

Abstract
Admixture mapping is a promising new tool for discovering genes that contribute to complex traits. This mapping approach uses samples from recently admixed populations to detect susceptibility loci at which the risk alleles have different frequencies in the original contributing populations. Although the idea for admixture mapping has been around for more than a decade, the genomic tools are only now becoming available to make this a feasible and attractive option for complex-trait mapping. In this article, we describe new statistical methods for analyzing multipoint data from admixture-mapping studies to detect “ancestry association.” The new test statistics do not assume a particular disease model; instead, they are based simply on the extent to which the sample’s ancestry proportions at a locus deviate from the genome average. Our power calculations show that, for loci at which the underlying risk-allele frequencies are substantially different in the ancestral populations, the power of admixture mapping can be comparable to that of association mapping but with a far smaller number of markers. We also show that, although “ancestry informative markers” (AIMs) are superior to random single-nucleotide polymorphisms (SNPs), random SNPs can perform quite well when AIMs are not available. Hence, researchers who study admixed populations in which AIMs are not available can perform admixture mapping with the use of modestly higher densities of random markers. Software to perform the gene-mapping calculations, “MALDsoft,” is freely available on the Pritchard Lab Web site.
Introduction
In most human populations, linkage disequilibrium (LD) decays rapidly with distance. As a result, genomewide association scans for complex-disease loci will need to type very large numbers of markers—probably 1 marker every few kb or so (Kruglyak 1999; Gabriel et al. 2002). However, there are some human populations in which weak LD extends over very large genetic distances because of recent population admixture. For example, in African Americans—who have ∼20% European ancestry, on average—significant LD has been observed over distances as large as 20 cM (Parra et al. 1998). As long ago as 1988, it was first proposed that this long-range “admixture LD” could enable efficient gene mapping with far fewer markers than would be required for conventional association mapping in an equilibrium population (Chakraborty and Weiss 1988; Stephens et al. 1994). To date, the applications of admixture mapping have been quite limited (Shriver et al. 2003), but the genomic tools have just now matured to the point at which admixture mapping is poised to make important contributions to the study of complex traits.
Admixture LD arises when two or more populations with divergent allele frequencies mix together. In subsequent generations, each individual has some proportion of his or her ancestry that is derived from each of the original contributing populations. Falush et al. (2003a) distinguished three types of LD that arise in such populations and that extend over different scales: (1) “mixture LD,” which occurs even between unlinked markers because of variation among individuals in ancestry proportions; (2) “admixture LD,” which occurs between markers on the same chromosome if they are frequently inherited together from a single ancestral chromosome in one of the original populations; and (3) “background LD,” which occurs over very short distances within populations. Although conventional association mapping makes use of background LD and aims to detect association between the phenotype and particular alleles, admixture mapping uses admixture LD to detect genomic regions with excess correlation between ancestry and phenotype.
The central premise of admixture mapping is that, since many diseases vary in frequency across populations, it is reasonable to hypothesize that the underlying genetic risk variants are also at substantially different frequencies in different populations (Halder and Shriver 2003). However, it should be noted that environmental and social factors correlated with race or ethnicity may also be important in disease etiology. Hence, the mere observation that disease risk correlates with ancestry does not immediately guarantee that there are underlying differences in risk-allele frequencies (Risch et al. 2002). Nonetheless, for numerous diseases, it seems highly plausible that the frequencies of risk alleles vary across the ancestral populations (Halder and Shriver 2003).
When risk-allele frequencies do vary across populations, then recently admixed individuals with a particular disease are likely to have high overall ancestry in the population in which the disease is common, relative to controls (Knowler et al. 1988). More important, near disease loci, affected individuals will have a yet-higher probability of having inherited their chromosomes from the population in which the risk alleles are more frequent. Admixture mapping aims to detect this latter signal, while controlling for the possibility of overall differences in ancestry between cases and controls.
To date, two main types of statistical tests have been proposed for admixture mapping. One class of test uses family data, such as data from parent/affected-offspring trios, and applies the transmission/disequilibrium test (TDT) framework. These tests screen for loci or chromosomal regions where there is overtransmission of chromosomes that derive from one population or another (as opposed to overtransmission of particular alleles, as in the standard TDT) (McKeigue 1997; Zheng and Elston 1999; Lee and Yen 2003). The other class of test, which makes use of unrelated affected individuals, was developed by Paul McKeigue and colleagues (McKeigue 1998; McKeigue et al. 2000). They describe their approach as testing “for association conditional on parental admixture” (McKeigue et al. 1998, p. 241). Their approach aims to find loci where the ancestry of affected individuals is skewed toward one of the ancestral populations, relative to what one would expect, given the estimated ancestry of the parents. Recently, Hoggart et al. (2004) and Patterson et al. (2004) have extended these approaches, using hidden Markov models (HMMs) to make full use of multipoint SNP data for detection of a signal (c.f., McKeigue 1998).
In this study, we describe a pair of new test statistics for admixture mapping. Like the recent methods of Hoggart et al. (2004) and Patterson et al. (2004), our approach uses HMMs to estimate the unobserved ancestry of chromosomes and is thus specifically designed to take advantage of the multipoint information that will be present in genomewide scans. Our approach is relatively nonparametric, in the sense that the test scans the genome for locations where there is an overall skew of ancestry proportions, rather than assuming a specific relationship among the penetrances at the disease locus. We also provide a simple simulation-based method for assessing genomewide significance.
Apart from statistical testing, another key outstanding issue in admixture mapping is how to choose the markers (Shriver et al. 1997; Smith et al. 2001; Collins-Schramm et al. 2002; Rosenberg et al. 2003) and what marker density is needed to capture most of the information about ancestry (McKeigue 1998; McKeigue et al. 2000; Patterson et al. 2004; Smith et al. 2004). To date, most of the discussion has centered on identifying so-called “ancestry informative markers” (AIMs). AIMs are markers that are unusually informative for distinguishing between the populations that have contributed to an admixed sample (Pfaff et al. 2001; Smith et al. 2004). Clearly, such markers will allow successful admixture mapping with fewer genotypes than would be needed if random markers were used. However, for some current genotyping technologies (e.g., chip-based genotyping), it may be easier to use standard predetermined marker sets than to create genotyping assays for new sets. Moreover, AIMs must be identified separately for every new combination of contributing populations. Our results indicate that admixture mapping with randomly selected markers is a feasible alternative to mapping with AIMs.
Software used to perform the calculations described in this article is available on the Pritchard Lab Web site.
Statement of the Problem
Consider the following problem. An investigator wishes to perform admixture mapping in a population that was formed by relatively recent mixing of distinct ancestral groups. The goal is to identify genetic variation that contributes to risk for a particular disease phenotype. The investigator collects (1) a sample of affected individuals from the admixed population, (2) a sample of unaffected or random control individuals, also from the admixed population, and (3) “learning samples” that consist of random individuals from each of the ancestral populations (or a close approximation thereof) and that are used to estimate the ancestral allele frequencies. As discussed below, it is preferable but not required to have both controls and learning samples. All of the sampled individuals are genotyped at a set of ∼1,000–20,000 marker loci spanning the genome. The primary objective of this study is to describe how to make efficient use of such data to identify chromosomal regions that contain disease susceptibility genes.
Models and Notation
Our approach is based on previous models for studying admixed populations developed by Pritchard et al. (2000a) and Falush et al. (2003a) and implemented in the linkage model of the program structure. We start by assuming that there are K distinct populations that contribute ancestry to the study sample. Individuals may have ancestors in more than one population, and we define the “ancestry” of each individual as the proportion of that individual’s genome that is inherited from each of the K populations. The ancestry of individual i is specified by a vector, q(i)={q(i)1, q(i)2, …, q(i)K}, where q(i)k is the proportion of ancestry of individual i from population k and where 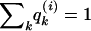 . We will use Q to denote the multidimensional vector containing all the values of q(i).
