

Abstract
Isovaleric acidemia (IVA) is an inborn error of leucine metabolism that can cause significant morbidity and mortality. Since the implementation, in many states and countries, of newborn screening (NBS) by tandem mass spectrometry, IVA can now be diagnosed presymptomatically. Molecular genetic analysis of the IVD gene for 19 subjects whose condition was detected through NBS led to the identification of one recurring mutation, 932C→T (A282V), in 47% of mutant alleles. Surprisingly, family studies identified six healthy older siblings with identical genotype and biochemical evidence of IVA. Our findings indicate the frequent occurrence of a novel mild and potentially asymptomatic phenotype of IVA. This has significant consequences for patient management and counseling.
With the implementation of newborn screening (NBS) by tandem mass spectrometry (MS/MS), >20 metabolic disorders can now be detected very early in life, which allows for presymptomatic initiation of treatment of affected individuals and prevention of mental retardation or even death. Expanded NBS, however, also has led to the identification of individuals with mild and benign variants of some disorders, which complicates the choice of treatment for these infants as well as the genetic counseling of families. To date, this has become an issue for medium-chain acyl-CoA dehydrogenase and 3-methylcrotonyl-CoA carboxylase deficiencies (Andresen et al. 2001; Koeberl et al. 2003).
Isovaleric acidemia (IVA [MIM 243500]) is one autosomal recessive inborn error of leucine metabolism that is detectable by NBS by use of MS/MS. IVA is caused by a deficiency of isovaleryl-CoA dehydrogenase (IVD [Enzyme Commission number 1.3.99.10]), which catalyzes the third step in the catabolism of leucine. IVD, a member of the family of acyl-CoA dehydrogenases, is a homotetrameric mitochondrial flavoenzyme. The IVD gene is located on chromosome 15q14-15 and consists of 12 exons that span ∼15 kb of DNA. IVA is considered a severe, potentially life-threatening disorder that manifests with acute neonatal encephalopathy in about half of affected individuals and with recurrent episodes of vomiting, lethargy, coma, and varying degrees of developmental delay in the other half of patients (Sweetman and Williams 2001). IVD deficiency results in the accumulation of isovaleryl-CoA derivatives, and the biochemical diagnosis is based on the detection of isovalerylglycine and other metabolites in urine and of isovalerylcarnitine in plasma and blood spots. In addition to dietary protein restriction, therapy with carnitine and/or glycine is recommended. Early diagnosis by NBS allows immediate initiation of treatment and close monitoring, particularly during times of illness. On the basis of recent NBS reports, the incidence of IVA has a range from 1/62,500 live births in parts of Germany (Schulze et al. 2003) to ∼1/250,000 in the United States (Zytkovicz et al. 2001; Chace et al. 2003).
To date, highly heterogeneous mutations in the IVD gene have been characterized in patients with IVA, including point mutations, frameshifts, and splice-site mutations (Parimoo and Tanaka 1993; Vockley et al. 2000; Ensenauer et al. 2003). A phenotype/genotype correlation, however, has not been established for IVA. In the present study, the spectrum of mutations in the IVD gene in patients who received diagnosis by NBS was investigated and correlated to the clinical and biochemical phenotype. All samples in this study were collected and analyzed with the approval of Mayo Clinic’s institutional review board.
Twelve patients with IVA from Germany and seven patients from the United States, born between 1998 and 2004, were investigated. Eleven patients are females and eight are males. Sixteen patients are white, two patients are Arabic, and one patient is Hispanic. All patients received diagnosis through expanded NBS by use of MS/MS, which revealed mildly to markedly elevated concentrations of C5 acylcarnitine (table 1). Treatment for IVA was begun upon diagnosis, and all patients (now 2 mo to 5 years of age [median age 1.9 years]) have developed appropriately. For all patients, follow-up biochemical genetic testing included (1) plasma acylcarnitine analysis and (2) urine organic acid and/or acylglycine analyses. These assays were performed using established procedures (Rinaldo et al., in press). Diagnostic values were observed for the characteristic IVA metabolites, and the extent of abnormalities showed a variable pattern similar to that of the NBS C5 acylcarnitine concentrations (table 1).
Table 1.
