New Y-Chromosomal Microsatellites[Note]
| Locus IDa | RepeatVariance | Diversity | LengthRange(bp) | RepeatCountRange | Primer 1 Sequence (5′–3′) | Primer 2 Sequence (5′–3′) |
| DYS570 | 5.125 | .857 | 246–274 | 14–21 | GAACTGTCTACAATGGCTCACG | TCAGCATAGTCAAGAAACCAGACA |
| DYS643 | 3.143 | .929 | 132–159 | 8–13 | AAGCCATGCCTGGTTAAACT | TGTAACCAAACACCACCCATT |
| DYS490 | 2.800 | .600 | 170–182 | 12–16 | CCTGGCAGGAATTATCCAGA | GCAGAGCTTGCACTGAGCT |
| DYS617 | 2.786 | .893 | 226–241 | 12–17 | AGCATGATGCCTTCAGCTTT | GGATTGGGGAGTGATAGCAT |
| DYF406S1 | 2.267 | .867 | 210–226 | NR | CCTGGGTGACACAGTGAGACT | TCCACCAAAATTCCATGACA |
| DYS485 | 2.214 | .786 | 270–282 | 12–16 | AAAGCAGACTTCGCCACTACA | AAAAATTAGCTGGGCCTGGT |
| DYS556 | 1.905 | .810 | 198–214 | 8–12 | TGCTGTCACATCACCAATGA | TTTGGTTGCTGAAGCATTGA |
| DYS522 | 1.714 | .821 | 350–366 | 9–13 | CCTTTGAAATCATTCATAATGC | TCATAAACAGAGGGTTCTGG |
| DYS549 | 1.411 | .786 | 229–245 | 10–14 | AACCAAATTCAGGGATGTACTGA | GTCCCCTTTTCCATTTGTGA |
| DYS641 | 1.411 | .643 | 207–222 | 7–11 | CTTGAGCCCAGGAAGCATAG | CCACACGATGCAATTTTGTC |
| DYS575 | 1.268 | .643 | 215–231 | 8–12 | GGTGGTGGACATCCGTAATC | AGTAATGGGATGCTGGGTCA |
| DYS589 | 1.268 | .821 | 271–286 | NR | CATCCACATTGTTGCAAAGG | TGACGAGTTAGTGGGTGCAG |
| DYS540 | 1.143 | .750 | 257–269 | 10–13 | GACCGTGTACTCTGGCCAAT | CAGGAGGCTAGCTCAGGAGA |
| DYS505 | 1.143 | .786 | 164–176 | 10–13 | TCTGGCGAAGTAACCCAAAC | TCGAGTCAGTTCACCAGAAGG |
| DYS594 | 1.143 | .464 | 251–266 | 8–11 | GATGTGCCTAATGCCACAGA | CCCTGGTGTTAATCGTGTCC |
| DYS495 | 1.125 | .821 | 211–220 | 14–17 | CCCAGCTATTCAGGAGGTTG | GCCAGAAAGTGTGAGTCATCC |
| DYS488 | 1.125 | .607 | 223–232 | 13–16 | GGGGAGGGATAGCATTAGGA | TACCCTGGTCCACTTCAACC |
| DYS578 | 1.071 | .821 | 164–176 | 7–10 | GAGGCGGAACTTTCAGTGAG | GCTTCAACAACCCTGGACAT |
| DYS576 | 1.071 | .821 | 182–194 | 16–19 | TTGGGCTGAGGAGTTCAATC | GGCAGTCTCATTTCCTGGAG |
| DYS494 | 1.071 | .679 | 171–180 | NR | TTGCAACACTGTTCATTTGGA | AACAAACCTGCATGTTCTTCAA |
| DYS533 | .982 | .750 | 202–214 | 9–12 | CATCTAACATCTTTGTCATCTACC | TGATCAGTTCTTAACTCAACCA |
| DYS525 | .982 | .750 | 302–314 | 9–12 | ATTCACACCATTGCACTCCA | CCATCTGTTTATCTTCCCATCA |
| DYS636 | .786 | .643 | 246–258 | 9–12 | AATCCCATTGCATTTAGCAGA | TGACACGTTAGTGGGTGCAG |
| DYS508 | .667 | .733 | 171–179 | NR | ACAATGGCAATCCCAAATTC | GAACAAATAAGGTGGGATGGAT |
| DYS531 | .571 | .714 | 113–121 | 11–13 | GACCCACTGGCATTCAAATC | TGCTCCCTTTCTTTGTAGACG |
| DYS638 | .571 | .714 | 256–264 | 10–12 | ACAATTTCCCTTGGGGCTAC | CATGGTGGTAGGCACCTGTA |
Note.— Each PCR amplicon contains a single homogeneous uninterrupted (simple) repetitive array. NR = no average repeat information (because no de novo sequence data were available).