. We will use Q to denote the multidimensional vector containing all the values of q(i).
The genome of an admixed individual can be visualized as being composed of a series of chromosomal segments or “chunks,” each of which descends as an intact unit, without recombination, from one of the ancestral populations (fig. 1). For individual i, each chromosomal chunk comes from population k independently with probability q(i)k. The breakpoints from one chunk to the next are assumed to occur as a Poisson process, with a rate of r per Morgan. Hence, the average size of chromosomal chunks is 100/r cM. Notice that r can be interpreted roughly as the average time since admixture (Falush et al. 2003a; Patterson et al. 2004).
Figure 1.

Schematic figure showing the mosaic structure of chromosomes in an admixed population. The shaded and unshaded boxes indicate chromosomal segments derived from different ancestral populations. If a susceptibility allele is at a higher frequency in the shaded population, then affected individuals will have increased ancestry from the shaded population at the locus of that gene (vertical line). Our method aims to detect this type of signal.
The data consist of a series of markers along each chromosome; these are used to infer the hidden pattern of chromosomal chunks. The notation z(i,a)l denotes the population of origin (1, …, K) of the ath copy of marker l in individual i. (Here, a distinguishes the two copies of a marker in a diploid individual.) Z refers to the multidimensional vector that contains all the values of z.
Each population is characterized by a list of the allele frequencies at each of the genotyped markers. P denotes the multidimensional vector that contains the allele frequencies at each marker in each population. The allele frequencies will be unknown in advance, but there will usually be samples of nonadmixed representatives from the original populations to assist in their estimation.
As described by Falush et al. (2003a), we use Markov chain–Monte Carlo to sample from the posterior distribution of P, Q, Z, and r, given the genotype data X. The algorithm has been implemented for phased, unphased, and partially phased data and can handle missing data and X-chromosome data. The posterior mean estimates of P, Q, and r will be denoted by 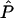 ,
, 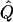 , and
, and 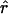 , respectively.
, respectively.
Finally, it will be useful for us to define some posterior average quantities. We use 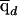 and
and 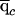 to denote the estimated average ancestry proportions of affected individuals and of controls, respectively. For example, if there are md cases, then
to denote the estimated average ancestry proportions of affected individuals and of controls, respectively. For example, if there are md cases, then
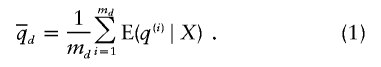 |
Notice that 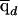 is a vector with K elements (as are the following quantities). Next, let
is a vector with K elements (as are the following quantities). Next, let 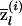 denote the posterior average ancestry of individual i at locus l, evaluated at
denote the posterior average ancestry of individual i at locus l, evaluated at 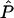 ,
, 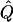 , and
, and 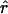 (see appendix A):
(see appendix A):
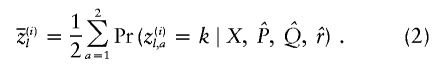 |
The posterior averages of z at locus l among cases and controls will be denoted by 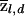 and
and 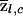 , respectively. For example,
, respectively. For example,
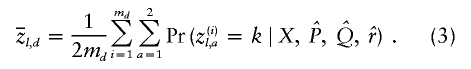 |
We will refer to 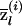 as the “locus-specific ancestry” of an individual (at locus l), and
as the “locus-specific ancestry” of an individual (at locus l), and 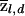 and
and 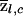 will be referred to as “average locus-specific ancestries” (at locus l).
will be referred to as “average locus-specific ancestries” (at locus l).
Notice that, in these models, the labeling of the K populations is typically arbitrary. When there are predefined learning samples, those can be used to attach numbers to the population samples, but, when there are not, the Monte Carlo algorithm assigns a set of labels at random. The average quantities defined above are intended to be computed with respect to particular labeling. See the article by Pritchard et al. (2000a) and the “Discussion” section for further comments.
Simulation Details
The results presented in this study are based on simulated data generated either under the linkage model described by Falush et al. (2003a) or under a Wright-Fisher model described below. We assumed biallelic markers, two ancestral populations (K=2), and r = 10. The ancestry proportion q(i)1 was modeled independently for each admixed individual, i, as a normally distributed random variable with parameters (0.2, 0.05); values of q(i)1 outside (0, 1) were rejected. Half the learning samples had ancestry proportions of (0, 1), and half had proportions of (1, 0). The values of r and the distribution of q were chosen to approximate the characteristics of the African American population (e.g., Parra et al. 1998; Falush et al. 2003a; Patterson et al. 2004).
For the linkage model simulations, the pattern of ancestry along each chromosome was then simulated in accordance with the linkage model (Falush et al. 2003a), conditional on q(i). For each chromosome in the sample, the ancestral state z(i,a)1 at the first marker was 0 with probability q(i)0, and, otherwise, the ancestral state was 1. The ancestral states at subsequent markers were simulated by
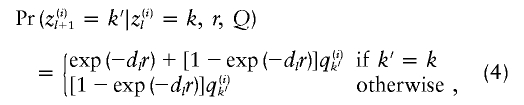 |
where dl denotes the genetic distance from locus l to locus l+1.
The population allele frequencies of markers were simulated under two models. The first model was used to generate AIMs with a prespecified absolute value of δ, the difference between the allele frequencies in the two ancestral populations. For the results presented, δ was set to 0.5 (Shriver et al. 1997). For each marker, the allele frequency of one allele in the first population was drawn from a uniform distribution in either the range [δ, 1] or the range [0, 1−δ], with probability 0.5; the frequency of the same allele in the second population was set so as to guarantee the distance δ. The second model was used to simulate random markers by a simple model of population divergence (Nicholson et al. 2002; Falush et al. 2003a). At each locus, l, the allele frequency, PA, of a hypothetical ancestral population is drawn from a uniform distribution in [0.1, 0.9]; then, conditional on PA, the allele frequency for each population, k, was generated from a beta distribution with parameters [fPA, f(1-PA)], where f is related to the common measure of population divergence, FST, as f=(1-FST)/FST.
The results presented here take either FST=0.1, which is roughly typical of the divergence between human populations on different continents, or FST=0.2, which is representative of the most divergent human populations. For instance, in a large SNP data set, the average three-way FST between African Americans, Asians, and Europeans was 0.12 (Akey et al. 2002). Under our model, at FST=0.1, ∼1.4% of random SNPs would qualify as AIMs (i.e., δ⩾0.5), and 8% would qualify at FST=0.2. For comparison, Rosenberg et al. (2003) reported that 1.9%, 4.6%, and 2.7% of SNPs qualified as AIMs in comparisons of African Americans and European Americans, African Americans and East Asians, and European Americans and East Asians, respectively (data from Akey et al. 2002). Divergence between Native Americans and Europeans (relevant for mapping with Hispanic samples) seems to be higher than that between Europeans and Africans (Rosenberg et al. 2002 [supplemental information]; Risch et al. 2002), but SNP data comparing those populations are currently sparse.
Some data sets were generated to evaluate the effects of misspecifying the allele frequencies. In those simulations, the learning samples were simulated with one set of allele frequencies, generated as described above. Then, the allele frequencies for the admixed individuals were obtained by resampling the allele frequencies from a normal variate centered at the original frequencies and with an SD of 0.05. Once Z and P were specified, the marker data were simulated as binomial draws from the appropriate allele-frequency distributions.
To simulate data under the alternative model, an additional disease locus was included in the simulation at a fixed position but was removed from the data prior to analysis. For illustrative purposes, we assumed relatively large effects: the high-risk allele was at frequencies of 0.01 and 0.60 in the two populations, respectively, and the three genotype penetrances were 0.050, 0.175, and 0.700. Below, we present a more general framework for describing the power of our methods.
