

Abstract
A powerful approach to mapping the genes for complex traits is to study isolated founder populations, in which genetic heterogeneity and environmental noise are likely to be reduced and in which extended genealogical data are often available. Using graph theory, we applied an approach that involved sampling from the large number of pairwise relationships present in an extended genealogy to reconstruct sets of subpedigrees that maximize the useful information for linkage mapping while minimizing calculation burden. We investigated, through simulation, the properties of the different sets in terms of bias in identity-by-descent (IBD) estimation and power decrease under various genetic models. We applied this approach to a small isolated population from Sardinia, the village of Talana, consisting of a unique large and complex pedigree, and performed a genomewide search through variance-components linkage analysis for serum lipid levels. We identified a region of significant linkage on chromosome 2 for total serum cholesterol and low-density lipoprotein (LDL) cholesterol. Through higher-density mapping, we obtained an increased linkage for both traits on 2q21.2-q24.1, with a LOD score of 4.3 for total serum cholesterol and of 3.9 for LDL cholesterol. A replication study was performed in an independent and larger set from a genetically differentiated isolated population of the same region of Sardinia, the village of Perdasdefogu. We obtained consistent linkage to the region for total serum cholesterol (LOD score 1.4) and LDL cholesterol (LOD score 2.2), with a level of concordance uncommon for complex traits, and refined the location of the quantitative-trait locus. Interestingly, the 2q21.1-22 region has also been linked to premature coronary heart disease in Finns, and, in the adjacent 2q14 region, significant linkage with triglycerides has been reported in Hutterites.
Introduction
An important route for complex disease mapping is through the study of normal physiological variation of quantitative risk factors. Intermediate measured phenotypes, which are assumed to be functionally related to or to potentially underlie a broader phenotype (or disease), may have stronger genetic determinants than the downstream phenotype they mediate.
Elevated levels of total cholesterol (TC), low-density lipoprotein (LDL) cholesterol (LDL-C), and triglyceride (TG), as well as decreased levels of high-density lipoprotein (HDL) cholesterol (HDL-C), are major risk factors for cardiovascular diseases (CVD) (Miller and Miller 1975; Rhoads et al. 1976; Gordon et al. 1977, 1989; Breslow 1988; Manninen et al. 1988; Criqui et al. 1993; Gardner et al. 1996; Hokanson and Austin 1996; Stampfer et al. 1996; Murray and Lopez 1997; Lamarche et al. 1998), the leading cause of morbidity and mortality in industrialized countries. Variation in serum lipid concentrations among individuals results from multiple genetic factors and their complex interactions with environmental factors, diet, and lifestyle. Different polymorphisms of genes encoding proteins involved in lipid metabolism have been associated with variations in lipid levels. The magnitude of the effect of each of these polymorphisms is generally small but, when combined, may lead to major changes. Occasionally, a single mutation capable of causing abnormalities of lipid metabolism is genetically transmitted as a familial dyslipidemia. Major mutations have been described in genes for the LDL receptor (LDLR [MIM 606945]) (Umans-Eckenhausen et al. 2001), apolipoprotein B (APOB [MIM 107730]) (Schonfeld 2003), and LDLR-adaptor protein (ARH [MIM (605747]) (Garcia et al. 2001b). Important but uncommon polymorphisms associated with variations in lipid levels have also been reported (Patsch et al. 1994; Hegele et al. 1995; Dallinga-Thie et al. 1996; Harris et al. 1998; Miettinen et al. 1998; Acton et al. 1999; Brooks-Wilson et al. 1999; Hagberg et al. 2000; Ikeda et al. 2001; Zambon et al. 2001; Shachter 2001; Evans and Kastelein 2002; Ledmyr et al. 2002; Boekholdt and Thompson 2003). Lipid concentrations in the general population, even in the absence of major mutations, are also highly heritable (Hunt et al. 1989; Perusse et al. 1997; Higgins 2000). The identification of new QTLs contributing to interindividual variation has proved difficult (Garcia et al. 2001a). Linkage analysis has the potential to identify regions of the genome not previously recognized to be involved in lipid metabolism, although these studies can be hampered by the small effects on phenotypes, as well as by the genetic heterogeneity of the underlying variants and their interactions with other genetic and environmental factors.
A promising strategy to aid in the search for genes is to focus on genetic isolates with small numbers of founders, in which the expected number of trait-influencing variants should be reduced compared with outbred populations (Lander and Schork 1994; Wright et al. 1999; Shifman and Darvasi 2001). Furthermore, the environment of the individuals in these populations is usually quite uniform.
We focus our studies on Ogliastra, a mountain area of central-eastern Sardinia. Ogliastra is an anciently populated region consisting of 23 small, isolated villages, whose founders originated from the same Neolithic population but that had little historical admixture, because of geographic and culture barriers. Isolation, small population size, high endogamy, and inbreeding have led to marked genetic differentiation among subpopulations within Ogliastra, as a consequence of founder effects and genetic drift (Fraumene et al. 2003). These villages offer great advantages for the study of complex traits—first, because of their expected increased genetic and environmental homogeneity, and, second, because of the opportunity for replication in one village of an initial finding obtained in another village, since they are very well genetically differentiated. The present study was performed on two villages of Ogliastra: Talana (1,200 inhabitants) and Perdasdefogu (2,400 inhabitants). These populations rank among the most genetically homogenous compared with other European populations (Fraumene et al. 2003).
An additional feature of these isolated populations is the availability of accurate genealogical records that allow us to trace the ancestry of each extant individual and to reconstruct a large and complex genealogy. We collected genealogical data for many of the Ogliastra villages and stored them in a database, which allowed us to place individuals from each village within large and complex genealogies. Although simulations and theoretical power calculations demonstrate that large pedigrees provide more power for quantitative-trait analyses than do smaller families or sib pairs (Wijsman and Amos 1997; Williams et al. 1997; Williams and Blangero 1999), complex pedigrees pose computational challenges for linkage methods based on identity-by-descent (IBD) sharing estimation. In particular, exact multipoint calculations are feasible only for pedigrees of moderate size (Lander and Green 1987; Kruglyak and Lander 1998; Gudbjartsson et al. 2000; Markianos et al. 2001, Abecasis et al. 2002), whereas, for large pedigrees, approximate IBD estimation, such as Markov-chain Monte Carlo (Sobel and Lange 1996; Heath 1997) and correlation-based (Almasy and Blangero 1998) methods, must be used. However, these methods are very time consuming for large genealogies, especially in the context of a genomewide search (GWS), and some are unable to deal with extremely complex pedigrees with many inbreeding loops—for example, the correlation-based method, which relies on predefined relationship classes and doesn’t support some of the unusual bilineal relationships that exist in isolated populations.
Abney et al. (2002) have proposed new methods for linkage and association mapping of QTLs in complex, inbred pedigrees; these methods make extensive use of the pedigree information while keeping the computations simple and efficient. Their methods are based on the existence of regions that are homozygous by descent in inbred individuals and, thus, are suitable for detection of QTLs that act recessively.
Although taking into account the exact relationships between all family members, particularly in inbred populations, increases information about linkage (Lander and Botstein 1987), a common approach to keep computation simple is to use only a subset of the genealogy, splitting the entire pedigree into more manageable subunits. The issue of pedigree breaking has already been investigated. Chapman and Wijsman (2001) showed that results are dependent on how the subpedigrees are chosen and often require arbitrary decisions concerning the lines of descent that are preserved (Chapman et al. 2001). Dyer et al. (2001) observed that pedigree simplification, through cutting the pedigree at different generations, requiring breaking original loops, and reducing the number of informative pairs, inevitably reduces the power to detect linkage. Pankratz and Iturria (2001) proposed a semiautomatic method that relies partly on human decisions and that therefore is not easily reproducible. A method that systematically does the dividing while identifying subpedigrees that are likely to provide the most utility to a linkage study has been absent. We propose an approach that samples from the large number of pairwise relationships that exist in an extended genealogy, such as the ones under study, that maximizes useful information for linkage while minimizing IBD calculation burden. Indeed, the availability of a sample with a complete genealogy provides a remarkable wealth of relative pairs when compared with a sample of the same size distributed in smaller families (e.g., a sample of n individuals in a complete pedigree provides as many as n(n-1)/2 relative pairs). We investigated, through simulation, the power and the accuracy of IBD estimation of different sampling schemes. Applying this approach, we performed a GWS for serum lipid levels, through variance-components linkage analysis on a sample collected from Talana, and replicated our findings in a larger sample derived from Perdasdefogu.
Material and Methods
Populations and Genealogical Data
Ogliastra is a sparsely populated area in central-eastern Sardinia, with a total of 60,000 inhabitants (34 inhabitants/km2) clustered into 23 villages. Mitochondrial analysis traced the original population back to the Neolithic era. Several Ogliastra villages slowly grew in isolation and had little or no admixture with the others, giving rise to high endogamy and inbreeding. Fraumene et al. (2003) observed a great deal of genetic differentiation among subpopulation isolates within Ogliastra, as a consequence of founder effects and genetic drift. Genetic diversity measures showed that these villages rank among the most genetically homogenous European populations.