Genotypes and Biochemical Phenotypes of Subjects with IVA
| Patients with IVA Diagnosed by NBS, with Genotypeb |
|||||
| Metabolite (Sample Type) | Reference Range | A282V/A282V | A282V/Non-A282V | Non-A282V/Non-A282V | “Symptomatic Patientswith IVA” |
| C5AC (NBS blood spots) (μmol/liter) | <.3–.99a | 3.0 (2.7–4.0) [n=4] | 2.4 (.8c–6.0) [n=8] | 10 (6.9–21.7) [n=5] | … |
| Isovalerylglycine (urine) (mmol/mol creatinine) | <10 | 51 (23–79) [n=2] | 133 (15–195) [n=8] | 630 (7–3,331) [n=5] | 1,219 (685–4,541) [n=9] |
| C5AC (plasma) (μmol/liter) | <.44 | 3.00 (.18–7.60) [n=4] | 2.85 (.66–11.50) [n=7] | 3.00 (.98–29.00) [n=5] | 1.01 (.30–48.20) [n=17] |
| Isovalerylcarnitine (fibroblasts) (μmol/g protein) | <.12 (n=88) | .25 (.24–.25) [n=2] | .55 (.15–.67) [n=3] | .23 [n=1] | 1.01 (.82–3.09) [n=8] |
| Isovalerylcarnitine (lymphoblasts) (μmol/g protein) | <.01 (n=11) | .29 (.22–.37) [n=2] | .14 (.03–.81) [n=4] | .57 (.39–.75) [n=2] | .71 (.31–1.3) [n=15] |
Range of cutoff values in the laboratories where the analyses were performed.
Genotypes and biochemical phenotypes of 19 patients with IVA diagnosed by NBS, compared with the biochemical phenotype of a group of patients with IVA diagnosed after they had presented clinically (“symptomatic patients”). Median-values; ranges are provided in parentheses. C5AC = C5 acylcarnitine.
For the patient with a C5AC concentration of .8 μmol/liter, the cutoff value of the screening laboratory was .6 μmol/liter.
To further characterize these patients, molecular genetic analysis of the IVD gene was undertaken, by use of genomic DNA extracted from fibroblasts, whole blood, or dried blood spots. All 12 exons, including part of the flanking intron sequences of the IVD gene, were amplified by use of PCR, with intron-located primers, under standard conditions. Mutation analysis was performed by direct sequencing of PCR-amplified fragments, by use of an ABI PRISM 3730 XL DNA Analyzer (96-capillary [Perkin Elmer Applied Biosystems]). Primer sequences are available on request. Twelve missense mutations, four splice-site mutations, and one frameshift mutation were identified (table 2). Three missense mutations—932C→T, 149G→C, and 125T→C—and 1 splice-site mutation—IVS4+2T→C—have been reported elsewhere in symptomatic patients (Mohsen et al. 1998), whereas the remaining 13 mutations were novel. Notably, the 932C→T (A282V) mutation was found in 18 (47%) of 38 of mutant alleles from the individuals identified by NBS, including 5 homozygous for this mutation (table 2). Eleven patients who carried the 932C→T mutation were white, and two patients were Arabic. Previous in vitro kinetic studies on purified recombinant A282V-mutant enzyme have demonstrated a significantly increased Km of 27 μM and a markedly decreased catalytic efficiency of 11.4 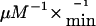 compared with wild-type enzyme (3.1 μM and 520
compared with wild-type enzyme (3.1 μM and 520 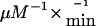 , respectively), which substantiates functional impairment of the mutant enzyme (Mohsen et al. 1998). Furthermore, the A282V mutant showed thermal instability, a finding that might indicate a potential risk for the development of symptoms, mainly during times of febrile illnesses (Nasser et al. 2004).
, respectively), which substantiates functional impairment of the mutant enzyme (Mohsen et al. 1998). Furthermore, the A282V mutant showed thermal instability, a finding that might indicate a potential risk for the development of symptoms, mainly during times of febrile illnesses (Nasser et al. 2004).
Table 2.