Finally, we used Wright-Fisher simulations to simulate a genomewide scan of data with random, unascertained SNPs. The allele frequencies in the two parental populations were simulated as described above, with FST=0.1 and no subsequent mutation. A new, third population was then established with 30,000 individuals whose genotypes were simulated in accordance with the allele frequencies in population 1. Next, we simulated five generations of migration from population 2 into the new population, at a rate of 5% per generation. Then, after another five generations of random mating with no gene flow, 500 cases and 500 controls were sampled from the admixed population. Furthermore, 200 individuals were simulated in accordance with the parental population allele frequencies to serve as learning samples. We simulated data for 23 chromosomes, each with 750 SNPs, at a spacing of 0.2 cM between each SNP. We assumed four disease loci, each with frequencies of the high-risk alleles of 0.05 and 0.60 in populations 1 and 2, respectively. The disease loci were simulated along with the other loci and then were deleted from the final data set prior to analysis. The disease loci were considered to be selectively neutral. Potential case individuals were simulated in the final generation and then were accepted with probability 5-n, where n is the number of low-risk alleles carried by that individual. Controls were sampled at random from the admixed population.
Measuring Variation in Ancestry across the Genome
As described above, in the “Models and Notation” section, the chromosomes of an admixed individual can be visualized as a mosaic of pieces from each of the K contributing populations (figs. 1 and 2). To perform admixture mapping, we need to use the marker data to reconstruct this mosaic structure of the chromosomes.
Figure 2.

Conditional independence structure of the data along a single chromosome. The chromosome is composed of a series of segments, each derived from one of the contributing populations. The zs indicate the population of origin of each marker along the chromosome; the sequence of zs forms a Markov chain with jump rate r. The genotype data (the Xs) are generated by drawing an allele at random from the appropriate population frequencies, given the zs. The genetic map distance between markers 1 and 2 is denoted d1. The model for diploid unphased data is analogous.
Figures 3 and 4 show examples of reconstruction of the locus-specific ancestry of a single individual with the use of AIMs and random markers, respectively (Falush et al. 2003a, 2003b; Patterson et al. 2004). The results illustrate several features of this approach: (1) with relatively dense markers, the data are essentially fully informative about ancestry for both phased and unphased data; (2) as expected, for low marker densities, the quality of the inference is lower for unphased data than for phased data, and it is lower for random markers than for AIMs; and (3) uncertainty in P, Q, and r is relatively minor and contributes very little to the uncertainty in Z (results not shown).
Figure 3.

Reconstruction of locus-specific ancestry for a single individual, using AIMs. The top plot shows the “true” simulated ancestry of a single individual (i.e., whether the individual has 0, 1, or 2 chromosomes inherited from population 1, as a function of position along a chromosome). The lower plots show the posterior mean estimates for this individual on the basis of marker data at different densities, as well as with and without known haplotype phase. These data were simulated under the assumption of an admixture time of 10 generations before the present.
Figure 4.

Reconstruction of locus-specific ancestry for a single individual, using random SNPs with average FST=0.1 between the two ancestral populations. See the legend to figure 3 and the “Simulation Details” section for more information.
To further explore the impact of marker density on the quality of the inference, figure 5 plots the mean square error (MSE) of the locus-specific ancestry estimates under a range of scenarios. Notice that, with AIMs (δ=0.5), relatively accurate estimates of locus-specific ancestry can be obtained by using ∼1 SNP/cM; similar accuracy can be achieved by using ∼3 random SNPs/cM when the FST between the ancestral populations is 0.2 and by using ∼5 random SNPs/cM when FST=0.1. The results plotted in this figure assume that admixture occurred 10 generations ago. If, instead, the admixture had occurred (on average) t generations ago, then the marker densities plotted on the X-axis would need to be multiplied by a factor of t/10. In summary, for a population such as African Americans, in which the average time of admixture is ∼7–10 generations and FST≈0.1, ∼3,000 AIMs—or 15,000 random SNPs—should permit accurate estimation of locus-specific ancestries across the human genome.
Figure 5.

Accuracy of locus-specific ancestry estimation as a function of marker density. The X-axis shows the number of SNPs per cM, and the Y-axis shows the MSE in the estimation of 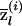 . The three lines correspond to an average FST between the ancestral populations of 0.1 (top line) and 0.2 (middle line) and to AIMs with δ=0.5 (bottom line). The data were treated as unphased. The values at zero density show the MSE when q(i) is known, but there is no additional information about Z at the locus of interest. These data were simulated under the assumption of an admixture time of 10 generations before the present, with mean q(i)=0.2 (see the “Simulation Details” section for specifics).
. The three lines correspond to an average FST between the ancestral populations of 0.1 (top line) and 0.2 (middle line) and to AIMs with δ=0.5 (bottom line). The data were treated as unphased. The values at zero density show the MSE when q(i) is known, but there is no additional information about Z at the locus of interest. These data were simulated under the assumption of an admixture time of 10 generations before the present, with mean q(i)=0.2 (see the “Simulation Details” section for specifics).
Having calculated the locus-specific ancestries for each individual in a sample, we can then plot the average ancestries in the sample as a function of genomic position (fig. 6). Notice that, across most of the region, the average ancestry in cases and controls fluctuates randomly around the average genomewide ancestry. Near the position of a disease mutation (fig. 6, vertical dashed line), the ancestry of cases spikes toward the population in which the risk variant is more common (fig. 6, top panel). Controls show no spike at that position (fig. 6, middle panel), and so if we compute the average ancestry of cases minus the average ancestry of controls at each position, this also shows an upward spike at the position of the disease mutation (fig. 6, bottom panel). In this example, the marker density was relatively low (1 AIM/2 cM), so there is moderate error in estimating the random variation in average ancestries. Nonetheless, the method clearly detects the outlier locus.
Figure 6.

Plots of average ancestry in a sample, as a function of chromosomal location. The gray lines plot the true values, and the black lines plot the estimated averages for cases (top), controls (middle), and the difference in the averages (bottom). The vertical dashed lines indicate the location of a simulated disease gene. Parameters: 800 cases, 800 controls, 200 learning samples, and 500 AIMs at a spacing of 2 cM.
Test Statistics
Figure 6 suggests that there are two types of signal in the data that would indicate the presence of disease variants. The first is that, near a disease locus, the local mean ancestry of cases should diverge from the genomewide mean ancestry of cases. To measure this signal, we define the following test statistic (T1), that uses only cases to test for ancestry association at locus l:
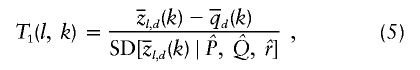 |
where SD(x) indicates the SD of a random variable, x, under the null hypothesis. The numerator of equation (5) computes the difference between the proportion of ancestry from population k at locus l and the overall genomewide proportion of ancestry from population k.
The second type of signal is that, near a disease locus, the local mean ancestry of cases should also diverge from the local mean ancestry of controls. This signal is captured by the case-control test statistic (T2):
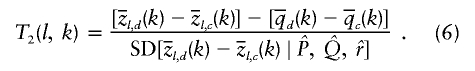 |
The term 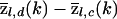 measures the local difference in ancestry between cases and controls. Overall, the numerator tests whether that is different from the genome-average difference in ancestry between cases and controls
measures the local difference in ancestry between cases and controls. Overall, the numerator tests whether that is different from the genome-average difference in ancestry between cases and controls 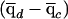 . Hence, this test corrects for the possibility that cases and controls might have different ancestry proportions on average (often referred to as “population stratification”). Indeed, it is to be expected that
. Hence, this test corrects for the possibility that cases and controls might have different ancestry proportions on average (often referred to as “population stratification”). Indeed, it is to be expected that 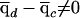 , if the underlying risk variants are at different frequencies in the different ancestral populations.
, if the underlying risk variants are at different frequencies in the different ancestral populations.