The GWS study was performed in a small isolated village of 1,200 inhabitants within Ogliastra, named Talana. Analyses of the Y chromosome and characterization of mtDNA haplogroups revealed that ∼80% of the Talana population derives from 8 paternal and 11 maternal ancestral lineages (Angius et al. 2001). The replication study was performed on a larger sample derived from another Ogliastra village, Perdasdefogu, located in the south of Ogliastra. Perdasdefogu has ∼2,400 residents, but during the past 50 years it has undergone a great immigration wave because of the opening of a military base nearby. mtDNA characterization has shown that only a small number of founders contributed to the actual female gene pool in Perdasdefogu native inhabitants (Fraumene et al. 2003).
Extensive historical and archival data allow an accurate reconstruction of the genealogy of each extant individual for as many as 16 generations in Talana and in Perdasdefogu. These data are structured in a relational database that includes phenotypic and genotypic information, and several algorithms allow database inquiry and data extraction for the analysis.
The Talana pedigree connecting a study sample of 875 extant individuals resulted in a complex 5,219-person pedigree spanning 16 generations, with multiple inbreeding loops, in which individuals are related to each other through multiple lines of descent. The median (25%–75% quartiles) kinship coefficient among the study sample of extant individuals is 0.014 (0.009–0.021); this value is between second cousins (0.0156) and second cousins once removed (0.0078). The median (25%–75% quartiles) inbreeding coefficient is 0.010 (0.007–0.011). The median number (25%–75% quartiles) of meiotic steps between each pair of extant individuals is 9 (7–10).
Given the large amount of immigration during the past 50 years in Perdasdefogu, we selected a subsample of extant individuals whose lineages could be traced to as many five generations before 1950. We extracted a 2,506-person, 15-generation inbred pedigree joining 821 extant individuals, with expected reduced genetic heterogeneity compared with the whole sample of residents. The median (25%–75% quartiles) kinship coefficient among them is 0.007 (0.004–0.011). The median (25%–75% quartiles) inbreeding coefficient is 0.005 (0.001–0.010). The median number (25%–75% quartiles) of meiotic steps between each pair of individuals is 10 (8–11).
Variance-Components Analysis
Statistical analyses were conducted using a variance-components approach. A linear mixed model is fit to the data so that the phenotypic variance about the trait mean is partitioned into a monogenic component (σ2QTL), representing the contribution of a QTL, a polygenic component (σ2R), attributable to residual additive genetic variance, and a residual component (σ2E), attributable to environmental effects unique to the individual. The phenotypic variance-covariance matrix (Ω) of the individuals in a pedigree may be written as 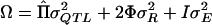 , where
, where 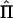 is a matrix of the proportion of alleles shared IBD estimated from the genotypic data at a point in the genome, 2Φ is a matrix of the expected proportion of alleles shared IBD over the genome, and I is an identity matrix. LOD scores are calculated as the difference between the maximum of the log10 likelihood of the full model, including estimates of σ2QTL, σ2R, and σ2E, and the maximum of the log10 likelihood of the reduced model in which σ2QTL is constrained to equal 0.
is a matrix of the proportion of alleles shared IBD estimated from the genotypic data at a point in the genome, 2Φ is a matrix of the expected proportion of alleles shared IBD over the genome, and I is an identity matrix. LOD scores are calculated as the difference between the maximum of the log10 likelihood of the full model, including estimates of σ2QTL, σ2R, and σ2E, and the maximum of the log10 likelihood of the reduced model in which σ2QTL is constrained to equal 0.
Simulation Study—Genealogy Partitioning
To explore the properties of our method for dividing large inbred genealogies, we simulated populations in which selection, immigration, bottlenecks, and other complicating factors were assumed to be absent. We generated, by simulation, 100 inbred genealogies, so that founding population size, expansion rate, and actual population size were comparable to those of the Ogliastra villages under study, which are well documented for the last 16 generations. Starting with an initial population size of 80 individuals, we randomly selected mating pairs in each of the 16 generations, allowing only one mate per individual. The mating partners were chosen among individuals more distant than first cousins, since marriages between first cousins have been discouraged and are rare in Ogliastra populations. If no partner was available, no mating occurred. The number of children per couple followed a Poisson distribution with mean 2.8. The resulting median (25%–75% quartiles) population size of the last three generations was 1,760 (1,269–2,146), whose median (25%–75% quartiles) kinship coefficient was 0.013 (0.009–0.020) and median (25%–75% quartiles) inbreeding level was 0.012 (0.007–0.018). The median (25%–75% quartiles) size of the genealogies was 3,629 (2,894–4,713) individuals.
Marker genotypes were simulated by gene dropping in each genealogy for four equally spaced markers spanning a region of 10 cM, to evaluate subsequent IBD-sharing estimation in a multipoint context. Each marker was simulated with eight equally frequent alleles, and founder haplotypes were assumed to be in linkage equilibrium. During simulations, the matrix of the true proportion of alleles shared IBD (Φ) between all individuals belonging to the last three generations was stored. Quantitative-trait phenotypes were simulated under different genetic models. The overall additive heritability (h2) was constrained to 0.5, attributable to a QTL and an additive polygenic effect. The QTL was assumed to be diallelic, with a fixed additive effect of moderate size on the individual phenotype and no dominance effect but with varying QTL-variant frequencies in the founder population. We simulated a QTL-variant with 0.05, 0.1, 0.3, and 0.5 frequency, yielding QTL-specific heritabilities (h2q) of 0.09, 0.15, 0.29, and 0.33, respectively. The remaining phenotypic variance was assigned to individual-specific effects. The QTL was positioned in the middle of the simulated chromosomal region.
Phenotypes and genotypes were assumed to be completely known only for individuals belonging to the last three generations. Power was calculated as the proportion of simulations that provided a multipoint LOD score ⩾3 in the region. The power of variance-components linkage analysis depends only on h2q, once the total heritability is fixed (Williams and Blangero 1999), and h2q depends only on the QTL frequency, when the additive and dominance effects are kept fixed. We investigated the probability of identifying, in the study sample, variants introduced in the ancestral genetic pool with different frequencies, thus also taking into account, in the evaluation of the power of our approach, genetic drift and founder effects, which greatly influence allele frequencies in an isolated inbred population and, consequently, h2q in the extant population. Therefore, instead of comparing the power on the basis of resulting QTL frequencies or h2q in the study sample, we focus on power evaluation for various QTL frequencies in the ancestral founder sample. Power calculations were adjusted to take into account those cases in which one of the QTL alleles was, in fact, lost.
From each simulated genealogy, different sets of subpedigrees were extracted using the maximum-cliques sampling approach described in the appendix. The approach is aimed at identifying the maximum number of individuals sharing a given characteristic—that is, a clique. We used the number of meiotic steps as a measure of relatedness to cluster individuals of the last three generations whose median number (25%–75% quartiles) of meiotic steps between pairs was 8 (7–10). After each replication, we extracted three sets of subpedigrees, named “S2,” “S3,” and “S4,” comprising individuals separated from each other by a maximum number of two, three, and four meiotic steps, respectively, and individuals belonging to the above generations who allowed us to preserve recent inbreeding loops. As larger numbers of individuals were included in the pedigrees from set S2 to S4, more generations were also included. The resulting mean numbers of individuals (mean numbers of generations) in the pedigrees of the three sets were 18 (5.5), 25 (6), and 62 (7), respectively. Larger numbers of meiotic steps generated extremely large, computationally heavy pedigrees and were not investigated. The mean number of the pairwise relationships among individuals of the study sample in the S2, S3, and S4 resulting sets were 1,677, 2,298, and 3,982, respectively.
We assessed the relative efficiency of the different subpedigree sets as the ratio between the power of the sets and the power of the complete pedigree, generated to include all the individuals of the sets and all their common ancestors. Since IBD-sharing estimation is not feasible in the general pedigree, the power of the complete pedigree was calculated using the exact IBD-sharing matrix. We also evaluated separately the reduction in power of the sets of subpedigrees derived from the misspecification of the null IBD-sharing probabilities, which influences the estimation of the polygenic component effect, as well as from the bias in IBD-estimation in the region, which influences the estimation of the QTL effect. The impact of misspecification of the null IBD-sharing probabilities was evaluated, comparing the power of the sets when assigning the true versus the estimated null IBD-sharing probabilities and using, in both cases, the true Π matrices. The effect on power reduction due to the bias in IBD estimation in the region was evaluated, comparing the power of the sets assigning the true Π versus the estimated 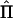 matrices, using the estimated null IBD-sharing probabilities in both cases. The estimated multipoint
matrices, using the estimated null IBD-sharing probabilities in both cases. The estimated multipoint 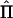 matrices were calculated from simulated genotypes by Simwalk2 (Sobel and Lange 1996) for the individuals in the last three generations and were imported into a framework for variance-components analysis. All calculations were performed for the individuals belonging to the last three generations of each simulated genealogy, for whom genotypes and phenotypes were assumed to be known. We did not consider the effects of missing data among these individuals, since it would have been beyond the scope of this article.
matrices were calculated from simulated genotypes by Simwalk2 (Sobel and Lange 1996) for the individuals in the last three generations and were imported into a framework for variance-components analysis. All calculations were performed for the individuals belonging to the last three generations of each simulated genealogy, for whom genotypes and phenotypes were assumed to be known. We did not consider the effects of missing data among these individuals, since it would have been beyond the scope of this article.