Genotypes of 19 Patients with IVA Diagnosed by NBS[Note]
| Patient | Allele 1 | Allele 2 |
| 1 | 932C→T (A282V) |
932C→T (A282V) |
| 2 | 932C→T (A282V) |
932C→T (A282V) |
| 3 | 932C→T (A282V) |
932C→T (A282V) |
| 4 | 932C→T (A282V) |
932C→T (A282V) |
| 5 | 932C→T (A282V) |
932C→T (A282V) |
| 6 | 932C→T (A282V) |
149G→A (R21H)a |
| 7 | 932C→T (A282V) |
149G→C (R21P) |
| 8 | 932C→T (A282V) |
262G→T (G59C)a |
| 9 | 932C→T (A282V) |
359G→A (G91E)a |
| 10 | 932C→T (A282V) |
436A→G (K117E)a |
| 11 | 932C→T (A282V) |
868G→A (G261R)a |
| 12 | 932C→T (A282V) |
870_871insGa |
| 13 | 932C→T (A282V) |
IVS4+2T→C |
| 14 | 125T→C (L13P) | IVS2-1G→Ca |
| 15 | 149G→C (R21P) | IVS8-2A→Ga |
| 16 | 149G→C (R21P) | 851G→A (R255Q)a |
| 17 | 943A→C (K286Q)a | IVS3+5G→Ta,b |
| 18 | 1136T→C (F350S)a | IVS3+5G→Tb |
| 19 | 358G→A (G91R)a | NI |
Note.— Nucleotide changes are shown; the corresponding amino acid change of the mature protein is in parentheses. The most common mutation (932C→T) is underlined. NI= not identified.
Novel mutations.
By comparison of the sequence environment with the wild-type sequence, by use of Splice Site Prediction by Neural Network, this intronic variant is expected to affect RNA splicing by compromising the splice-site consensus sequence.
Because of the high frequency of this mutation in our study population, we genotyped 100 randomly selected subjects born at the Mayo Clinic in Rochester, MN, who had normal NBS results, by use of DNA extracted from the original NBS blood spots. For this purpose, we developed a mutation-specific site-directed mutagenesis assay for the 932C→T allele on the basis of creation of a new restriction endonuclease recognition site for BsaJI. After amplification by use of the forward primer (5′-CTGGGATTCTGGCCTTCCCACGCTAGCATTTTGCCACCACACCCGGTGGTGGGATGAGGAGGTGCTCAC-3′) at 0.3 μM and a mutagenic reverse primer (the altered base is underlined) (5′-CAGTTGAAGAGTACATGTCAGCAAAAGCACTCGGCTCACCTGGAAGTGGCCGATCTTCTGGCCCAA-3′) at 0.3 μM, a restriction site for BsaJI is introduced into the wild-type IVD sequence that results in cleavage of the 213-bp product by BsaJI into two fragments, of 146 bp and 67 bp. The mutated sequence remains undigested by this enzyme. Fragments were separated by gel electrophoresis on a 3% Nusieve/agarose gel. The assay was validated by DNA from dried blot spots previously shown by sequence analysis to harbor the 932C→T mutation on one or both IVD gene alleles (fig. 1). The 932C→T mutation was not detected in any of the 100 randomly selected individuals with normal NBS test results.
Figure 1.

Restriction-enzyme analysis for the detection of the 932C→T (A282V) mutation. The 213-bp product is cleaved into two fragments, of 146 bp and 67 bp, in the presence of the wild-type sequence (CC). The mutated sequence is not cleaved (see lanes CT and TT).