When there are just two populations, it does not matter whether these test statistics are computed with respect to one population or the other; only the sign of the test statistic will change. If there are more than two ancestral populations, then the test statistics can be computed separately with respect to each ancestral population. For both of these test statistics, we treat P and r as if they are known without error. In simulations (not shown), we have found that the error in P and r tends to be small and that the vast majority of the uncertainty in 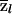 is due to the limited information in the marker data. Test 2 is similar in concept to the “case-control” test proposed in the recent study by Patterson et al. (2004).
is due to the limited information in the marker data. Test 2 is similar in concept to the “case-control” test proposed in the recent study by Patterson et al. (2004).
Both test statistics are constructed in such a way that we can expect them to be asymptotically distributed as standard normals under the null hypothesis (and this is confirmed by the simulations described below). Although we may often have a prior hypothesis that disease loci will produce excess ancestry in the population in which the disease is common, it seems foolhardy to assume for a multifactorial disease that all disease loci will produce excesses in that direction. That is, we will be interested in departures of 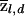 both above and below the expectation; hence, we treat these tests as two-sided.
both above and below the expectation; hence, we treat these tests as two-sided.
The next issue is how to compute the SD terms in the denominators of equations (5) and (6). If the marker data were perfectly informative about ancestry, then, since we assume that Q is known, it would be straightforward to compute the variance of  and
and 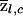 . (These variances would be
. (These variances would be 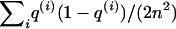 , where n is the number of cases [in T1] or cases plus controls [in T2].) However, the marker data normally leave some ambiguity about ancestry, and this makes the true variances smaller than would be obtained with perfect information. Hence, plugging in the variance computed under the assumption of perfect information would be conservative. (Notice that an analogous problem arises in nonparametric linkage mapping [Kruglyak et al. 1996; Kong and Cox 1997].)
, where n is the number of cases [in T1] or cases plus controls [in T2].) However, the marker data normally leave some ambiguity about ancestry, and this makes the true variances smaller than would be obtained with perfect information. Hence, plugging in the variance computed under the assumption of perfect information would be conservative. (Notice that an analogous problem arises in nonparametric linkage mapping [Kruglyak et al. 1996; Kong and Cox 1997].)
Instead, our solution is to estimate the appropriate SDs by a parametric bootstrapping approach. Specifically, we resimulate marker data with the estimated values 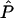 ,
, 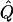 , and
, and 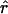 under the null hypothesis. As for the real data, each simulated data set is run through a single iteration of the forward-backward algorithm, described in appendix A, to obtain the posterior mean of
under the null hypothesis. As for the real data, each simulated data set is run through a single iteration of the forward-backward algorithm, described in appendix A, to obtain the posterior mean of
Each iteration of the forward-backward algorithm is quite fast, so it is computationally convenient to perform many replicate simulations. From these, we obtain empirical estimates of 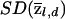 and
and 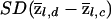 that are then plugged into equations (5) and (6). The estimated SDs vary across markers in accordance with how much information there is at different positions across the genome, and they are bounded between 0 (no information about ancestry) and the SD for the full-information case. We point out that, as an alternative to the normal approximation, the empirical distribution of both tests can be computed by Monte Carlo simulation, and the corresponding empirical P values can be used for hypothesis testing. However, as shown below, the normal approximation turns out to be extremely accurate, so there seems to be little gain in using the empirical distribution. Finally, this parametric bootstrapping approach also provides a convenient method for assessing genomewide significance of the largest signals in the data, as discussed below.
that are then plugged into equations (5) and (6). The estimated SDs vary across markers in accordance with how much information there is at different positions across the genome, and they are bounded between 0 (no information about ancestry) and the SD for the full-information case. We point out that, as an alternative to the normal approximation, the empirical distribution of both tests can be computed by Monte Carlo simulation, and the corresponding empirical P values can be used for hypothesis testing. However, as shown below, the normal approximation turns out to be extremely accurate, so there seems to be little gain in using the empirical distribution. Finally, this parametric bootstrapping approach also provides a convenient method for assessing genomewide significance of the largest signals in the data, as discussed below.
The statistical tests that we have proposed here are relatively nonparametric. The tests are designed to look for regions of significant departure from the normal background variation in average ancestry. This approach differs from the more parametric approaches recently taken by other researchers (Hoggart et al. 2004; Patterson et al. 2004), which implicitly or explicitly assume a particular genetic model at the unobserved disease locus. Parametric approaches will often be more powerful when the assumed model is correct but may perform badly if the genetic model is wrong. Similar issues arise in linkage mapping; our method is somewhat analogous to nonparametric linkage methods that simply test for increased sharing among affected individuals (e.g., Kruglyak et al. 1996).
Distributions of the Test Statistics
We have performed a series of simulations to assess the validity and power of our proposed test. Figure 7 shows an example of both the cases-only and case-control test statistics, for the same data shown in figure 6. As expected, both test statistics lie between −2 and 2 (i.e., the central 95% of the normal distribution) across most of the region. At the position of the disease locus, both tests show highly significant signals.
Figure 7.

Plots of the test-statistic values, as a function of chromosomal location. The gray line plots T1 (cases only), and the black line plots T2 (cases vs. controls). The vertical dashed line indicates the location of a disease gene. As is typical, the signal in this example is larger when the cases-only test is used. The genotype data are the same as those used in figure 6.
Furthermore, we have checked that the test statistics follow the correct distribution by simulating many data sets under the model described above. Figure 8 shows the distribution of both the cases-only and case-control test statistics under the null and alternative models. Under the null model, both tests show an excellent fit to the normal distribution. Under the alternative model, both distributions are substantially shifted away from the null. Notice that, in this example, the cases-only test is substantially more powerful than the case-control test. As discussed below, this result holds in general, although we believe that the case-control test may be more robust to model misspecification (see the “Discussion” section).
Figure 8.

Simulated distributions of the test statistics under the null and alternative hypotheses for the cases-only (black line) and case-control (gray line) strategies. The dotted lines show the theoretical normal density. Parameters: 100 AIMs at a spacing of 2 cM, 350 cases, and 350 controls. See the “Simulation Details” section for further details on the simulations.
We have also conducted simulations to assess whether misspecifying the allele frequencies in the ancestral populations could inflate the type 1 error rate (see the “Simulation Details” section). These simulations were designed to model the situation in which there is fine-scale population structure within the ancestral population (e.g., within West Africans for admixture mapping in African Americans). In that case, the learning samples used to estimate the ancestral population allele frequencies may not be ideal representatives of the ancestral populations. For the parameters we used, the results were indistinguishable from those obtained under a correct model, as in figure 8 (results not shown). This seems to be because most of the information about locus-specific ancestry comes jointly from many markers, so random errors of this type tend to cancel out. We would be much more concerned about the effect of misspecified allele frequencies in a study using a low-density marker map. Patterson et al. (2004) suggested that, by deleting the most significant marker in a peak, one could test whether a signal is overly reliant on one outlier locus. This seems a sensible test of data quality, particularly in sparse maps.
Genomewide Significance
So far, we have discussed how to evaluate the significance of a signal for ancestry association at a single point in the genome. But, for a genomewide scan, it is most common to report the highest peaks, so one needs a method of assessing the genomewide significance of those peaks that takes into account the large number of statistical tests that have been performed.
The “genomewide significance” of a test statistic value, t, is defined as follows. Suppose that the genome-scan experiment were repeated, in the absence of any genuine signal, and that the maximum absolute value of the test statistic anywhere in the genome was t*. The genomewide significance of t is defined as the probability that |t*|⩾|t|.
The traditional approach to multiple testing in linkage analysis applies analytical theory to predict the probability that the maximum signal in a genome scan exceeds a certain value (e.g., Lander and Kruglyak 1995). It seems likely that such theory could be extended to the present situation. Alternatively, the false-discovery–rate approach to multiple testing is robust to dependence among tests and may provide a convenient alternative solution for admixture mapping (Sabatti et al. 2003; Efron 2004). However, the approach that we have developed thus far makes use of a simulation approach to multiple testing, as follows.