We also evaluated the type I error rates, estimating the number of times a given LOD-score threshold was exceeded by chance in each set of subpedigrees. For this study, we assumed the same QTL models described above and simulated conditions in which the QTL variant was completely unlinked to the chromosomal region. LOD scores were computed using the estimated 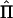 matrices.
matrices.
Subjects and Phenotypic Data
All individuals participating in the study signed informed consent forms, and all samples were collected in accordance with the Declaration of Helsinki (World Medical Association Web site). All participants were given a physical exam, and total blood was drawn for phenotype and genotype determination.
TC levels, HDL-C levels, TG levels, and BMI were measured in 572 subjects from Talana and in 1,443 subjects from Perdasdefogu, all of ages ⩾18 and ⩽90 years. We determined TC, HDL-C, and TG levels with an automated Vitros 250 Chemistry System (Orthoclinical Diagnostics, Johnson & Johnson Gateway SM). Quantitative measures of TC and HDL-C concentration were evaluated by use of the Vitros Chemistry Products Magnetic HDL Cholesterol Reagent and Vitros Chol Slides, following the manufacturer’s instructions (Allain et al. 1974; Warnick et al. 1983). LDL-C levels were determined by the Friedewald formula: LDL-C = TC-(HDL-C+TG/5). This formula was used only for subjects presenting with TG <400 mg/dl (i.e., 4.4 mmol/liter), as recommended. The BMI was calculated as weight divided by the square of height (kg/m2). Extreme outliers (i.e., trait value below the mean minus 3 SD or above the mean plus 3 SD) were excluded from the analysis.
Genotyping and Genetic Maps
Genomic DNA was extracted from 7 ml of EDTA-treated blood, as described by Ciulla et al. (1988). Genotyping of 875 individuals from Talana was performed in collaboration the National Heart, Lung, and Blood Institute Mammalian Genotyping Service of the Marshfield laboratory, under the direction of J. L. Weber. A total of 654 highly informative markers, distributed over the autosomal chromosomes, were genotyped. The data were checked for typing inconsistent with the pedigree structure through use of PedCheck (O’Connell and Weeks 1998), employing semiautomatic procedures to investigate and remove errors. Instances that could not be resolved were treated as missing data.
The Talana-specific genetic maps were constructed using CRI-MAP (Lander and Green 1987), which was run on an extended pedigree in which the number of informative meioses were maximized (as many as 900 informative meioses). Allele frequencies were estimated from the whole sample of genotyped subjects, and average marker heterozygosity was 0.71 (SD 0.09).
In the identified region on chromosome 2, we typed eight additional markers (D2S347, D2S112, D2S150, D2S132, D2S151, D2S2277, D2S2241, and D2S142) in 352 individuals of the Talana family set used in the GWS, thus allowing us to obtain an average marker density of 2.8 cM. PCR was performed according to standard protocols. Microsatellite products were loaded on an ABI PRISM 3730 DNA Analyzer (Applied Biosystems), and data were processed by GENEMAPPER v3.0 software. These eight markers, plus the seven markers belonging to the GWS set (D2S1328, D2S442, D2S1399, D2S1388, D2S1353, GATA126A06, and D2S1776), were subsequently genotyped in a larger sample of 821 individuals from Perdasdefogu, to replicate the original result. The mean heterozygosity in Perdasdefogu for these 15 markers was 0.74 (SD 0.06).
For the markers typed in the candidate region of chromosome 2, population-specific allele frequencies in the Talana and Perdasdefogu samples were estimated, considering typed individuals as independent and also taking into account their relatedness. For the latter, we used the procedure recently proposed by McPeek et al. (2004), which allowed us to derive the best linear unbiased estimator (BLUE) of the allele frequencies in the two large pedigrees—one for each of these population samples—that connect all genotyped individuals of the respective samples. Using these pedigrees, we also used CRI-MAP to estimate the combined Talana and Perdasdefogu–specific map of the chromosome 2 region. This genetic map was used for calculations, although in the figures we report the deCODE genetic map (Kong et al. 2002), for consistency. For two markers (D2S1388 and GATA126A06), the deCODE genetic locations were not available and were interpolated from the genetic and physical positions of flanking markers. The genetic map estimated in the combined sample was ∼10 cM longer than the deCODE map, even after rechecking genotypes that caused apparent tight double-recombination events.
Statistical Analysis
Narrow trait heritabilities were estimated in the complete genealogies by use of SOLAR (Almasy and Blangero 1998), after accounting for the effects of relevant covariates. Variance-components analysis was performed, on a Pentium IV 3.2 GHz/4 Gb computer, using Merlin (Abecasis et al. 2002), which performs fast and exact multipoint IBD calculation, incorporating a simultaneous correction for sex, age, age-squared, and BMI. Inclusion of these covariates improves the ability to detect linkage, by both reducing the unexplained residual variance in lipid levels and increasing the relative genetic signal. We did not derive empirical P values adjusted to account for the scanning of the whole genome, since it would have required prohibitively time-consuming simulations.
Results
Simulation Study—Genealogy Partitioning
We have investigated the effects of genealogy partitioning on expected power in variance-components linkage analysis by simulation. For a QTL introduced in the founders' generation with different allele frequencies (0.5, 0.30, 0.10, 0.05), a range of QTL allele frequencies is observed in the study sample, with a mean value equal to the original value and a nonzero probability of being lost for the two rarer variants (namely, 0.10 and 0.05) (table 1). To assess the relative efficiency of the different subpedigree sets compared with the whole pedigree, we first estimated the power in the general pedigree, computed using the exact IBD-sharing matrix and excluding the replicates in which the QTL allele was lost. The power of the whole pedigree was extremely high: 100% for QTL frequencies of 0.50 and 0.30, 86% for a QTL frequency of 0.10, and 70% for the rarest QTL frequency, 0.05, with a fixed additive effect of moderate size. We then evaluated separately (1) the decrease in power compared with the whole pedigree for the sets due only to lack of information in the ancestral generations and sample reduction, (2) the effect of misspecification of the null IBD-sharing probabilities, and (3) the effect due to the bias in IBD-estimation in the region (fig. 1). We observed a marked decreased relative power for all the sets as the gene frequency of the QTL decreased while constraining additive heritability, which indicates that increasingly larger samples and numbers of informative generations should be needed to identify rarer variants. On the contrary, the relative reduction in power of each set of subpedigrees due to the bias in IBD estimation in the region was relatively modest, indicating that bias in IBD estimations only weakly influenced the power of variance-components linkage studies of the sets. The misspecification of the null IBD-sharing probabilities had a negligible effect on LOD scores and did not influence power for all the sets. As expected, the power increased as the number of pairwise relationships in the sets also increased, although these differences were not particularly strong. We observed that the LOD scores were highly concordant among the sets (mean correlation coefficient [R] 0.84; P<.0001). Within each set, we evaluated the relative contribution of different types of relative pairs to the overall power of the set, by computing the relative increase in power when more distantly related pairs were added in the sets. We observed that, as expected, as the QTL variant became less common, the relative contribution of distant relationships to the overall power increased.
Table 1.
Fluctuations of the QTL Allele Frequencies in the Individuals from the Last Three Generations (Study Population) Due to Random Genetic Drift and Founder Effects, Obtained through Simulations of the 100 Inbred Genealogies Described in the Text
| Founder PopulationQTL AlleleFrequency | Study Population Mean QTL Allele Frequency (25%–75% Quartiles) | % Lost |
| .5 | .49 (.40–.59) | 0 |
| .3 | .29 (.20–.38) | 0 |
| .1 | .09 (.04–.15) | 8 |
| .05 | .05 (.01–.04) | 16 |
Figure 1.

Relative power for the S2, S3, and S4 sets compared with the power of the whole pedigree. Segments above the bars indicate the relative power of the sets once bias in IBD estimation in the region was removed by use of the true-IBD matrix. The different colors show the relative contribution to power of the different types of relative pairs in the sets (blackened bars, four meiotic steps between the pair; striped bars, three meiotic steps; unblackened bars, two meiotic steps). Power was evaluated through simulation of 100 inbred genealogies, with a fixed-effect QTL variant entering the population at different frequencies indicated on the X-axis, excluding replicates in which the QTL variant was lost.
We examined in more detail the effects of pedigree simplification on accuracy in IBD estimation in the chromosomal region. We compared the true proportion of alleles shared IBD, πj, stored during simulation for all pairs j of individuals, with the estimated proportion, 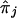 , within each set. The true and estimated proportions were calculated as the average proportion of alleles shared IBD at the four genotyped marker loci. Each set showed a similar slight underestimation of πj, and the mean deviation over the sets was 0.04 (SD 0.11). The mean deviations of the estimated IBD probabilities were of the same magnitude as the mean deviations of the null IBD-sharing probabilities estimated only from pedigree data. Figure 2 shows that the accurateness of the posterior IBD-sharing probabilities for the same type of relative pairs was increased when the number of ancestral generations in the pedigrees increased from S2 to S4. Since more distantly related individuals were included in the sets from S2 to S4, the mean accuracy resulted of the same magnitude in the three sets. We investigated whether the means and the variances of the IBD deviations depend on the genotypic status of the relative pairs and found the most significant correlation (P<.0001) with the homozygous genotype rate, evaluated across all four markers, which yielded an adjusted coefficient of determination (adjusted R2) between 0.25 and 0.41. This indicates a moderate but significant correlation between the homozygous genotype rate in the sample and the accuracy and fluctuation of IBD-sharing estimations.