Locus IDs correspond to those used in the Genome Database (see the Genome Database Web site).
The availability of such a large number of loci creates new possibilities, particularly for genealogical research. For example, if the average mutation rate for all Y microsatellites were the same as that of the known loci, namely 2.8 × 10−3 (Kayser et al. 2000), then approximately half of meioses would show one or more mutations when all 216 loci were typed, so very closely related Y chromosomes could often be distinguished. More-precise estimates of the time back to the most recent common ancestor of any pair of Y chromosomes could also be obtained (Walsh 2001). For example, if two Y chromosomes match at 9/10 microsatellites, their most recent common ancestor is expected to have lived 26 generations ago, with a 95% CI of 6–147 generations; if they match at 90/100 loci, the CI is reduced to 15–49 generations.
The variation of a set of microsatellites observed in a population sample usually depends on both locus structure and population history. In this study, each locus has the same genealogy and population history because the loci are never separated by interchromosomal recombination. We can therefore investigate the effects of locus structure on variance in a straightforward way. Previous work has documented the decreased variability that results from base substitution within the repetitive array (i.e., complexity) (Jin et al. 1996) and the positive correlation between repeat count and population variability for human dinucleotide microsatellites (Weber 1990). Our work now allows these effects to be quantified. We find a highly significant positive correlation of repeat variance with repeat count at simple and complex microsatellites. In complex microsatellites, the largest contribution comes from the number of repeats at the longest homogeneous array, which is normally the polymorphic part of the complex microsatellite (and not from the total repeat count). However, the presence of repeats in addition to the longest homogeneous array increases the variance above that expected for a simple microsatellite matching the repeat count of the homogeneous array but below that for a simple locus with the repeat count of the total complex locus. In other words, a complex microsatellite consisting of the arrays 8,4 (e.g., [GATA]8 [GACA]4) is typically more variable than a simple microsatellite with a homogeneous repeat count of 8 but less variable than a simple microsatellite with a homogeneous repeat count of 12.
It should be possible to take advantage of the stable Y phylogeny, already established by use of binary markers, to infer further aspects of the microsatellite mutational processes, such as the relative rates of gains and losses. Information about gene-conversion events can be obtained, and regions of the chromosome that are involved in duplication or deletion events can be identified. Many of the findings should be applicable to loci throughout the genome. In conclusion, we thus anticipate that there will be many uses for the new Y-chromosomal microsatellites described here.
Supplementary Material
Acknowledgments
We wish to thank the donors of the DNA samples, Gunter Weiss for statistical advice, Eve Karvinen and Kirsti Höök for technical assistance, and Conover Talbot for help in making the new markers publicly available via the Genome Database (GDB). We are grateful to Qasim Ayub and John Mitchell for useful comments on the work. M.K. is a Heisenberg Fellow of the German Research Council (Deutsche Forschungsgemeinschaft [DFG grant Ka 1413/3-1]). R.K. and A.E. were supported by the German National Merit Foundation (Studienstiftung des Deutschen Volkes). M.A.J. was supported by a Wellcome Trust Senior Fellowship in Basic Biomedical Science (grant 057559). M.K., R.K., A.E., and M.S. all thank the Max Planck Society for support, and A.C.L., A.M., S.Q.M., and C.T.-S. similarly all thank The Wellcome Trust.