To determine the enzymatic activity of IVD, transformed lymphocytes of five patients (homozygosity for 932C→T in two patients, compound heterozygosity for 932C→T and another mutation in one patient, and compound heterozygosity for different mutations in two patients) were investigated using the anaerobic electron-transferring flavoprotein fluorescence reduction assay (Frerman and Goodman 1985). In all cases, no enzyme activity was detected. To further delineate the biochemical effect of the IVD gene mutations in our patients, the accumulation of isovalerylcarnitine was analyzed in a culture medium of fibroblasts or transformed lymphocytes, after incubation with palmitic acid, l-carnitine, and isotopically labeled l-valine and l-isoleucine. Similar to assays described elsewhere (Roe and Roe 1999; Shen et al. 2000), skin fibroblast or Epstein-Barr virus–transformed lymphocyte cultures from the patients, a healthy control, and an IVD-deficient control individual were grown in triplicate in T25 flasks to ∼90% confluency. Culture medium was then removed, and 3 ml of modified MEM (Modified RPMI 1640 with l-glutamine and without isoleucine and valine [JRH Biosciences]) was added. This incubation medium contains fatty acid–free BSA (200 mg/dl), palmitic acid (0.2 mmol/liter), l-carnitine (0.4 mmol/liter [Sigma]), and isotopically labeled l-valine (0.8 mmol/liter U-13C-l-valine) and l-isoleucine (U-13C-l-isoleucine [Cambridge Isotope Laboratories]). After 72 h of incubation at 37°C, the medium was collected from each flask for acylcarnitine analysis (Shen et al. 2000). The remaining cells were gently washed in PBS (Dulbecco’s PBS [Cellgro by Mediatech]) and were harvested by trypsinization (0.25 ml 0.25% trypsin-EDTA [Life Technologies]). Protein content was measured in all cell pellets after sonication, by use of the method described by Lowry et al. (1951), and the concentration of isovalerylcarnitine in the cell medium was normalized to the protein concentration.
All patient cell lines accumulated increased levels of isovalerylcarnitine in the cell medium, as compared with normal controls (table 1), regardless of whether fibroblasts or lymphoblasts were used. Four newborns who were homozygous for the 932C→T mutation had isovalerylcarnitine concentrations with a range of 0.2–0.4 μmol/g protein (table 1). In contrast, fibroblast cultures available from patients who had previously received diagnoses of IVA after clinical manifestation (“symptomatic patients”) (n=8) produced isovalerylcarnitine concentrations >0.4 μmol/g protein, whereas 93% of lymphoblast cultures from such patients (n=15) exceeded that value (table 1). No patient in this group carried the 932C→T mutation. Overall, the differences in isovalerylcarnitine concentrations between the group of newborns who were either homozygous or compound heterozygous for the 932C→T mutation and the symptomatic patients were statistically significant for both fibroblasts (Wilcoxon rank sum test; P<.0020) and lymphoblasts (P<.0091).
Review of the initial NBS results also revealed a correlation with the genotype. The C5 acylcarnitine concentration in the dried blood spots of newborns carrying the 932C→T mutant allele, in either homozygous or compound heterozygous fashion, was at least 1.3× above the cutoff value of the individual screening laboratories (table 1). However, the C5 acylcarnitine concentration in NBS bloods spots of these patients was significantly lower than in those of newborns with two different IVD gene mutations (P<.0019) (table 1).
Individuals who harbored the 932C→T mutation also had an abnormal urinary excretion of isovalerylglycine; however, this was much less pronounced than in the group of symptomatic patients (n=9) (P<.0001) (table 1). All patients were in a stable clinical condition when specimens were collected. To avoid treatment artifacts, all patients who received glycine supplementation were excluded from this analysis. Plasma C5 acylcarnitine concentrations did not reliably differentiate between the patients carrying the 932C→T mutation and the symptomatic patient group (P<.2037) (table 1), whereas this parameter generally allowed differentiation between unaffected individuals and patients with IVA. It is important to note that all plasma samples were collected when the patients were already being treated with oral l-carnitine supplements at variable doses (15–150 mg/kg body weight per day); assessment of correlation between plasma C5 acylcarnitine concentration and patient genotype was not possible.
These findings raise the possibility that IVD deficiency can be associated with a mild or asymptomatic phenotype that is now detected by NBS. We are aware of two patients, aged 4.5 and 14 years, who are compound heterozygous (the 932C→T mutation on one allele and different mutations on the second allele). They received diagnoses at age 4 years, after a metabolic work-up for mild developmental delay, but have not experienced acute episodes of metabolic decompensation. It is uncertain whether the presence of the 932C→T mutation and a second mutation in the IVD gene explains their clinical phenotype or whether there is an underlying ascertainment bias. On the basis of previous in vitro studies of this variant enzyme (Mohsen et al. 1998; Nasser et al. 2004), we understand that it is possible that individuals homozygous for this allele or compound mutant for it and another defective allele are at risk of having acute metabolic crises when exposed to common stressors such as fever and prolonged fasting.