, within each set. The true and estimated proportions were calculated as the average proportion of alleles shared IBD at the four genotyped marker loci. Each set showed a similar slight underestimation of πj, and the mean deviation over the sets was 0.04 (SD 0.11). The mean deviations of the estimated IBD probabilities were of the same magnitude as the mean deviations of the null IBD-sharing probabilities estimated only from pedigree data. Figure 2 shows that the accurateness of the posterior IBD-sharing probabilities for the same type of relative pairs was increased when the number of ancestral generations in the pedigrees increased from S2 to S4. Since more distantly related individuals were included in the sets from S2 to S4, the mean accuracy resulted of the same magnitude in the three sets. We investigated whether the means and the variances of the IBD deviations depend on the genotypic status of the relative pairs and found the most significant correlation (P<.0001) with the homozygous genotype rate, evaluated across all four markers, which yielded an adjusted coefficient of determination (adjusted R2) between 0.25 and 0.41. This indicates a moderate but significant correlation between the homozygous genotype rate in the sample and the accuracy and fluctuation of IBD-sharing estimations.
Figure 2.

Mean deviations of the true proportion of alleles shared IBD, πj, and the estimated proportion, 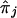 , grouped for the same type of relative pairs j present in each of the S2 (unblackened bar), S3 (striped bars), and S4 (blackened bars) sets. In S2, all relative pairs are separated by as many as two meiotic steps; in S3, 62% of the pairs are separated by as many as two meiotic steps; in S4, 36% of the pairs are separated by as many as two meiotic steps, and 32% are separated by three meiotic steps. IBD estimations in the chromosomal region were calculated as the mean proportion of alleles shared IBD over the four genotyped marker loci.
, grouped for the same type of relative pairs j present in each of the S2 (unblackened bar), S3 (striped bars), and S4 (blackened bars) sets. In S2, all relative pairs are separated by as many as two meiotic steps; in S3, 62% of the pairs are separated by as many as two meiotic steps; in S4, 36% of the pairs are separated by as many as two meiotic steps, and 32% are separated by three meiotic steps. IBD estimations in the chromosomal region were calculated as the mean proportion of alleles shared IBD over the four genotyped marker loci.
In the evaluation of the type I error rates, we did not observe, in any set, any LOD score >3 in 100 replicates simulated for each of the considered models, which yielded an upper-bound 95% CI of 0.0295. We observed only once a LOD score >2 in the S4 set for a 0.5 QTL-variant frequency, yielding an upper-bound 95% CI of 0.0545.
In the light of these results, we applied the clique partitioning approach to the extended Talana and Perdasdefogu pedigrees, using a range of one to four meiotic steps between individuals (analogous to set S4 in the simulation study) to generate the sets. Any larger number of meiotic steps as partitioning criteria generated pedigrees that were too extended. The characteristics of the sets used in the analyses are shown in table 2. A total of 991 and 2,167 relative pairs were included in the Talana and Perdasdefogu family sets, respectively. The varying degrees of genetic relationship in the sets are shown in table 3.
Table 2.
Characteristics of the Family Sets Used in the Analyses[Note]
|
No. of Individualsb |
||||||||
| Population | No. ofFamilies | Mean FamilySize (Range) | Mean No. ofGenerations (Range) | MedianKinship coefficient(25%–75% Quartiles)a | Founder | Nonfounder | Male | Female |
| Talana | 44 | 17.6 (9–32) | 4.0 (3–6) | .0625 (0–.1563) | 278 | 497 | 344 | 431 |
| Perdasdefogu | 96 | 15.4 (5–31) | 3.9 (2–7) | .0645 (0–.2500) | 541 | 938 | 714 | 765 |
Note.— We utilized the maximum-cliques approach described in the appendix, using a maximum number of four meiotic steps between individuals, to generate these sets of subpedigrees.
The kinship coefficients are estimated for the phenotyped and genotyped individuals of the sets.
The total numbers of individuals were 775 for Talana and 1,479 for Perdasdefogu.
Table 3.
Pairwise Relationships with Number of Occurrences in the Talana and Perdasdefogu Family Sets
|
No. of Pairs |
||
| Pairwise Relationship(s) | Talana | Perdasdefogu |
| Avuncular | 210 | 466 |
| Sib | 205 | 521 |
| 1st cousin | 189 | 327 |
| Parent-offspring | 147 | 439 |
| 1st cousin plus 2nd or 3rd cousin | 52 | 45 |
| Other 1st cousin | 29 | 42 |
| Grand avuncular and other grand avuncular | 24 | 24 |
| Other avuncular | 23 | 61 |
| Half 1st cousin | 17 | 30 |
| Other parent-offspring | 15 | 42 |
| Double 1st cousin or double 2nd cousin | 14 | 6 |
| 2nd cousin or other 2nd cousin | 14 | 60 |
| Half avuncular | 12 | 21 |
| Other sib | 9 | 16 |
| Grandparent-grandchild | 6 | 41 |
| 3rd cousin or other 3rd cousin | 4 | 11 |
| Other | 21 |
15 |
| Total | 991 | 2,167 |
Phenotypic Analyses
The phenotypic characteristics of the 572 individuals from Talana and of the 1,443 from Perdasdefogu used in the analysis are summarized in table 4. In Talana, the average age of men was 53 years (range 26–92 years), and the average age of women was 52 years (range 23–90 years). In Perdasdefogu, the average age of men was 49 years (range 18–90 years), and the average age of women was 48 years (range 18–89 years). The average BMI in Talana was 27.0 kg/m2 for men and 24.8 kg/m2 for women; in Perdasdefogu, it was 26.6 kg/m2 for men and 25.7 kg/m2 for women. BMI was rather elevated, since individuals in these populations are short with a stocky build (mean height is 160 cm in men and 150 cm in women). Although these populations have a fat-rich diet, given the lack of suitable land for cultivating vegetables and their taste for meat, lipid levels were, on average, not elevated compared with standard guidelines (National Cholesterol Education Program 2001), and only TC showed borderline values in the middle age classes (table 4). A normal distribution for all variables was checked using the Kolmogorov-Smirnov goodness-of-fit test: although TC, HDL-C, and LDL-C followed a normal distribution, TG concentration values were markedly skewed and were logarithmically transformed to improve normality. Lipid levels were significantly correlated with each other (table 5), except for HDL-C and LDL-C.
Table 4.
Lipid Levels and BMI by Age for the 572 Subjects from Talana and for the 1,443 Subjects from Perdasdefogu[Note]
|
Median Lipid Level (25%–75% Quartiles)(mg/dl) |
||||||
| Populationand AgeClass (inYears) | No. of Subjects | TC | HDL-C | LDL-C | TG | Median BMI (25%–75% Quartiles)(kg/m2) |
| Talana: | ||||||
| <41 | 166 | 186 (166–212) | 61 (52–70) | 106.7 (89.7–124.9) | 89 (67–123) | 23.8 (21.4–26.6) |
| 41–65 | 242 | 199.0 (178–222) | 59 (51–68) | 118.0 (100.1–137.4) | 98 (76–130) | 27.2 (23.9–30.5) |
| >65 | 164 | 200 (175–228) |
60 (53–70) | 117.5 (97.9–135.6) | 94 (73–123) | 27.0 (24.0–30.9) |
| All | 572 | 196 (175–219) | 60 (51–70) | 114.0 (96.9–134.6) | 94 (73–128) | 25.9 (23.1–29.4) |
| Perdasdefogu: | ||||||
| <41 | 575 | 183 (164–209) | 62 (53–71) | 102.8 (84.7–124.6) | 76 (58–108) | 23.1 (20.9–26.1) |
| 41–65 | 571 | 208 (182–236) |
61 (52–70) | 123.8 (101.9–148.6) | 93 (72–129) | 26.9 (24.3–30.0) |
| >65 | 297 | 208 (184–231) |
64 (56–75) | 122.0 (100.4–141.6) | 100 (79–132) | 27.9 (24.7–31.2) |
| All | 1,443 | 198 (173–226) | 62 (53–71) | 115.0 (93.6–138.4) | 89 (67–122) | 25.6 (22.5–28.9) |
Note.— According to National Cholesterol Education Program (2001) guidelines, the normal ranges for each phenotype are as follows: TC, 120–199 mg/dl; HDL-C, 40–80 mg/dl; LDL-C, 60–129 mg/dl; TG, 30–149 mg/dl; and BMI, <25 kg/m2. Median values that exceed normal ranges are underlined.
Table 5.
Correlations among Lipid Levels Estimated Independently in 572 Subjects from Talana (above the Diagonal) and in 1,443 Subjects from Perdasdefogu (below the Diagonal)
| TC | HDL-C | LDL-C | lnTGa | |
| TC | … | .257 (P<.001) | .913 (P<.001) | .380 (P<.001) |
| HDL-C | .253 (P<.001) | … | −.020 (P=.643) | −.279 (P<.001) |
| LDL-C | .912 (P<.001) | −.018 (P=.494) | … | .206 (P<.001) |
| lnTGa | .423 (P<.001) | −.257 (P<.001) | .254 (P<.001) | … |
log-transformed TG concentration values.