Using our parametric bootstrapping approach described above, we can directly estimate the genomewide significance of a signal. That is, each replicate simulation, given 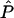 ,
, 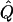 , and
, and 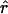 , simulates a genome scan with the appropriate marker spacings and values of marker informativeness. For each simulation, we can simply record the maximum absolute value, t*, and thus obtain an empirical distribution against which each signal, t, can be compared (see fig. 9). Hence, this procedure provides a correction for multiple testing with no additional simulation beyond what is required for all the single-point tests.
, simulates a genome scan with the appropriate marker spacings and values of marker informativeness. For each simulation, we can simply record the maximum absolute value, t*, and thus obtain an empirical distribution against which each signal, t, can be compared (see fig. 9). Hence, this procedure provides a correction for multiple testing with no additional simulation beyond what is required for all the single-point tests.
Figure 9.

Distribution over replicate simulations of the most extreme value of the test statistic T1 in a 400-cM region with no disease loci. This type of simulated distribution can be used to quantify empirical genomewide significance for the most extreme signals observed in a data set.
Simulation of a Genomewide Scan
As described in the “Simulation Details” section, we also used Wright-Fisher simulations to generate data under a more realistic model of continuous admixture over a period of five generations (followed by five generations of random mating before the present). We simulated ∼17,000 markers across a genome of 23 chromosomes, with an intermarker spacing of 0.2 cM. The markers were randomly ascertained, with FST=0.1 between the parental populations.
Figure 10 shows results of the tests for these data. Both tests clearly pick out the four “true” disease loci (but note that the assumed effect sizes are relatively large for these). The threshold for genomewide significance is about ±4. Apart from the four true signals, there are no regions that reach genomewide significance, although two loci approach −4 when the cases-only test is used. (For these plots, we used the genomewide median of 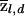 , in place of
, in place of 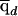 , in computing equations [5] and [6], because the four “true” loci produce a slight upward bias in the estimated values of qd.)
, in computing equations [5] and [6], because the four “true” loci produce a slight upward bias in the estimated values of qd.)
Figure 10.

Mapping results for a simulated genome scan of 500 cases and 500 controls, with four true disease loci. The upper and lower plots show results for the cases-only and case-control tests, respectively. The four large upward peaks on each plot correspond to the four simulated disease loci; for most of the remainder of the genome, the test statistics lie within the dotted lines at ±1.96, corresponding to the central 95% of the null distribution.
However, in additional Wright-Fisher simulations that used smaller population sizes in the admixed population, we found that the cases-only test has a tendency to be anticonservative (results not shown). This appears to result from genetic drift in the admixed population. Even rather small amounts of genetic drift create some extra variance in the test statistic that is not accounted for by the model. Since both cases and controls are similarly affected by drift, the case-control test continues to be reasonably robust. This effect may be important, in practice, unless the admixed population has been large throughout its history.
The Power of Admixture Mapping Compared with the Power of Linkage and Association
This section describes the theoretical performance of the proposed tests in the situation in which there is perfect information about ancestry. We compare the performance of these admixture tests with the performance of linkage and association mapping under similarly idealized conditions.
Theory
Consider a disease susceptibility locus with alleles A and a, which confer different levels of disease risk. Let p1 be the frequency of the A allele in population 1, and let p2 be the frequency of A in population 2. Suppose that all sampled individuals in the admixed population have a fraction of their ancestry from population 1 (q1) and a fraction of their ancestry from population 2 (q2=1-q1). Furthermore, we assume that the marker data are completely informative about ancestry at the disease locus and that q1 is known; hence, our calculations will represent an upper bound on the power that can be achieved in practice.
Under these conditions (i.e., q1 constant across all individuals and perfectly informative marker data), the two tests that we have proposed can be rewritten more simply as
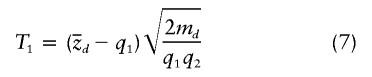 |
and
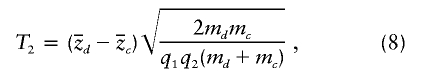 |
where 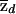 is the sample proportion of case chromosomes (and
is the sample proportion of case chromosomes (and 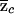 is the sample proportion of control chromosomes) that derive from population 1 at a particular locus and where md and mc are the total numbers of case and control individuals, respectively. The square root terms on the right-hand side of expressions (7) and (8) are the inverses of the SDs of
is the sample proportion of control chromosomes) that derive from population 1 at a particular locus and where md and mc are the total numbers of case and control individuals, respectively. The square root terms on the right-hand side of expressions (7) and (8) are the inverses of the SDs of 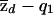 and
and 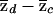 , respectively, under the null hypothesis. Under the null hypothesis, both tests are asymptotically normal, with mean 0 and variance 1.
, respectively, under the null hypothesis. Under the null hypothesis, both tests are asymptotically normal, with mean 0 and variance 1.
To study the performance of the tests when the A and a alleles confer different risks, we assume a multiplicative model of disease risk. In this model, individuals with genotypes AA, Aa, and aa have the disease with probabilities γ2f, γf, and f, respectively. For simplicity, we assume that the control individuals are sampled randomly with respect to phenotype.
Now, let q*1 represent the probability that a chromosome from a case individual is from population 1, at the disease locus. Our test aims to detect that q*1≠q1. After some algebra, it can be shown that
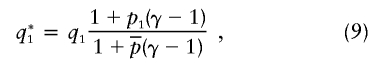 |
where 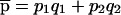 is the overall frequency of A in the admixed population. Under the multiplicative model, the populations of origin of the two chromosomes in an affected individual are independent. As expected, if p1=p2, if γ=1, or if q1=0 or 1, then q*1=q1, in which case the disease locus produces no signal. Under the alternative hypothesis, the two tests are asymptotically normal, with means and variances as follows:
is the overall frequency of A in the admixed population. Under the multiplicative model, the populations of origin of the two chromosomes in an affected individual are independent. As expected, if p1=p2, if γ=1, or if q1=0 or 1, then q*1=q1, in which case the disease locus produces no signal. Under the alternative hypothesis, the two tests are asymptotically normal, with means and variances as follows:
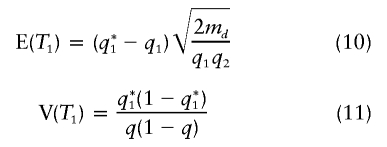 |
and
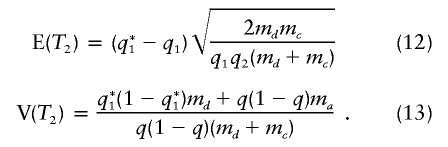 |
Notice that, for md=mc, the expected value of the test statistic T2 is smaller than that of T1 by a factor of 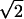 , despite the genotyping of twice as many individuals.
, despite the genotyping of twice as many individuals.
We will report power in terms of the sample size required to achieve a two-sided significance level α with probability β. To do this, we solve 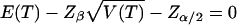 for md and mc (where T stands for either T1 or T2) (Risch and Merikangas 1996). The required sample size will be a function of p1, p2, q1, and γ.
for md and mc (where T stands for either T1 or T2) (Risch and Merikangas 1996). The required sample size will be a function of p1, p2, q1, and γ.
In table 1, we report the sample sizes required to achieve β=80% power (Z0.8=0.84) at a P value of α=2.5×10-5 (Z2.5×10-5=4.06). This P value was arrived at by supposing that we aim to reach genomewide significance at the .05 level in a two-sided test and by assuming that the genome contains ∼1,000 independent tests (i.e., that the correlation between admixture tests decays over distances of ∼3 cM). The results reported in table 1 do not consider the possibility that some genotyping effort might also be spent on learning samples.
Table 1.