To determine whether older siblings of our patients might have the same genotype and biochemical findings, 24 asymptomatic siblings from 11 families were evaluated by urine acylglycine and/or plasma acylcarnitine analyses followed by mutation analysis. Eight of these families had a newborn who was either homozygous (n=2) or compound heterozygous (n=6) for the 932C→T mutation. One additional child with a similar biochemical phenotype and identical genotype was identified in each of the two families with newborns homozygous for the 932C→T mutation and in four families with newborns carrying one copy of the mutation and a second mutation in the other allele. Each of the siblings, aged 3, 4, 6, 7, 8, and 11 years at the time of identification, had normal development and remained asymptomatic during episodes of common pediatric illnesses. Study of the families without at least one copy of the 932C→T mutation did not disclose additional siblings with IVA.
Overall, our results suggest that individuals with IVD deficiency discovered by NBS and who have at least one copy of a 932C→T (A282V) mutant allele can exhibit a mild phenotype or be free of symptoms throughout childhood. This is a major departure from our previous understanding of the natural history of IVA; there are obvious implications for the management and genetic counseling of individuals identified through NBS, which include the need for genotyping of the IVD gene in the early stages of follow-up. The expansion of NBS has led to the identification of mild and potentially benign forms of disorders heretofore considered to have more consistently serious implications. Our findings for IVD deficiency lead to a novel variation of this problem. Whereas appropriate and specific information on the nature of a disorder identified by NBS leads to a higher rate of parental satisfaction and to reduced stress levels, the opposite is true for false-positive results (Waisbren et al. 2003). Accordingly, dealing with situations in which long-term outcome is uncertain requires particular care in management and counseling, to avoid unnecessary stress and anxiety. This is complicated further by the fact that C5 acylcarnitine represents a mixture of isomers—namely, isovalerylcarnitine, 2-methylbutyrylcarnitine, and pivaloylcarnitine—that cannot be differentiated by MS/MS analysis, as applied in the NBS setting. 2-Methylbutyrylcarnitine is elevated in patients with 2-methylbutyrylglycinuria, a recently discovered inborn error of isoleucine catabolism (Andresen et al. 2000; Gibson et al. 2000), whereas pivaloylcarnitine is derived from pivalic acid, a component of several antibiotics (Abdenur et al. 1998).
The present study shows that a mild elevation of the C5 acylcarnitine concentration in NBS blood spots does not rule out a diagnosis of IVA, as previously hypothesized (Matern et al. 2003). Urine acylglycine and organic acid analyses are necessary first steps in the differentiation between these inborn errors of branched-chain amino acid (BCAA) metabolism and a false-positive result due to medication, as illustrated in the algorithm in figure 2. In vitro study of BCAA metabolism on either fibroblasts or lymphoblasts is a useful and sometimes necessary next step for confirmation of the diagnosis and for detection of functional differences between individuals carrying the 932C→T mutation and symptomatic patients. These investigations are ultimately key for definition of the clinical and biochemical phenotype in individuals carrying the 932C→T mutant IVD gene allele.
Figure 2.

Algorithm for the diagnostic evaluation of newborns with an elevated C5 acylcarnitine concentration in an NBS blood spot. An asterisk (*) indicates that NBS was not available as a clinical test (research only). AC = acylcarnitine analysis; AG = acylglycine analysis; C5AC = C5 acylcarnitine; H = abnormally elevated; IVC = isovalerylcarnitine; IVG = isovalerylglycine; MBC = 2-methylbutyrylcarnitine; MBG = 2-methylbutyrylglycine; N = normal; ND = 932C→T mutation not detected; OA = organic acid analysis; PVA = pivalic acid (a component of several antibiotic medications [Abdenur et al. 1998]); SBCAD-D = short/branched-chain acyl-CoA dehydrogenase deficiency (2-methylbutyrylglycinuria).
In summary, our findings suggest the existence of a new, mild phenotype of IVA, detected by NBS, that is associated with a 932C→T mutation in the IVD gene; however, the long-term implications of this mutation require further study. To this end, patients who receive diagnosis of mild IVA outside of infancy should be evaluated for the presence of the 932C→T mutation and should be characterized carefully at the molecular and biochemical levels. We recommend early genotyping and sibling evaluation of all new cases of IVA that are diagnosed by NBS.