Quantitative genetic analysis was performed by use of SOLAR on an extended 12-generation pedigree of 1,198 individuals connecting all 572 phenotyped subjects from Talana. The estimated heritabilities in Talana, allowing for sex, age, age-squared as covariates, were 0.40 (SE 0.10) for TC, 0.49 (SE 0.09) for HDL-C, 0.38 (SE 0.10) for LDL-C, and 0.43 (SE 0.13) for log-transformed TG values (lnTG), all highly significant when compared with the sporadic model. Adding BMI slightly increased the estimated heritabilities, to 0.42 (SE 0.10) for TC, 0.50 (SE 0.09) for HDL-C, 0.41 (SE 0.10) for LDL-C, and 0.45 (SE 0.13) for lnTG. These estimates closely reflect the genetic component in the traits, since confounding effects of household environment are minimized in this large pedigree, because relationships between all individuals are considered.
GWS in Talana
Multipoint linkage analyses for TC, HDL-C, LDL-C, and lnTG concentrations in subjects from Talana were performed across autosomes by use of 654 highly informative markers. Results of the GWS are shown in figure 3. We found strong evidence of linkage, for a QTL influencing TC level, to chromosome 2, in a 36-cM region flanked by markers D2S1328 and D2S1776 (peak LOD score 3.40 at D2S1388, 164 cM from the p-ter). The region seems to harbor a major locus for variation in LDL-C level, for which a peak LOD score of 2.65 was also obtained at marker D2S1388, whereas LOD scores <1 were observed for HDL-C and lnTG. Tentative evidence of a QTL influencing variation in HDL-C was found on the q-ter of chromosome 5, at marker ATT079 (LOD score 2.05), but we did not further investigate this locus.
Figure 3.

Results of the GWS for TC (red line), HDL-C (green line), LDL-C (orange line), and log-transformed TG (blue line) obtained in Talana sample by use of variance-components analysis, incorporating sex, age, age-squared, and BMI as covariates. X-axis values are in centimorgans (Talana-specific genetic maps).
To confirm our major finding on chromosome 2, we added eight polymorphic markers to the seven previously used in the genome scan set in the region that contains our peak linkage signal for TC and LDL-C. Typing the additional markers in 352 individuals from the Talana family set, we obtained an increased peak LOD score of 3.73 for TC and of 3.67 for LDL-C at marker D2S2241, located ∼1 cM proximal to D2S1388. Removing BMI as a covariate had the effect of increasing the peak signal to 4.25 for TC and 3.87 for LDL-C (fig. 4A).
Figure 4.

Multipoint LOD scores on chromosome 2q21.2-q24.1 for TC and LDL-C in Talana (A) and in Perdasdefogu (B), obtained through variance-components analysis, incorporating sex, age, age-squared (thin line) and BMI (bold line) as covariates. X-axis values are in centimorgans (deCODE map).
For these markers, we also estimated allele frequencies by use of the procedure recently proposed by McPeek et al. (2004), which allowed us to take into account the relationships among individuals. We obtained similar multipoint LOD scores over the region, with a peak LOD score of 3.76 for TC and of 3.71 for LDL-C at D2S2241, and peak LOD scores of 4.29 and of 3.91, respectively, when BMI was removed as covariate.
Replication Study in Perdasdefogu
As a replication study, we conducted multipoint linkage analyses of TC and LDL-C levels with the 15 markers on chromosome 2 by use of a large sample of 821 individuals clustered in 96 extended families from Perdasdefogu, another isolated village of Ogliastra. We found evidence of a locus influencing variation in TC level on chromosome 2, between markers D2S1328 and D2S132 (peak LOD scores 1.44 for TC and 2.21 for LDL-C at D2S442; fig. 4B). Removing BMI as a covariate had different, small effects on the linkage signals for the two traits (LOD score increased to 1.61 for TC and decreased to 1.93 for LDL-C). Estimating allele frequencies by use of the method of McPeek et al. (2004), we obtained similar multipoint LOD scores over the region, with peaks of 1.42 for TC and of 2.20 for LDL-C. Linkage analysis performed in the larger replication set of families provides evidence for the presence of a QTL underlying TC and LDL-C variations in a smaller interval flanked by markers D2S1328 and D2S132, which are ∼18 cM apart. The highest linkage peaks in the replication set were associated with nominal P values of .005 for TC and .0007 for LDL-C, which are below the suggested threshold of significance required for a replication study (Lander and Kruglyak 1995).
The distance between the linkage peaks obtained for TC and LDL-C in the two populations is ∼15 cM, although the identified regions overlap, since a LOD score of 2.71 is observed for TC in the Talana sample at marker D2S150, which is only 2.3 cM distal to the highest peak obtained in the Perdasdefogu sample, at D2S442 (fig. 4B).
Discussion
The reduction in power that is associated with gene discovery efforts for complex traits may be ameliorated by careful attention to population selection—for example, by use of population isolates, which reduce the possible confounding effects of genetic and environmental heterogeneity. We focused our study in Ogliastra, an ancient isolated region of Sardinia, whose population, clustered into 23 villages or small towns, originated from the same Neolithic population. The availability of extensive genealogical information, the willingness of the populations of these villages to participate in genetic studies, and the geographic isolation and enhanced genetic homogeneity within each village (Angius et al. 2001; Fraumene et al. 2003) make these populations suitable for gene-mapping studies for complex traits. Furthermore, the availability of accurate genealogical records for these isolated populations allows us to place extant individuals from each village within large and complex genealogies. To make the most of the genealogical information while minimizing the computational burden of linkage analysis, we developed a new sampling approach from the large number of pairwise relationships existing among individuals. Through a maximum-cliques partitioning approach based on measures of relatedness between the subjects, we extracted various sets of subpedigrees of different complexity and size, whose members share a higher degree of relatedness within the family than they do among families.
We explored by simulation the feasibility of this approach, investigating the power of the sets—extracted from genealogies with characteristics similar to the ones under study—for identifying a QTL variant with a fixed additive effect but varying founder frequencies. We assumed a QTL with a fixed effect of moderate size, to mimic a complex-trait genetic model. We constrained the overall narrow heritability, attributable to the QTL additive effects and to unlocalized additive polygenic effects, to 0.5 and explored the power for identifying the variant in the last generations, in which its frequency is associated with high random variation, determined by the probability that that specific gene has been transmitted through generations. Indeed, gene frequencies in small isolated populations are affected by both founder effect and random genetic drift. Rare alleles are more likely to be lost, whereas common alleles are more likely to be fixed, with a probability proportional to their initial frequencies. Therefore, fluctuations in the QTL frequency lead to varying QTL heritability in the extant population.
We assessed the relative power of the different subpedigree sets compared with the power that would be obtained analyzing the complete genealogy. Although the analysis of the whole pedigree would also allow us to identify rare variants (frequency <0.05), the different sets of pedigrees provide strong power for common variants (frequency >0.10), whereas power was extremely low (<10%) in all sets in the case of a rare variant (frequency 0.05). Rare variants are most likely to be recent in origin and, hence, to be specific to a single founder population, whereas common alleles are more likely to be found globally and to be of importance in other populations as well.
We also investigated the extent of the bias due to pedigree breaking in the estimation of the IBD-sharing probabilities for each set of subpedigrees and the effect of these misspecifications on power of the analysis. The mean IBD-sharing accuracy was found to be of the same magnitude in the three sets, with an increased accurateness for closely related individuals mediated by the introduction of more distant pairs in the larger sets. We observed that IBD-estimation accuracy is particularly diminished by the presence of an increased number of homozygous individuals, who are more likely to be less informative for linkage, especially in simple pedigrees with less inbreeding. In particular, the bias in the IBD estimation tends toward that expected on the basis of genealogical relationships. Identification and removal of uninformative pairs, as recently suggested Schork and Greenwood (2004) to remove bias from the analysis and to increase power for sib-pair data, is not achievable in extended pedigrees, since each individual might present several relationships with different members of the same pedigree, some of which could be informative. To evaluate how IBD estimation affects the result of variance-components analysis in large inbred pedigrees, instead of identifying and removing potentially uninformative pairs, we assigned the true IBD-sharing values to all individuals of the study sample in our simulations and evaluated the power decrease obtained with the estimated ones. As was recently shown for sib-pair data (Cordell 2004), we observed that bias in IBD-sharing estimation influences only slightly the power of variance-components linkage studies, even in complex inbred pedigrees. We also inspected the consequences on power of the misspecification of the null IBD-sharing probabilities when breaking might lead to an underestimation of inbreeding. We observed that, through our approach, which is apt to preserve all the most informative genealogical connections, the extent of inbreeding underestimation had negligible effects on LOD scores and, therefore, on power.