Electronic-Database Information
The URLs for data presented herein are as follows:
- BLAST, http://www.ncbi.nlm.nih.gov/genome/seq/page.cgi?F=HsBlast.html&=Hs (for comparing sequences with the human genome sequence)
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for sequence information of the human Y chromosome)
- Genome Database, http://www.gdb.org/ (for locus information, including details of new loci)
- MacClade, http://macclade.org/macclade.html (for inference of direction of mutational changes)
- Primer3, http://www-genome.wi.mit.edu/cgi-bin/primer/primer3_www.cgi (for designing PCR primers)
- RepeatMasker Web Server, http://woody.embl-heidelberg.de/repeatmask/ (for identifying retroposons adjacent to microsatellites)
- Tandem Repeats Finder, http://c3.biomath.mssm.edu/trf.html (for identifying microsatellites in sequence data)
References
- Amos W, Sawcer SJ, Feakes RW, Rubinsztein DC (1996) Microsatellites show mutational bias and heterozygote instability. Nat Genet 13:390–391 [DOI] [PubMed] [Google Scholar]
- Ayub Q, Mohyuddin A, Qamar R, Mazhar K, Zerjal T, Mehdi SQ, Tyler-Smith C (2000) Identification and characterization of novel human Y-chromosomal microsatellites from sequence database information. Nucleic Acids Res 28:e8 10.1093/nar/28.2.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson G (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27:573–580 10.1093/nar/27.2.573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch E, Jobling MA (2003) Duplications of the AZFa region of the human Y chromosome are mediated by homologous recombination between HERVs and are compatible with male fertility. Hum Mol Genet 12:341–347 10.1093/hmg/ddg031 [DOI] [PubMed] [Google Scholar]
- Bosch E, Lee AC, Calafell F, Arroyo E, Henneman P, de Knijff P, Jobling MA (2002) High resolution Y chromosome typing: 19 STRs amplified in three multiplex reactions. Forensic Sci Int 125:42–51 10.1016/S0379-0738(01)00627-2 [DOI] [PubMed] [Google Scholar]
- Butler JM, Schoske R, Vallone PM, Kline MC, Redd AJ, Hammer MF (2002) A novel multiplex for simultaneous amplification of 20 Y chromosome STR markers. Forensic Sci Int 129:10–24 10.1016/S0379-0738(02)00195-0 [DOI] [PubMed] [Google Scholar]
- Callinan PA, Hedges DJ, Salem AH, Xing J, Walker JA, Garber RK, Watkins WS, Bamshad MJ, Jorde LB, Batzer MA (2003) Comprehensive analysis of Alu-associated diversity on the human sex chromosomes. Gene 317:103–110 10.1016/S0378-1119(03)00662-0 [DOI] [PubMed] [Google Scholar]
- Chen H, Lowther W, Avramopoulos D, Antonarakis SE (1994) Homologous loci DXYS156X and DXYS156Y contain a polymorphic pentanucleotide repeat (TAAAA)n and map to human X and Y chromosomes. Hum Mutat 4:208–211 [DOI] [PubMed] [Google Scholar]
- Dib C, Faure S, Fizames C, Samson D, Drouot N, Vignal A, Millasseau P, Marc S, Hazan J, Seboun E, Lathrop M, Gyapay G, Morissette J, Weissenbach J (1996) A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature 380:152–154 10.1038/380152a0 [DOI] [PubMed] [Google Scholar]
- Forster P, Röhl A, Lunnemann P, Brinkmann C, Zerjal T, Tyler-Smith C, Brinkmann B (2000) A short tandem repeat-based phylogeny for the human Y chromosome. Am J Hum Genet 67:182–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedman M, Pimenoff V, Lukka M, Sistonen P, Sajantila A (2004) Analysis of 16 Y STR loci in the Finish population reveals a local reduction in the diversity of male lineages. Forensic Sci Int 142:37–43 [DOI] [PubMed] [Google Scholar]
- Iida R, Tsubota E, Matsuki T (2001) Identification and characterization of two novel human polymorphic STRs on the Y chromosome. Int J Legal Med 115:54–56 10.1007/s004140100207 [DOI] [PubMed] [Google Scholar]