Comparison of the Power of Admixture Mapping with the Power of Association and Linkage Analyses[Note]
|
Admixture Mapping |
Association and Linkage |
|||||||
| Sample SizeRequired for |
Sample SizeRequired for |
|||||||
| Alleleswith | p1 | p2 | q1 | Test 1 | Test 2 | p | Association | Linkage |
| γ=4.0 | ||||||||
| .05 | .60 | .8 | 65 | 251 | .05 | 512 | 1,070 | |
| .05 | .30 | .8 | 236 | 924 | .10 | 298 | 552 | |
| .05 | .60 | .2 | 152 | 644 | .30 | 179 | 479 | |
| .05 | .30 | .2 | 387 | 1,593 | .60 | 237 | 1,414 | |
| γ=2.0 | ||||||||
| .05 | .60 | .8 | 347 | 1,362 | .05 | 2,604 | 46,756 | |
| .05 | .30 | .8 | 1,483 | 5,873 | .10 | 1,430 | 16,169 | |
| .05 | .60 | .2 | 526 | 2,155 | .30 | 715 | 6,073 | |
| .05 | .30 | .2 | 1,834 | 7,420 | .60 | 771 | 10,367 | |
Note.— Sample sizes (total numbers of individuals) required to achieve 80% power in genomewide studies. For admixture mapping, the models are parameterized by the subpopulation risk-allele frequencies p1 and p2 and the admixture proportions q1 and 1-q1; for linkage and association, we assume a single nonadmixed population with risk-allele frequency p. Hence, the left- and right-hand sides of the table are not exactly comparable, but they do allow a loose comparison of power to detect alleles with γ=2.0 and γ=4.0, respectively, under these different types of mapping strategies. The results show the required numbers of cases plus controls; the linkage column reports twice the required number of sib pairs. The significance levels required for genomewide significance are lowest for linkage and highest for association, as described in the text. The admixture tests have no power if p1=p2.
Table 1 also displays a comparison of the power of admixture mapping with the power of linkage studies using affected sib pairs and case-control studies of association in nonadmixed populations. Our calculations follow those of Risch and Merikangas (1996). For all three study designs, we assume the same underlying disease model. The linkage calculations assume that the marker data are completely informative about inheritance. The association calculations assume that there is only one variant in the region that affects susceptibility and that this variant is genotyped. Following Risch and Merikangas (1996), we require significance at P=10-4 for linkage and at P=5×10-8 for association. The linkage results presented here correct a computational error in the original study by Risch and Merikangas (1996) (see Risch and Merikangas 1997). The required number of cases plus random controls for a case-control study to achieve suitable power in a panmictic population is approximately (Zα+Zβ)2(p*+p)(2-p*-p)(p*-p)-2/4, where p is the frequency of the risk allele, p*=pγ(pγ+1-p)-1 is the frequency of the risk allele in cases, and Zα=5.45.
Predicted Power and Comparison with Linkage and Association
Table 1 describes the power of four types of study design under idealized conditions: (1) cases-only admixture mapping, (2) case-control admixture mapping, and two standard approaches for nonadmixed populations—namely, (3) linkage mapping using sib pairs and (4) case-control association. One result of these analyses is that the case-control test is always less powerful than the cases-only test, requiring ∼4-fold more individuals to achieve comparable power. This is because the cases-only test compares the local ancestry proportion (which is moderately variable) with the genome average ancestry (which is known quite accurately), whereas the case-control test compares two local ancestry proportions, both of which are variable.
However, the case-control test is more robust when there is genetic drift or selection or when the population allele frequencies are not well estimated. Therefore, it seems that a sensible compromise that minimizes genotyping costs is to screen the genome by use of cases only and then to check regions with promising signals by use of control individuals as well.
The power comparisons across study designs are less straightforward, because the different study types differ both in their underlying assumptions and in the cost and feasibility of genotyping and sample collection. First, admixture mapping is only powerful when there are substantial differences in disease-allele frequencies between the ancestral populations; for many diseases, the existence of such genes seems quite plausible but is unproven at this time. Association mapping will perform well when there is a single variant affecting susceptibility but may perform poorly for genes with multiple variants. Furthermore, most current plans for association mapping aim to genotype a subset of the markers and to detect causative variants by LD, which will further reduce power from the theoretical maximum.
Second, the amount of genotyping required for these studies ranges from ∼103 markers for a genomewide linkage scan to ∼104 markers for admixture mapping to ∼106 markers for a moderately complete genomewide association scan. With currently available genotyping technologies, admixture mapping is already within reach for medium-sized studies, whereas genomewide association is still too expensive to be routine.
These caveats aside, it is still interesting to compare across the study types. As pointed out by Risch and Merikangas (1996) and as illustrated in table 1, under ideal conditions association mapping is far more powerful than linkage. When the population disease-allele frequencies are very different, admixture mapping shares the same advantageous statistical properties as association mapping and can be substantially better than linkage.
In general, one might expect admixture mapping to have lower power in a single-point test than association mapping, because normally ancestry only provides incomplete information about whether the underlying disease mutation is present; in contrast, we assume for these calculations that, in association mapping, the actual disease marker is typed. Indeed, our test 2 (cases vs. controls) does always have lower power than association mapping, but, for certain parameter combinations, our test 1 (cases only) can actually perform better than association in a genomewide scan. Test 1 enjoys the advantage of comparing the case ancestry at each locus with the average case ancestry, which can be estimated very accurately, whereas test 2 and association mapping both look for a difference between two estimated frequencies (in cases and controls, respectively). Furthermore, the penalty for multiple testing is substantially smaller in admixture mapping than in association mapping.
In summary, for disease alleles with frequencies that differ greatly across populations, admixture mapping is much more powerful than linkage mapping and can have power that is comparable to association mapping. However, admixture mapping will have little or no power to find disease alleles with frequencies that are relatively uniform across populations. The genotyping effort required for admixture mapping is slightly more than that required for linkage mapping and far less than that required for association mapping.
Discussion
In this study, we have described two tests for detecting “ancestry association” in admixed populations. The cases-only test and, to a lesser extent, the case-control test can potentially deliver much of the power of genomewide association mapping at a small fraction of the cost. These tests are potentially far more powerful than the widely used affected sib pairs study design for linkage analysis.
Of course, the caveat with admixture mapping is that this method will only work well if the underlying risk variants are at substantially different frequencies in the original populations. At the time of writing, there are not enough data on complex-trait variants to know how often this will be true. However, population variation in risk-allele frequencies seems a sensible working hypothesis for many diseases with prevalences that vary substantially across ethnic groups. Additional preliminary evidence might be obtained by testing whether phenotype status is correlated with ancestry within the admixed group, as seems to be the case for prostate cancer (Kittles et al. 2002). In any case, there is now great interest within the human genetics community in admixture mapping, and there will soon be hard data to start addressing this question.
As we have shown here, the cases-only study design is far more powerful than the case-control design. This raises the question of whether there is any point in collecting and genotyping controls. Although the models suggest that there is no benefit in having the controls, in practice we believe that the controls provide an important check that the test is performing correctly in the regions where there are signals. In particular, showing a difference between cases and controls can help rule out the possibilities that misspecified allele frequencies have produced a signal or that a shift in ancestry is due to some other factor, such as natural selection or genetic drift. Our simulations suggest that the cases-only test may be surprisingly sensitive to genetic drift, unless the admixed population is reasonably large. Controls can also help improve the allele-frequency estimates in the cases-only test. An economical genotyping strategy may be to type the controls only in regions where the cases show signals, plus enough additional markers to estimate the ancestries of the controls accurately.
An important issue for admixture mapping is to decide how many markers to genotype and which markers to choose. Smith et al. (2004) have developed a SNP map of some 3,000 unusually informative markers for use with African American samples. Certainly, this will be an important resource for admixture mapping in that population, but we wish to point out that admixture mapping is already feasible in other admixed populations where such resources are not yet available. In admixed populations for which FST⩾0.1 between the ancestral populations, as few as 10,000–15,000 random SNPs will capture most of the information about locus-specific ancestry. The cost of genotyping this many markers is becoming increasingly reasonable.