We applied the partitioning scheme that provided the largest number of informative pairs while not overburdening computations to the genealogy of one of these small founder subpopulations within Ogliastra—namely, the village of Talana—and performed a GWS for QTLs influencing serum lipid concentrations in the resulting set of families. In the GWS performed with 654 markers distributed over the genome, the region on chromosome 2 stands out significantly against the genomic background for TC level (LOD score 3.4) and for LDL-C variations (LOD score 2.7). Through genotyping of eight additional markers in the identified region, we obtained an increased LOD score of 4.3 for TC and 3.9 for LDL-C in 2q21.2-q24.1. The addition of BMI as a covariate has the effect of reducing the linkage signal to 3.7 for TC and LDL-C, which suggests that the underlying variant may have a broader effect on metabolism.
Replication of initial linkages to putative loci in independent samples is an essential step in the genetic dissection of complex human traits and provides vital confirmation of the original findings. We extended our study to another subisolate within Ogliastra to confirm the initial results and to fine map the genomic associated region with the traits under study. The replication study was performed on a larger sample derived from the village of Perdasdefogu, located in the south of Ogliastra. We genotyped an approximately threefold larger sample of individuals, since the sample size required to replicate a linkage finding should always be larger than the one required for initial detection. We genotyped 821 individuals for 15 markers spanning the chromosome 2 region and clustered them into 96 extended families extracted from the whole genealogy, by use of the same partitioning approach used in Talana. We obtained evidence of a locus influencing variation in TC level on 2q21.2-q24.1 (LOD scores 1.4 for TC and 2.2 for LDL-C). When BMI was excluded as a covariate, the LOD score increased to 1.6 for TC and decreased to 1.9 for LDL-C. The associated nominal P values for the TC and LDL-C LOD scores were .005 and .0007, respectively. In accordance with published guidelines regarding the level of significance necessary to declare significant replication (Lander and Kruglyak 1995), since replication requires a lower threshold for significance, our findings constitute a replication of the linkage signal on chromosome 2 and add further evidence that the 2q21.2-q24.1 locus contains a TC and LDL-C gene of widespread importance. The replication peak was obtained at a distance of 15 cM from the highest linkage peak observed in the original population, although the identified regions overlapped, since a LOD score of 2.7 is observed for TC in the Talana sample at marker D2S150, which is only 2.3 cM from the replication peak. The variation in the localization of this locus is modest compared with the width of the linkage signal in subjects from Talana and with the chance variation in location estimates, which are expected to be high for independent replication of an initial linkage (Hauser and Boehnke 1997; Roberts et al. 1999). Furthermore, the larger replication set allowed us to narrow the associated interval to ∼18 cM, since no evidence for linkage was observed in the flanking distal region. In any case, the presence of an independent QTL in the Talana set cannot be excluded.
Despite these differences, it is significant that linkage to 2q21.2-q24.1 is seen for these traits in both populations. The fact that the same locus was identified in two founder populations of old, common ancestry indicates that the variant underlying the traits, common in both populations, is probably evolutionarily old and, therefore, more likely to be found in other populations as well. Interestingly, a GWS for premature coronary heart disease in Finns (Pajukanta et al. 2000) revealed significant evidence for linkage to the 2q21.1-22 region, yielding a multipoint LOD score of 3.0. Since a disadvantageous plasma lipoprotein profile is an important risk factor for atherogenesis, it is tempting to hypothesize that allelic variants of the same gene may influence both traits. Furthermore, our results agree with a reported genome scan, conducted in another isolated population (Hutterites) enriched for cardiovascular disease, in which a locus with a multipoint LOD score of 3.40 was identified for TG in the adjacent 2q14 region (Newman et al. 2003). Although the critical interval identified by Newman and colleagues slightly overlaps the critical region identified in the present study, we might have independently replicated their finding, since TC and TG levels are highly correlated traits, and this putative locus may have influence on both phenotypes. For instance, VLDL is the primary carrier for the delivery of endogenous TC and TG to extrahepatic tissues; thus, factors involved in VLDL secretion and receptor-mediated clearance of VLDL remnants may have effects on both phenotypes.
The present study provides evidence for the existence of a QTL on 2q21.2-q24.1 that influences lipid phenotypes fundamental to common diseases, such as cardiovascular diseases, and, to our knowledge, represents the first replication for a QTL in isolated populations. The population sample; the selection of families through informative members; the consistent linkage to another larger sample, with a level of concordance uncommon for complex phenotypes; and the magnitude of the LOD scores emphasize the significance of this locus. Differences in environmental and genetic exposure and sample sizes, as well as differences in gene frequencies, may explain observed variations in the location of the QTL. Nevertheless, the same variant might have different phenotypic effects in different populations, because of background factors (genetic and otherwise) differentiating populations that can modify the expression of a variant and lead to different levels of association.
Our study provides new information about genomic regions in humans that influence interindividual variation in TC and LDL-C lipid levels. Fine-mapping studies are currently under way to identify the QTL involved in lipid metabolism, since the region is still too large to undertake candidate-gene studies. Identification of genes that influence lipid metabolism will lead to a better understanding of the etiology of CVD and provide suggestions for the development of new therapies for the treatment and prevention of disease.
Acknowledgments
We are grateful to the populations of Talana and Perdasdefogu and to all the individuals who participated in this study. We thank James L. Weber and the National Heart, Lung, and Blood Institute Mammalian Genotyping Service. We thank Marcella Devoto and Michael Whalen for their valuable suggestions. Finally, we thank the two anonymous reviewers for their helpful comments and suggestions. M.F. is a Doctorate fellow at the University of Modena, Department of Genetics. The power-evaluation and partitioning tools described in this article are available from M.F. or at the Shardna Web site.
Appendix
By use of graph theory, a pedigree can be represented by an undirected graph whose vertices (V) correspond to individuals, and the edges (E) connecting two vertices are weighted on the basis of the pairwise measure of relatedness between the two individuals, such as their kinship coefficient or the number of meiotic steps separating them. A graph G=(V;E) is complete if each vertex is connected with each of the others (all vertices are pairwise adjacent). A clique, C, is a subset of V, such that the induced graph is complete. Given a graph G=(V;E) and a positive integer K⩽|V|, the clique partitioning problem is to partition the vertices of G into k⩽K disjoint sets V1,V2,…,Vk such that, for 1⩽i⩽k, the subgraph induced by Vi is a complete graph. When we are directly solving the clique partitioning problem, a solution can be determined by iteratively searching for the maximum clique of the graph and deleting it from the graph, until there are no more vertices left. Thus, the problem is reduced to that of finding the maximum clique. A maximum clique corresponds to a maximum independent set of the complement of a graph (the maximum subset of vertices that are pairwise nonadjacent). Several approximations and search algorithms have been developed to solve the clique partitioning problem. Bron and Kerbosch (1973) identified all cliques in an undirected graph by use of two backtracking algorithms based on a branch-and-bound technique. The branch-and-bound technique is the basis for most recent clique algorithms (Babel 1991; Balas and Niehaus 1996). We can therefore partition the whole pedigree by use of cliques in which individuals share a desired amount of relatedness with each other.
We customized the original version of the Bron-Kerbosch algorithm to partition the large Talana and Perdasdefogu genealogies. Specifically, we restricted the search of cliques on user-defined ranges of edge weights, to include in the cliques all individuals whose mutual relationship was “up to” a specific value. Furthermore, to avoid excessive imbalance in family size, we constrained on the consented range of clique size, or cardinality. Once the clusters of individuals that maximize the number of pairwise relationships were identified for specific user-defined characteristics, the families were reconstructed around the individuals in each clique through use of the information from the genealogical database. An algorithm based on joining binary trees has been created to reconstruct pedigrees from an arbitrary set of individuals, according to various rules such as the maximum allowed number of nongenotyped individuals between family members or the maximum number of generations.
Figure A1 shows an example of the way a pedigree can be divided through the maximum-cliques approach by use of the meiotic-steps matrix of the family members. This example is based on an extremely simple pedigree for illustration, which does not represent the real complexity of an extended genealogy. The 37 pedigree members provide 451 pairwise relationships, under the assumption that they all have complete data on phenotypes and genotypes. The mutual distance among family members ranges from one to seven meiotic steps. The dotted-line boxes represent the subpedigrees generated by a maximum-cliques selection of as many as two meiotic steps between individuals of the clique (S2), whereas the continuous-line boxes represent the subpedigrees generated by a maximum-cliques selection of as many as four meiotic steps (S4). The resulting sets contain 47 (S2) and 112 (S4) relative pairs. Given the pedigree structure, the generated sets maximize the number of individuals belonging to the same family (maximum cardinality of the cliques) and the overall number of individuals present in the specific set. The degree of overlap of pairwise relationships between the two sets are only 30% in S2 and 14% in S4. This is because the maximum cliques develop in the region of the graph in which the mutually adjacent vertices are more dense, and these regions are different when sampled using different degrees of relationship.
Figure A1.

Simplified example of the way a pedigree can be divided through the maximum-cliques approach using the meiotic steps matrix of the family members. The dotted lines surround the S2 subpedigrees, and the continuous lines surround the S4 subpedigrees. See the appendix for details.
The graph-partitioning algorithm also allows us to constrain for the minimum and maximum size of the cliques (the cardinality of the cliques), instead of maximizing the number of individuals belonging to the same clique. If, for example, in S2 we accept a minimum cardinality of the cliques of 3, a clique including individuals 12, 25, and 28 will be extracted, and individual 12 will not be included in the clique of individuals 3, 4, 26, 5, and 11.