- Iida R, Tsubota E, Sawazaki K, Masuyama M, Matsuki T, Yasuda T, Kishi K (2002) Characterization and haplotype analysis of the polymorphic Y-STRs DYS443, DYS444, and DYS445 in a Japanese population. Int J Legal Med 116:191–194 10.1007/s00414-001-0266-7 [DOI] [PubMed] [Google Scholar]
- Jin L, Macaubas C, Hallmayer J, Kimura A, Mignot E (1996) Mutation rate varies among alleles at a microsatellite locus: phylogenetic evidence. Proc Natl Acad Sci USA 93:15285–15288 10.1073/pnas.93.26.15285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobling MA (2001) In the name of the father: surnames and genetics. Trends Genet 17:353–357 10.1016/S0168-9525(01)02284-3 [DOI] [PubMed] [Google Scholar]
- Jobling MA, Pandya A, Tyler-Smith C (1997) The Y chromosome in forensic analysis and paternity testing. Int J Legal Med 110:118–124 10.1007/s004140050050 [DOI] [PubMed] [Google Scholar]
- Jobling MA, Samara V, Pandya A, Fretwell N, Bernasconi B, Mitchell RJ, Gerelsaikhan T, Dashnyam B, Sajantila S, Salo PJ, Nakahori Y, Disteche CM, Thangaraj K, Singh L, Crawford MH, Tyler-Smith C (1996) Recurrent duplication and deletion polymorphisms on the long arm of the Y chromosome in normal males. Hum Mol Genet 5:1767–1775 10.1093/hmg/5.11.1767 [DOI] [PubMed] [Google Scholar]
- Jobling MA, Tyler-Smith C (2003) The human Y chromosome: an evolutionary marker comes of age. Nat Rev Genet 4:598–612 10.1038/nrg1124 [DOI] [PubMed] [Google Scholar]
- Kayser M, Brauer S, Willuweit S, Schadlich H, Batzer MA, Zawacki J, Prinz M, Roewer L, Stoneking M (2002) Online Y-chromosomal short tandem repeat haplotype reference database (YHRD) for U.S. populations. J Forensic Sci 47:513–519 [PubMed] [Google Scholar]
- Kayser M, Caglià A, Corach D, Fretwell N, Gehrig C, Graziosi G, Heidorn F, Herrmann S, Herzog B, Hidding M, Honda K, Jobling MA, Krawczak M, Leim K, Meuser S, Meyer E, Oesterreich W, Pandya A, Parson W, Penacino G, Perez-Lezaun A, Piccinini A, Prinz M, Schmitt C, Roewer L (1997) Evaluation of Y-chromosomal STRs: a multicenter study. Int J Legal Med 110:125–133, 141–149 10.1007/s004140050051 [DOI] [PubMed] [Google Scholar]
- Kayser M, Krawczak M, Excoffier L, Dieltjes P, Corach D, Pascali V, Gehrig C, Bernini LF, Jespersen J, Bakker E, Roewer L, de Knijff P (2001) An extensive analysis of Y-chromosomal microsatellite haplotypes in globally dispersed human populations. Am J Hum Genet 68:990–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayser M, Roewer L, Hedman M, Henke L, Henke J, Brauer S, Kruger C, Krawczak M, Nagy M, Dobosz T, Szibor R, de Knijff P, Stoneking M, Sajantila A (2000) Characteristics and frequency of germline mutations at microsatellite loci from the human Y chromosome, as revealed by direct observation in father/son pairs. Am J Hum Genet 66:1580–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayser M, Sajantila A (2001) Mutations at Y-STR loci: implications for paternity testing and forensic analysis. Forensic Sci Int 118:116–121 10.1016/S0379-0738(00)00480-1 [DOI] [PubMed] [Google Scholar]
- Kittler R, Erler A, Brauer S, Stoneking M, Kayser M (2003) Apparent intrachromosomal exchange on the human Y chromosome explained by population history. Eur J Hum Genet 11:304–314 10.1038/sj.ejhg.5200960 [DOI] [PubMed] [Google Scholar]
- Maddison WP, Maddison DR (1992) MacClade: analysis of phylogeny and character evolution. Version 3.0. Sinauer Associates, Sunderland, MA [Google Scholar]
- Mathias N, Bayés M, Tyler-Smith C (1994) Highly informative compound haplotypes for the human Y chromosome. Hum Mol Genet 3:115–123 [DOI] [PubMed] [Google Scholar]
- Mohyuddin A, Ayub Q, Qamar R, Zerjal T, Helgason A, Mehdi SQ, Tyler-Smith C (2001) Y-chromosomal STR haplotypes in Pakistani populations. Forensic Sci Int 118:141–146 10.1016/S0379-0738(01)00382-6 [DOI] [PubMed] [Google Scholar]