We have not considered microsatellites in this study, because high-throughput SNP genotyping seems to be becoming more widespread than microsatellite genotyping. However, microsatellites tend to be much more informative than SNPs for ancestry estimation (Rosenberg et al. 2003) and therefore may represent a sensible study approach in some situations—especially for studies of admixture and hybridization in nonmodel organisms (e.g., Rieseberg et al. 1999) for which high-density SNP maps may not be available.
Although we have focused here on discrete binary traits, our general framework can also handle quantitative traits in a natural way. Suppose that xi is the trait value of individual i and that 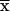 is the mean of xi across m sampled individuals. Then one test statistic is
is the mean of xi across m sampled individuals. Then one test statistic is
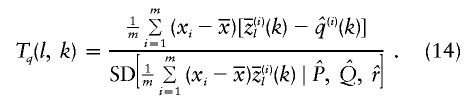 |
This test is also asymptotically normal, and, again, both positive and negative tails of the distribution are of potential interest.
As with linkage analysis, positional cloning of admixture mapping peaks would normally be followed by dense marker association mapping across the region. Admixture mapping peaks will normally be much narrower than linkage peaks, suggesting that fine mapping should be easier. One plausible concern about fine mapping in admixed populations is that, as discussed above, admixture LD can extend over very large distances in such populations. Does this mean that it might be difficult to localize the mutations? In fact, at least for African Americans, the strength of short-range LD is quite similar to that in Africans (Gabriel et al. 2002). Thus, there would seem to be no problem with proceeding to fine mapping, at least in African Americans. The apparent discrepancy between LD at short and long scales is presumably because background LD is very strong at short distances but decays very rapidly, whereas admixture LD is relatively weak at all distances but decays slowly. In African Americans, admixture LD contributes little to the total LD at short distances but produces measurable LD at cM distances, where there is no background LD. Nonetheless, association tests in admixed populations are potentially subject to false positives due to the variation in ancestry. Therefore, it is important to use methods that can control for this effect (e.g., Pritchard et al. 2000b; Hoggart et al. 2003).
We turn now to a technical issue related to our approach. Hoggart et al. (2003) criticized an earlier study by Pritchard et al. (2000b) for using a two-stage test analogous to the one used here, in which ancestry estimates from the program structure were “plugged in” to a test of association. Their first criticism was that this procedure does not account for uncertainty in the ancestry estimates. Second, they worried that, in the absence of learning samples, there is nonidentifiability of the population labels. The nonidentifiability means that, in theory at least, the labels might switch during a run of the Markov chain, in which case mean ancestry estimates would not be meaningful. Although these concerns are theoretically plausible, extensive simulations of the admixture mapping tests presented here, as well as simulations of the STRAT test (Pritchard et al. 2000b), show that, in practice, the statistical tests are indeed correctly calibrated under the null hypothesis. Moreover, we have a great deal of experience with the program structure and we have found that label switching is not a concern, in practice, for informative data sets. Besides, there are some practical advantages to the two-stage process. First, the two-stage process makes the output much more transparent and interpretable for the end user. Second, it makes it much easier for users to take the ancestry estimates and develop other tests of association that are appropriate for their own data (e.g., Thornsberry et al. 2001).
In summary, we have presented powerful multipoint methods for detecting ancestry association in admixed populations. Now that dense genomewide SNP panels are available in humans and SNP genotyping costs are becoming increasingly reasonable, we believe that admixture mapping is poised to make an important contribution to the dissection of complex traits.
Acknowledgments
We thank our colleagues Daniel Falush and Matthew Stephens, who helped to create the linkage model of structure used here; Noah Rosenberg, for helpful discussion; and two anonymous reviewers, for their careful reading of the manuscript. This work was supported by the National Institutes of Health (grant GR 2772 to J.K.P.).
Appendix A : HMM
To compute the admixture mapping test statistic, estimates of parameters P, Q, and r are obtained from structure under the linkage model. The estimation of the hidden states of the Markov chain for Z is then performed independently for each individual by use of the Baum-Welch algorithm on the basis of the probabilities defined below. These computations are similar to those described by Falush et al. (2003a), except that the goal here is to compute the marginal posterior assignment probabilities at each locus rather than to sample a single realization from the joint posterior distribution.
For each chromosome from each individual, we define the forward probabilities as βlk=Pr(x1, …, xl, zl=k|P, r, Q) and the backward probabilities as αlk=Pr(xl+1, …, xL|zl=k, P, r, Q), which are defined for all states k and for all loci from 1 to L. It follows that βlkαlk=Pr(x1, …, xL, zl=k|P, r, Q), and the likelihood can be computed as
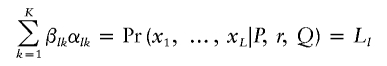 |
for each given l. The algorithm used here differs slightly from the one implemented under the linkage model in structure, as the interest here is in computing the conditional probabilities,
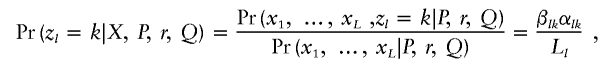 |
for all loci l and all populations k. We start by providing the algorithm details for the case of complete phase information. Recalling that the equation
defines the transition probabilities of the Markov chain (eq. [4]) and that pklj is the frequency of allele j at locus l in population k, we find that
for k=1, …, K, and β(l+1)k is obtained recursively from βlk as
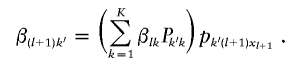 |
The computation of βlk for l=1, …, L and k=1, …, K allows us to obtain the forward probabilities. Starting with αLk=1, the backward probabilities are then computed as
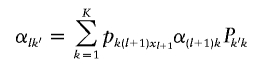 |
for l=L-1, …, 1 and k=1, …, K.
When phase information is missing or only partially known, the forward probabilities need to be expressed as
where the superscript (1) refers to the first allele copy and the superscript (2) refers to the second allele copy at each locus. Analogously, the backward probability at each locus becomes
and the resulting joint conditional probability of the ancestral states in the two allele copies is
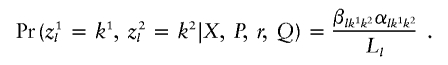 |
The algorithm is implemented both for fully phased data and for unphased data. Let bl represent the probability that the first alleles of adjacent loci l and l+1 are on the same chromosome. For unphased data, the order of the allele copies is random, and so bl is set to 0.5. Under this scenario, we obtain the forward probability at the first locus as
for k1=1, …, K and k2=1, …, K, and the full forward recursion is then computed as
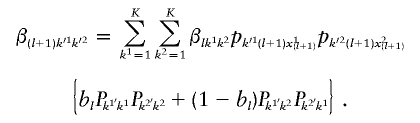 |
As for the backward probabilities, we obtain αlk1′k2′=1 and
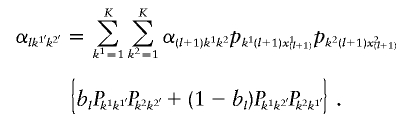 |
The actual implementation of this algorithm is slightly more complicated, since we rescale the probabilities periodically during the forward and backward steps, to avoid numerical underflow.