Also, the reconstruction of pedigrees from the cliques of individuals can be accomplished using different rules, such as the number of generations in the family. In general, we want to include recent inbreeding loops. For instance, the S4 pedigree on the right side of figure 5 would include later generations (individuals 3, 4, 6, and 7) to increase information about linkage.
Electronic-Database Information
The URLs for data presented herein are as follows:
- Declaration of Helsinki, World Medical Association, http://www.wma.net/e/policy/b3.htm
- Mammalian Genotyping Service, Center for Medical Genetics, http://research.marshfieldclinic.org/genetics/
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for LDLR, APOB, and ARH)
- Shardna, http://www.shardna.it/falchi/ (for the power-evaluation and partitioning tools described in this article)
References
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR (2002) Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 30:97–101 [DOI] [PubMed] [Google Scholar]
- Abney M, Ober C, McPeek MS (2002) Quantitative-trait homozygosity and association mapping and empirical genome-wide significance in large, complex pedigrees: fasting serum-insulin level in the Hutterites. Am J Hum Genet 70:920–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acton S, Osgood D, Donoghue M, Corella D, Pocovi M, Cenarro A, Mozas P, Keilty J, Squazzo S, Woolf EA, Ordovas JM (1999) Association of polymorphisms at the SR-BI gene locus with plasma lipid levels and body mass index in a white population. Arterioscler Thromb Vasc Biol 19:1734–1743 [DOI] [PubMed] [Google Scholar]
- Allain CC, Poon LS, Chan CS, Richmond W, Fu PC (1974) Enzymatic determination of total serum cholesterol. Clin Chem 4:470–475 [PubMed] [Google Scholar]
- Almasy L, Blangero J (1998) Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet 62:1198–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angius A, Melis PM, Morelli L, Petretto E, Casu G, Maestrale GB, Fraumene C, Bebbere D, Forabosco P, Pirastu M (2001) Archival, demographic and genetic studies define a Sardinian sub-isolate as a suitable model for mapping complex traits. Hum Genet 109:198–209 [DOI] [PubMed] [Google Scholar]
- Babel L (1991) Finding maximum cliques in arbitrary and in special graphs. Computing 46:321–341 [Google Scholar]
- Balas E, Niehaus W (1996) Finding large cliques in arbitrary graphs by bipartite matching. In: Johnson DS, Trick MA (eds): DIMACS series in discrete mathematics and theoretical computer science 26: Cliques, coloring, and satisfiability. American Mathematical Society, Providence, RI, pp 29–52 [Google Scholar]
- Boekholdt SM, Thompson JF (2003) Natural genetic variation as a tool in understanding the role of CETP in lipid levels and disease. J Lipid Res 44:1080–1093 [DOI] [PubMed] [Google Scholar]
- Breslow J (1988) Apolipoprotein genetic variation and human disease. Physiol Rev 68:85–132 [DOI] [PubMed] [Google Scholar]
- Bron C, Kerbosch J (1973) Finding all cliques of an undirected graph—algorithm 457. Communication of the ACM 16:575–577 [Google Scholar]
- Brooks-Wilson A, Marcil M, Clee SM, Zhang LH, Roomp K, van Dam M, Yu L, et al (1999) Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat Genet 22:336–345 [DOI] [PubMed] [Google Scholar]
- Chapman NH, Leutenegger AL, Badzioch MD, Bogdan M, Conlon EM, Daw EW, Gagnon F, Li N, Maia JM, Wijsman EM, Thompson EA (2001) The importance of connections: joining components of the Hutterite pedigree. Genet Epidemiol 21:S230–S235 [DOI] [PubMed] [Google Scholar]
- Chapman NH, Wijsman EM (2001) Introduction: linkage analyses in the Hutterites. Genet Epidemiol 21:S222–S223 [DOI] [PubMed] [Google Scholar]
- Ciulla TA, Sklar RM, Hauser SL (1988) A simple method for DNA purification from peripheral blood. Anal Biochem 174:485–488 [DOI] [PubMed] [Google Scholar]
- Cordell HJ (2004) Bias toward the null hypothesis in model-free linkage analysis is highly dependent on the test statistic used. Am J Hum Genet 74:1294–1302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criqui MH, Heiss G, Cohn R, Cowan LD, Chirayath MS, Bangdiwala S, Kritchevsky S, Jacobs DR Jr, O’Grady HK, Davis CE (1993) Plasma triglyceride level and mortality from coronary heart disease. N Engl J Med 328:1220–1225 [DOI] [PubMed] [Google Scholar]
- Dallinga-Thie GM, Bu XD, van Linde-Sibenius Trip M, Rotter JI, Lusis AJ, de Bruin TW (1996) Apolipoprotein A-I/C-III/A-IV gene cluster in familial combined hyperlipidemia: effects on LDL-cholesterol and apolipoproteins B and C-III. J Lipid Res 37:136–147 [PubMed] [Google Scholar]
- Dyer TD, Blangero J, Williams JT, Goring HH, Mahaney MC (2001) The effect of pedigree complexity on quantitative trait linkage analysis. Genet Epidemiol 21:S236–S243 [DOI] [PubMed] [Google Scholar]
- Evans V, Kastelein JJ (2002) Lipoprotein lipase deficiency—rare or common? Cardiovasc Drugs Ther 16:283–287 [DOI] [PubMed] [Google Scholar]
- Fraumene C, Petretto E, Angius A, Pirastu M (2003) Striking differentiation of sub-populations within a genetically homogeneous isolate (Ogliastra) in Sardinia as revealed by mtDNA analysis. Hum Genet 114:1–10 [DOI] [PubMed] [Google Scholar]
- Garcia CK, Mues G, Liao Y, Hyatt T, Patil N, Cohen JC, Hobbs HH (2001a) Sequence diversity in genes of lipid metabolism. Genome Res 11:1043–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia CK, Wilund K, Arca M, Zuliani G, Fellin R, Maioli M, Calandra S, Bertolini S, Cossu F, Grishin N, Barnes R, Cohen JC, Hobbs HH (2001b) Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein. Science 292:1394–1398 [DOI] [PubMed] [Google Scholar]
- Gardner CD, Fortmann SP, Krauss RM (1996) Association of small low-density lipoprotein particles with the incidence of coronary artery disease in men and women. JAMA 276:875–881 [PubMed] [Google Scholar]
- Gordon DJ, Probstfield JL, Garrison RJ, Neaton JD, Castelli WP, Knoke JD, Jacobs DR Jr, Bangdiwala S, Tyroler HA (1989) High-density lipoprotein cholesterol and cardiovascular disease: four prospective American studies. Circulation 79:8–15 [DOI] [PubMed] [Google Scholar]
- Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR (1977) High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am J Med 62:707–714 [DOI] [PubMed] [Google Scholar]
- Gudbjartsson DF, Jonasson K, Frigge ML, Kong A (2000) Allegro, a new computer program for multipoint linkage analysis. Nat Genet 25:12–13 [DOI] [PubMed] [Google Scholar]
- Hagberg JM, Wilund KR, Ferrell RE (2000) APO E gene and gene-environment effects on plasma lipoprotein-lipid levels. Physiol Genomics 4:101–108 [DOI] [PubMed] [Google Scholar]
- Harris MR, Bunker CH, Hamman RF, Sanghera DK, Aston CE, Kamboh MI (1998) Racial differences in the distribution of a low density lipoprotein receptor-related protein (LRP) polymorphism and its association with serum lipoprotein, lipid and apolipoprotein levels. Atherosclerosis 137:187–195 [DOI] [PubMed] [Google Scholar]
- Hauser ER, Boehnke M (1997) Confirmation of linkage results in affected-sib-pair linkage analysis for complex genetic traits. Am J Hum Genet Suppl 61:A278 [Google Scholar]
- Heath SC (1997) Markov chain Monte Carlo segregation and linkage analysis for oligogenic models. Am J Hum Genet 61:748–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegele RA, Brunt JH, Connelly PW (1995) Multiple genetic determinants of variation of plasma lipoproteins in Alberta Hutterites. Arterioscler Thromb Vasc Biol 15:861–871 [DOI] [PubMed] [Google Scholar]
- Higgins M (2000) Epidemiology and prevention of coronary heart disease in families. Am J Med 108:387–395 [DOI] [PubMed] [Google Scholar]
- Hokanson JE, Austin MA (1996) Plasma triglyceride level is a risk factor for cardiovascular disease independent of high-density lipoprotein cholesterol level: a meta-analysis of population-based prospective studies. J Cardiovasc Risk 3:213–219 [PubMed] [Google Scholar]
- Hunt SC, Hasstedt SJ, Kuida H, Stults BM, Hopkins PN, Williams RR (1989) Genetic heritability and common environmental components of resting and stressed blood pressures, lipids, and body mass index in Utah pedigrees and twins. Am J Epidemiol 129:625–638 [DOI] [PubMed] [Google Scholar]