Acknowledgments
Financial support was provided by the Barbara Woodward Lipps fund of the Mayo Foundation (to J.V.) and by Public Health Service grant R01-DK45482 (to J.V.). The funding sources had no involvement in study design; in the collection, analysis, and interpretation of data; in the writing of the report; or in the decision to submit the article for publication. The authors thank Jennifer L. Winters and W. Edward Highsmith Jr., Ph.D., for assistance with molecular genetic analysis and are grateful to all physicians, genetic counselors, and screening laboratories (Drs. J. Charrow, J. W. Ellison, T. Freese, D. K. Grange, C. Korenke, W. Lehnert, M. Leichsenring, M. Lindner, L. A. Schimmenti, J. P. Schubert, K. O. Schwab, L. Sweetman, D. A. H. Whiteman, and D. A. Pond, M.S.) who provided patient samples and/or data.
Electronic-Database Information
The URLs for data presented herein are as follows:
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for IVA) [PubMed]
- Splice Site Prediction by Neural Network, http://www.fruitfly.org/seq_tools/splice.html
References
- Abdenur JE, Chamoles NA, Guinle AE, Schenone AB, Fuertes AN (1998) Diagnosis of isovaleric acidaemia by tandem mass spectrometry: false positive result due to pivaloylcarnitine in a newborn screening programme. J Inherit Metab Dis 21:624–630 10.1023/A:1005424331822 [DOI] [PubMed] [Google Scholar]
- Andresen BS, Christensen E, Corydon TJ, Bross P, Pilgaard B, Wanders RJA, Ruiter JPN, Simonsen H, Winter V, Knudsen I, Schroeder LD, Gregersen N, Skovby F (2000) Isolated 2-methylbutyrylglycinuria caused by short/branched-chain acyl-CoA dehydrogenase deficiency: identification of a new enzyme defect, resolution of its molecular basis, and evidence for distinct acyl-CoA dehydrogenases in isoleucine and valine metabolism. Am J Hum Genet 67:1095–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andresen BS, Dobrowolski SF, O’Reilly L, Muenzer J, McCandless SE, Frazier DM, Udvari S, Bross P, Knudsen I, Banas R, Chace DH, Engel P, Naylor EW, Gregersen N (2001) Medium-chain acyl-CoA dehydrogenase (MCAD) mutations identified by MS/MS-based prospective screening of newborns differ from those observed in patients with clinical symptoms: identification and characterization of a new, prevalent mutation that results in mild MCAD deficiency. Am J Hum Genet 68:1408–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chace DH, Kalas TA, Naylor EW (2003) Use of tandem mass spectrometry for multianalyte screening of dried blood specimens from newborns. Clin Chem 49:1797–1817 10.1373/clinchem.2003.022178 [DOI] [PubMed] [Google Scholar]
- Ensenauer R, Grünert S, Willard J, Matern D, Wendel U, Lehnert W, Schwab KO, Brandis M, Mohsen A-W, Vockley J (2003) Natural history of isovaleric acidemia (IVA). J Inherit Metab Dis Suppl 2 26:38 [Google Scholar]
- Frerman FE, Goodman SI (1985) Fluorometric assay of acyl-CoA dehydrogenases in normal and mutant human fibroblasts. Biochem Med 33:38–44 10.1016/0006-2944(85)90124-3 [DOI] [PubMed] [Google Scholar]
- Gibson KM, Burlingame TG, Hogema B, Jakobs C, Schutgens RB, Millington D, Roe CR, Roe DS, Sweetman L, Steiner RD, Linck L, Pohowalla P, Sacks M, Kiss D, Rinaldo P, Vockley J (2000) 2-Methylbutyryl-coenzyme A dehydrogenase deficiency: a new inborn error of L-isoleucine metabolism. Pediatr Res 47:830–833 [DOI] [PubMed] [Google Scholar]
- Koeberl DD, Millington DS, Smith WE, Weavil SD, Muenzer J, McCandless SE, Kishnani PS, McDonald MT, Chaing S, Boney A, Moore E, Frazier DM (2003) Evaluation of 3-methylcrotonyl-CoA carboxylase deficiency detected by tandem mass spectrometry newborn screening. J Inherit Metab Dis 26:25–35 10.1023/A:1024015227863 [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275 [PubMed] [Google Scholar]