Electronic-Database Information
The URL for data presented herein is as follows:
- Pritchard Lab Web site, http://pritch.bsd.uchicago.edu
References
- Akey JM, Zhang G, Zhang K, Jin L, Shriver MD (2002) Interrogating a high-density SNP map for signatures of natural selection. Genome Res 12:1805–1814 10.1101/gr.631202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty R, Weiss KM (1988) Admixture as a tool for finding linked genes and detecting that difference from allelic association between loci. Proc Natl Acad Sci USA 85:9119–9123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins-Schramm HE, Phillips CM, Operario DJ, Lee JS, Weber JL, Hanson RL, Knowler WC, Cooper R, Li H, Seldin MF (2002) Ethnic-difference markers for use in mapping by admixture linkage disequilibrium. Am J Hum Genet 70:737–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efron B (2004) Large-scale simultaneous hypothesis testing: the choice of a null hypothesis. J Am Stat Assoc 99:96–104 [Google Scholar]
- Falush D, Stephens M, Pritchard JK (2003a) Inference of population structure: extensions to linked loci and correlated allele frequencies. Genetics 164:1567–1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falush D, Wirth T, Linz B, Pritchard JK, Stephens M, Kidd M, Blaser MJ, Graham DY, Vacher S, Perez-Perez GI, Yamaoka Y, Megraud F, Otto K, Reichard U, Katzowitsch E, Wang XY, Achtman M, Suerbaum S (2003b) Traces of human migrations in Helicobacter pylori populations. Science 299:1582–1585 10.1126/science.1080857 [DOI] [PubMed] [Google Scholar]
- Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly MJ, Altshuler D (2002) The structure of haplotype blocks in the human genome. Science 296:2225–2229 10.1126/science.1069424 [DOI] [PubMed] [Google Scholar]
- Halder I, Shriver MD (2003) Measuring and using admixture to study the genetics of complex diseases. Human Genomics 1:52–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoggart CJ, Parra EJ, Shriver MD, Bonilla C, Kittles RA, Clayton DG, McKeigue PM (2003) Control of confounding of genetic associations in stratified populations. Am J Hum Genet 72:1492–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoggart CJ, Shriver MD, Kittles RA, Clayton DG, McKeigue PM (2004) Design and analysis of admixture mapping studies. Am J Hum Genet 74:965–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittles RA, Chen WD, Panguluri RK, Ahaghotu C, Jackson A, Adebamowo CA, Griffin R, Williams T, Ukoli F, Adams-Campbell L, Kwagyan J, Isaacs W, Freeman V, Dunston GM (2002) CYP3A4-V and prostate cancer in African Americans: causal or confounding association because of population stratification? Hum Genet 110:553–560 10.1007/s00439-002-0731-5 [DOI] [PubMed] [Google Scholar]
- Knowler WC, Williams RC, Pettitt DJ, Steinberg AG (1988) Gm3-5,13,14 and type 2 diabetes mellitus—an association in American Indians with genetic admixture. Am J Hum Genet 43:520–526 [PMC free article] [PubMed] [Google Scholar]
- Kong A, Cox NJ (1997) Allele-sharing models: LOD scores and accurate linkage tests. Am J Hum Genet 61:1179–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruglyak L (1999) Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nat Genet 22:139–144 10.1038/9642 [DOI] [PubMed] [Google Scholar]
- Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES (1996) Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet 58:1347–1363 [PMC free article] [PubMed] [Google Scholar]
- Lander E, Kruglyak L (1995) Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11:241–247 10.1038/ng1195-241 [DOI] [PubMed] [Google Scholar]
- Lee WC, Yen YC (2003) Admixture mapping using interval transmission/disequilibrium tests. Ann Hum Genet 67:580–588 10.1046/j.1529-8817.2003.00052.x [DOI] [PubMed] [Google Scholar]
- McKeigue PM (1997) Mapping genes underlying ethnic differences in disease risk by linkage disequilibrium in recently admixed populations. Am J Hum Genet 60:188–196 [PMC free article] [PubMed] [Google Scholar]
- ——— (1998) Mapping genes that underlie ethnic differences in disease risk: methods for detecting linkage in admixed populations, by conditioning on parental admixture. Am J Hum Genet 63:241–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeigue PM, Carpenter JR, Parra EJ, Shriver MD (2000) Estimation of admixture and detection of linkage in admixed populations by a Bayesian approach: application to African-American populations. Ann Hum Genet 64:171–186 10.1046/j.1469-1809.2000.6420171.x [DOI] [PubMed] [Google Scholar]
- Nicholson G, Smith AV, Jónsson F, Gústafsson O, Stefansson K, Donnelly P (2002) Assessing population differentiation and isolation from single nucleotide polymorphism data. J R Stat Soc [Ser B] 64:695–715 [Google Scholar]
- Parra EJ, Marcini A, Akey J, Martinson J, Batzer MA, Cooper R, Forrester T, Allison DB, Deka R, Ferrell RE, Shriver MD (1998) Estimating African American admixture proportions by use of population-specific alleles. Am J Hum Genet 63:1839–1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N, Hattangadi N, Lane B, Lohmueller KE, Hafler DA, Oksenberg JR, Hauser SL, Smith MW, O'Brien SJ, Altshuler D, Daly MJ, Reich D (2004) Methods for high-density admixture mapping of disease genes. Am J Hum Genet 74:979–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaff CL, Parra EJ, Bonilla C, Hiester K, McKeigue PM, Kamboh MI, Hutchinson RG, Ferrell RE, Boerwinkle E, Shriver MD (2001) Population structure in admixed populations: effect of admixture dynamics on the pattern of linkage disequilibrium. Am J Hum Genet 68:198–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P (2000a) Inference of population structure using multilocus genotype data. Genetics 155:945–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Rosenberg NA, Donnelly P (2000b) Association mapping in structured populations. Am J Hum Genet 67:170–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieseberg LH, Whitton J, Gardner K (1999) Hybrid zones and the genetic architecture of a barrier to gene flow between two sunflower species. Genetics 152:713–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risch N, Burchard E, Ziv E, Tang H (2002) Categorization of humans in biomedical research: genes, race and disease. Genome Biol 3:comment2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risch N, Merikangas K (1996) The future of genetic studies of complex human diseases. Science 273:1516–1517 [DOI] [PubMed] [Google Scholar]
- ——— (1997) Genetic analysis of complex diseases: response. Science 275:1329–1330 [Google Scholar]
- Rosenberg NA, Pritchard JK, Weber JL, Cann HM, Kidd KK, Zhivotovsky LA, Feldman MW (2002) Genetic structure of human populations. Science 298:2381–2385 10.1126/science.1078311 [DOI] [PubMed] [Google Scholar]
- ——— (2003) Response to comment on “Genetic structure of human populations.” Science 300:1877 [DOI] [PubMed] [Google Scholar]
- Sabatti C, Service S, Freimer N (2003) False discovery rate in linkage and association genome screens for complex disorders. Genetics 164:829–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shriver MD, Parra EJ, Dios S, Bonilla C, Norton H, Jovel C, Pfaff C, Jones C, Massac A, Cameron N, Baron A, Jackson T, Argyropoulos G, Jin L, Hoggart CJ, McKeigue PM, Kittles RA (2003) Skin pigmentation, biogeographical ancestry and admixture mapping. Hum Genet 112:387–399 [DOI] [PubMed] [Google Scholar]
- Shriver MD, Smith MW, Jin L, Marcini A, Akey JM, Deka R, Ferrell RE (1997) Ethnic-affiliation estimation by use of population-specific DNA markers. Am J Hum Genet 60:957–964 [PMC free article] [PubMed] [Google Scholar]
- Smith MW, Lautenberger JA, Shin HD, Chretien JP, Shrestha S, Gilbert DA, O’Brien SJ (2001) Markers for mapping by admixture linkage disequilibrium in African American and Hispanic populations. Am J Hum Genet 69:1080–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MW, Patterson N, Lautenberger JA, Truelove AL, McDonald GJ, Waliszewska A, Kessing BD, et al (2004) A high-density admixture map for disease gene discovery in African Americans. Am J Hum Genet 74:1001–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens JC, Briscoe D, O’Brien SJ (1994) Mapping admixture linkage disequilibrium in human populations: limits and guidelines. Am J Hum Genet 55:809–824 [PMC free article] [PubMed] [Google Scholar]
- Thornsberry JM, Goodman MM, Doebley J, Kresovich S, Nielsen D, Buckler ES (2001) Dwarf8 polymorphisms associate with variation with flowering time. Nat Genet 28:286–289 10.1038/90135 [DOI] [PubMed] [Google Scholar]
- Zheng C, Elston RC (1999) Multipoint linkage disequilibrium mapping with particular reference to the African-American population. Genet Epidemiol 17:79–101 [DOI] [PubMed] [Google Scholar]