- Ikeda Y, Takagi A, Nakata Y, Sera Y, Hyoudou S, Hamamoto K, Nishi Y, Yamamoto A (2001) Novel compound heterozygous mutations for lipoprotein lipase deficiency. A G-to-T transversion at the first position of exon 5 causing G154V missense mutation and a 50 splice site mutation of intron 8. J Lipid Res 42:1072–1081 [PubMed] [Google Scholar]
- Kong A, Gudbjartsson DF, Sainz J, Jonsdottir GM, Gudjonsson SA, Richardsson B, Sigurdardottir S, Barnard J, Hallbeck B, Masson G, Shlien A, Palsson ST, Frigge ML, Thorgeirsson TE, Gulcher JR, Stefansson K (2002) A high-resolution recombination map of the human genome. Nat Genet 31:241–247 [DOI] [PubMed] [Google Scholar]
- Kruglyak L, Lander ES (1998) Faster multipoint linkage analysis using Fourier transforms. J Comput Biol 5:1–7 [DOI] [PubMed] [Google Scholar]
- Lamarche B, Tchernof A, Mauriege P, Cantin B, Dagenais GR, Lupien PJ, Despres JP (1998) Fasting insulin and apolipoprotein B levels and low density lipoprotein particle size as risk factors for ischemic heart disease. JAMA 279:1955–1961 [DOI] [PubMed] [Google Scholar]
- Lander ES, Botstein D (1987) Homozygosity mapping: a way to map human recessive traits with the DNA of inbred children. Science 236:1567–1570 [DOI] [PubMed] [Google Scholar]
- Lander ES, Green P (1987) Construction of multilocus genetic linkage maps in humans. Proc Natl Acad Sci USA 84:2363–2367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander E, Kruglyak L (1995) Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11:241–247 [DOI] [PubMed] [Google Scholar]
- Lander ES, Schork NJ (1994) Genetic dissection of complex traits. Science 265:2037–2048 [DOI] [PubMed] [Google Scholar]
- Ledmyr H, Karpe F, Lundahl B, McKinnon M, Skoglund-Andersson C, Ehrenborg E (2002) Variants of the microsomal triglyceride transfer protein gene are associated with plasma cholesterol levels and body mass index. J Lipid Res 43:51–58 [PubMed] [Google Scholar]
- Manninen V, Elo MO, Frick MH, Haapa K, Heinonen OP, Heinsalmi P, Helo P, Huttunen JK, Kaitaniemi P, Koskinen P (1988) Lipid alterations and decline in the incidence of coronary heart disease in the Helsinki Heart Study. JAMA 260:641–651 [PubMed] [Google Scholar]
- Markianos K, Daly MJ, Kruglyak L (2001) Efficient multipoint linkage analysis through reduction of inheritance space. Am J Hum Genet 68:963–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPeek MS, Wu X, Ober C (2004) Best linear unbiased allele-frequency estimation in complex pedigrees. Biometrics 60:359–367 [DOI] [PubMed] [Google Scholar]
- Miettinen HE, Gylling H, Tenhunen J, Virtamo J, Jauhiainen M, Huttunen JK, Kantola I, Miettinen TA, Kontula K (1998) Molecular genetic study of Finns with hypoalphalipoproteinemia and hyperalphalipoproteinemia: a novel Gly230 Arg mutation (LCAT[Fin]) of lecithin:cholesterol acyltransferase (LCAT) accounts for 5% of cases with very low serum HDL cholesterol levels. Arterioscler Thromb Vasc Biol 18:591–598 [DOI] [PubMed] [Google Scholar]
- Miller GJ, Miller NE (1975) Plasma-high-density-lipoprotein concentration and development of ischaemic heart-disease. Lancet 1:16–19 [DOI] [PubMed] [Google Scholar]
- Murray CJL, Lopez AD (1997) Mortality by cause for eight regions of the world: global burden of disease study. Lancet 349:1269–1276 [DOI] [PubMed] [Google Scholar]
- National Cholesterol Education Program (2001) Third report of the National Cholesterol Education Program Expert Panel on Detection, Evaluation and Treatment of High Blood Cholesterol in Adults. Department of Health and Human Services, Public Health Service, National Institutes of Health, National Heart Lung and Blood Institute, Bethesda [Google Scholar]
- Newman DL, Abney M, Dytch H, Parry R, McPeek MS, Ober C (2003) Major loci influencing serum triglyceride levels on 2q14 and 9p21 localized by homozygosity-by-descent mapping in a large Hutterite pedigree. Hum Mol Genet 12:137–144 [DOI] [PubMed] [Google Scholar]
- O’Connell JR, Weeks DE (1998) PedCheck: a program for identifying genotype incompatibilities in linkage analysis. Am J Hum Genet 63:259–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajukanta P, Cargill M, Viitanen L, Nuotio I, Kareinen A, Perola M, Terwilliger JD, Kempas E, Daly M, Lilja H, Rioux JD, Brettin T, Viikari JSA, Rönnemaa T, Laakso M, Lander ES, Peltonen L (2000) Two loci on chromosomes 2 and X for premature coronary heart disease identified in early- and late-settlement populations of Finland. Am J Hum Genet 67:1481–1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankratz VS, Iturria SJ (2001) A pedigree partitioning approach to quantitative trait loci mapping of IgE serum level in the GAW12 Hutterite data. Genet Epidemiol 21:S258–S263 [DOI] [PubMed] [Google Scholar]
- Patsch W, Sharrett R, Chen LY, Lin-Lee Y, Spencer AB, Gotto AM Jr, Boerwinkle E (1994) Association of allelic differences at the A-I/C-111/A-IV gene cluster with carotid artery intima-media thickness and plasma lipid transport in hypercholesterolemic-hypertriglyceridemic humans. Arterioscler Thromb 14:874–883 [DOI] [PubMed] [Google Scholar]
- Perusse L, Rice T, Despres JP, Bergeron J, Province MA, Gagnon J, Leon AS, Rao DC, Skinner JS, Wilmore JH, Bouchard C (1997) Familial resemblance of plasma lipids, lipoproteins and postheparin lipoprotein and hepatic lipases in the HERITAGE Family Study. Arterioscler Thromb Vasc Biol 17:3263–3269 [DOI] [PubMed] [Google Scholar]
- Rhoads GG, Gulbrandsen CL, Kagan A (1976) Serum lipoproteins and coronary heart disease in a population study of Hawaii Japanese men. N Engl J Med 294:293–298 [DOI] [PubMed] [Google Scholar]
- Roberts SB, MacLean CJ, Neale MC, Eaves LJ, Kendler KS (1999) Replication of linkage studies of complex traits: an examination of variation in location estimates. Am J Hum Genet 65:876–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonfeld G (2003) Familial hypobetalipoproteinemia: a review. J Lipid Res 44:878–883 [DOI] [PubMed] [Google Scholar]
- Schork NJ, Greenwood TA (2004) Inherent bias toward the null hypothesis in conventional multipoint nonparametric linkage analysis. Am J Hum Genet 74:306–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shachter NS. (2001) Apolipoproteins C-I and C-III as important modulators of lipoprotein metabolism. Curr Opin Lipidol 12:297–304 [DOI] [PubMed] [Google Scholar]
- Shifman S, Darvasi A (2001) The value of isolated populations. Nat Genet 28:309–310 [DOI] [PubMed] [Google Scholar]
- Sobel E, Lange K (1996) Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. Am J Hum Genet 58:1323–1337 [PMC free article] [PubMed] [Google Scholar]
- Stampfer MJ, Krauss RM, Ma J, Blanche PJ, Holl LG, Sacks FM, Hennekens CH (1996) A prospective study of triglyceride level, low-density lipoprotein particle diameter, and risk of myocardial infarction. JAMA 276:882–888 [PubMed] [Google Scholar]
- Umans-Eckenhausen MA, Defesche JC, Sijbrands EJ, Scheerder RL, Kastelein JJ (2001) Review of first 5 years of screening for familial hypercholesterolaemia in the Netherlands. Lancet 357:165–168 [DOI] [PubMed] [Google Scholar]
- Warnick Gr, Benderson J, Albers JJ (1983) Dexstran sulfate-Mg+2 precipitation procedure for quantitation of high density lipoprotein cholesterol. In: Cooper GR (ed) Selected methods of clinical chemistry, vol 10. American Association for Clinical Chemistry, Washington DC, pp 91–99 [Google Scholar]
- Wijsman EM, Amos CI (1997) Genetic analysis of simulated oligogenic traits in nuclear and extended pedigrees: summary of GAW10 contributions. Genet Epidemiol 14:719–735 [DOI] [PubMed] [Google Scholar]
- Williams JT, Blangero J (1999) Power of variance component linkage analysis to detect quantitative trait loci. Ann Hum Genet 63:545–563 [DOI] [PubMed] [Google Scholar]
- Williams JT, Duggirala R, Blangero J (1997) Statistical properties of a variance components method for quantitative trait linkage analysis in nuclear families and extended pedigrees. Genet Epidemiol 14:1065–1070 [DOI] [PubMed] [Google Scholar]
- Wright AF, Carothers AD, Pirastu M (1999) Population choice in mapping genes for complex diseases. Nat Genet 23:397–404 [DOI] [PubMed] [Google Scholar]
- Zambon A, Deeb SS, Brown BG, Hokanson JE, Brunzell JD (2001) Common hepatic lipase gene promoter variant determines clinical response to intensive lipid-lowering treatment. Circulation 103:792–798 [DOI] [PubMed] [Google Scholar]