- Mohyuddin A, Ayub Q, Siddiqi S, Carvalho-Silva DR, Mazhar K, Rehman S, Firasat S, Dar A, Tyler-Smith C, Mehdi SQ (2004) Genetic instability in EBV-transformed lymphoblastoid cell lines. Biochim Biophys Acta 1670:81–83 [DOI] [PubMed] [Google Scholar]
- Nadir E, Margalit H, Gallily T, Ben-Sasson SA (1996) Microsatellite spreading in the human genome: evolutionary mechanisms and structural implications. Proc Natl Acad Sci USA 93:6470–6475 10.1073/pnas.93.13.6470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York [Google Scholar]
- Redd AJ, Agellon AB, Kearney VA, Contreras VA, Karafet T, Park H, de Knijff P, Butler JM, Hammer MF (2002) Forensic value of 14 novel STRs on the human Y chromosome. Forensic Sci Int 130:97–111 10.1016/S0379-0738(02)00347-X [DOI] [PubMed] [Google Scholar]
- Roewer L, Krawczak M, Willuweit S, Nagy M, Alves C, Amorim A, Anslinger K et al. (2001) Online reference database of European Y-chromosomal short tandem repeat (STR) haplotypes. Forensic Sci Int 118:106–113 10.1016/S0379-0738(00)00478-3 [DOI] [PubMed] [Google Scholar]
- Rolf B, Keil W, Brinkmann B, Roewer L, Fimmers R (2001) Paternity testing using Y-STR haplotypes: assigning a probability for paternity in cases of mutations. Int J Legal Med 115:12–15 10.1007/s004140000201 [DOI] [PubMed] [Google Scholar]
- Rozen S, Skaletsky H (2000) Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 132:365–386 [DOI] [PubMed] [Google Scholar]
- Rozen S, Skaletsky H, Marszalek JD, Minx PJ, Cordum HS, Waterston RH, Wilson RK, Page DC (2003) Abundant gene conversion between arms of palindromes in human and ape Y chromosomes. Nature 423:873–876 10.1038/nature01723 [DOI] [PubMed] [Google Scholar]
- Santos FR, Rodriguez-Delfin L, Pena SD, Moore J, Weiss KM (1996) North and South Amerindians may have the same major founder Y chromosome haplotype. Am J Hum Genet 58:1369–1370 [PMC free article] [PubMed] [Google Scholar]
- Skaletsky H, Kuroda-Kawaguchi T, Minx PJ, Cordum HS, Hillier L, Brown LG, Repping S, et al (2003) The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 423:825–837 10.1038/nature01722 [DOI] [PubMed] [Google Scholar]
- Stumpf MP, Goldstein DB (2001) Genealogical and evolutionary inference with the human Y chromosome. Science 291:1738–1742 10.1126/science.291.5509.1738 [DOI] [PubMed] [Google Scholar]
- Subramanian S, Mishra RK, Singh L (2003) Genome-wide analysis of microsatellite repeats in humans: their abundance and density in specific genomic regions. Genome Biol 4:R13 10.1186/gb-2003-4-2-r13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MG, Bradman N, Flinn HM (1999) High throughput analysis of 10 microsatellite and 11 diallelic polymorphisms on the human Y-chromosome. Hum Genet 105:577–581 10.1007/s004390051148 [DOI] [PubMed] [Google Scholar]
- Walsh B (2001) Estimating the time to the most recent common ancestor for the Y chromosome or mitochondrial DNA for a pair of individuals. Genetics 158:897–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber JL (1990) Informativeness of human (dC-dA)n.(dG-dT)n polymorphisms. Genomics 7:524–530 [DOI] [PubMed] [Google Scholar]
- White PS, Tatum OL, Deaven LL, Longmire JL (1999) New, male-specific microsatellite markers from the human Y chromosome. Genomics 57:433–437 10.1006/geno.1999.5782 [DOI] [PubMed] [Google Scholar]
- Y Chromosome Consortium (2002) A nomenclature system of the tree of human Y-chromosomal binary haplogroups. Genome Res 12:339–348 10.1101/gr.217602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerjal T, Xue Y, Bertorelle G, Wells RS, Bao W, Zhu S, Qamar R, Ayub Q, Mohyuddin A, Fu S, Li P, Yuldasheva N, Ruzibakiev R, Xu J, Shu Q, Du R, Yang H, Hurles ME, Robinson E, Gerelsaikhan T, Dashnyam B, Mehdi SQ, Tyler-Smith C (2003) The genetic legacy of the Mongols. Am J Hum Genet 72:717–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.