- Matern D, He M, Berry SA, Rinaldo P, Whitley CB, Madsen PP, Van Calcar SC, Lussky RC, Andresen BS, Wolff JA, Vockley J (2003) Prospective diagnosis of 2-methylbutyryl-CoA dehydrogenase deficiency in the Hmong population by newborn screening using tandem mass spectrometry. Pediatrics 112:74–78 10.1542/peds.112.1.74 [DOI] [PubMed] [Google Scholar]
- Mohsen AW, Anderson BD, Volchenboum SL, Battaile KP, Tiffany K, Roberts D, Kim JJ, Vockley J (1998) Characterization of molecular defects in isovaleryl-CoA dehydrogenase in patients with isovaleric acidemia. Biochemistry 37:10325–10335 10.1021/bi973096r [DOI] [PubMed] [Google Scholar]
- Nasser I, Mohsen AW, Jelesarov I, Vockley J, Macheroux P, Ghisla S (2004) Thermal unfolding of medium chain acyl-CoA dehydrogenase and iso(3)valeryl-CoA dehydrogenase: study of the effect of genetic defects on enzyme stability. Biochim Biophys Acta 1690:22–32 [DOI] [PubMed] [Google Scholar]
- Parimoo B, Tanaka K (1993) Structural organization of the human isovaleryl-CoA dehydrogenase gene. Genomics 15:582–590 10.1006/geno.1993.1111 [DOI] [PubMed] [Google Scholar]
- Rinaldo P, Hahn SH, Matern D. Inborn errors of amino acid, organic acid, and fatty acid metabolism. In: Ashwood ER, Bruns DE, Burtis CA (eds) Tietz textbook of clinical chemistry. W. B. Saunders (in press) [Google Scholar]
- Roe CR, Roe DS (1999) Recent developments in the investigation of inherited metabolic disorders using cultured human cells. Mol Genet Metab 68:243–257 10.1006/mgme.1999.2911 [DOI] [PubMed] [Google Scholar]
- Schulze A, Lindner M, Kohlmüller D, Olgemöller K, Mayatepek E, Hoffmann GF (2003) Expanded newborn screening for inborn errors of metabolism by electrospray ionization-tandem mass spectrometry: results, outcome, and implications. Pediatrics 111:1399–1406 10.1542/peds.111.6.1399 [DOI] [PubMed] [Google Scholar]
- Shen JJ, Matern D, Millington DS, Hillman S, Feezor MD, Bennett MJ, Qumsiyeh M, Kahler SG, Chen YT, Van Hove JL (2000) Acylcarnitines in fibroblasts of patients with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency and other fatty acid oxidation disorders. J Inherit Metab Dis 23:27–44 10.1023/A:1005694712583 [DOI] [PubMed] [Google Scholar]
- Sweetman L, Williams JC (2001) Branched chain organic acidurias. In: Scriver CR, Beaudet AL, Valle D, Sly WS, Childs B, Kinzler KW, Vogelstein B (eds) The metabolic and molecular bases of inherited disease. Vol 2. McGraw-Hill, New York, pp 2125–2163 [Google Scholar]
- Vockley J, Rogan PK, Anderson BD, Willard J, Seelan RS, Smith DI, Liu W (2000) Exon skipping in IVD RNA processing in isovaleric acidemia caused by point mutations in the coding region of the IVD gene. Am J Hum Genet 66:356–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waisbren SE, Albers S, Amato S, Ampola M, Brewster TG, Demmer L, Eaton RB, Greenstein R, Korson M, Larson C, Marsden D, Msall M, Naylor EW, Pueschel S, Seashore M, Shih VE, Levy HL (2003) Effect of expanded newborn screening for biochemical genetic disorders on child outcomes and parental stress. JAMA 290:2564–2572 [DOI] [PubMed] [Google Scholar]
- Zytkovicz TH, Fitzgerald EF, Marsden D, Larson CA, Shih VE, Johnson DM, Strauss AW, Comeau AM, Eaton RB, Grady GF (2001) Tandem mass spectrometric analysis for amino, organic, and fatty acid disorders in newborn dried blood spots: a two-year summary from the New England Newborn Screening Program. Clin Chem 47:1945–1955 [PubMed] [Google Scholar]