

Abstract
White-rot fungi are efficient organisms for the mineralization of lignin and polysaccharides into CO2 and H2O. Despite their biotechnological potential, WRF metabolism remains underexplored. Building on recent findings regarding the utilization of lignin-related aromatic compounds as carbon sources by WRF, we aimed to gain further insights into these catabolic processes. For this purpose, Trametes versicolor and Gelatoporia subvermispora were incubated in varying conditions – in static and agitation modes and different antioxidant levels – during the conversion of 4-hydroxybenzoic acid (a lignin-related compound) and cellobiose. Their metabolic responses were assessed via transcriptomics, proteomics, lipidomics, metabolomics, and microscopy analyses. These analyses reveal the significant impact of cultivation conditions on sugar and aromatic catabolic pathways, as well as lipid composition of the fungal mycelia. Additionally, this study identifies biosynthetic pathways for the production of extracellular fatty acids and phenylpropanoids – both products with relevance in biotechnological applications – and provides insights into carbon fate in nature.
Subject terms: Systems analysis, Multicellular systems
Cultivation of white-rot fungi in distinct incubation conditions and in the presence of a lignin-related aromatic compound induces significant metabolic changes on aromatic catabolic pathways in the transcriptome, proteome, lipidome, and metabolome.
Introduction
Plant-derived biomass stands as the preeminent biogenic carbon source on earth and is predominantly comprised of cellulose, hemicellulose, and lignin. While the proportions of these components vary across plant types, lignin—an energy-rich polyaromatic molecule—is the second most abundant source of organic carbon on the planet1,2. Although numerous bacteria and fungi can depolymerize cellulose and hemicellulose in natural environments, white-rot fungi (WRF) are the most efficient organisms depolymerizing lignin3. The mechanisms for depolymerizing lignin have been extensively studied, revealing laccases, peroxidases, and other enzymes and metabolites responsible for breaking down lignin into smaller components4. In general, lignin depolymerization can result in a randomized pool of aromatics such as 4-hydroxybenzoate (4-HBA), syringate, and vanillate5–7. However, the conversion of these compounds by WRF remains underexplored. Recent investigations have revealed the utilization of 4-HBA as a carbon source in two species of WRF6 (Trametes versicolor and Gelatoporia (Ceriporiopsis) subvermispora) and have elucidated a catabolic pathway from 4-HBA towards central carbon metabolism via hydroxyquinol8. These discoveries elevate these WRF as promising candidates for ‘lignin consolidated bioprocesses’ to enable simultaneous depolymerization of lignin and conversion of aromatic compounds to valued-added products, which is a critical path to increase the feasibility of lignocellulosic biorefineries9,10. Despite these advances, the overall intracellular metabolism of WRF remains understudied, yet it is crucial to strategically and efficiently conducting future genetic manipulations in these organisms.
The selection of adequate fungal cultivation conditions become imperative for biotechnological applications. Notably, typical WRF growth conditions that influence aeration, such as the choice between cultivations conducted in static or agitation mode, can create distinct gradients of oxygen and other redox-active species11,12, thereby impacting the uptake and catabolism of carbon sources such as aromatic compounds derived from lignin. Discrepancies in fungal cell morphology and variations in cell wall structure under different cultivation conditions may also affect the permeability of the cell wall, altering the uptake and efflux of these compounds13,14. Another major challenge associated with the utilization of monomeric aromatic compounds by WRF lie in the susceptibility to repolymerization catalyzed by secreted fungal ligninolytic enzymes15. In nature, various factors prevent repolymerization, including enzymes like aryl alcohol oxidases within the lignin-fungal matrix, other ecosystem organisms acting as carbon sinks, and natural antioxidants scavenging reactive oxygen species (ROS)16–18. Hence, supplementing WRF with antioxidants (AO) could prove instrumental in preventing the repolymerization of monomeric aromatic compounds and enhancing their uptake. In addition, AO have demonstrated the capacity to alleviate the detrimental effects of ROS and reinforce cell walls by promoting ergosterol biosynthesis in the yeast Candida albicans19,20. Therefore, understanding the effect of cultivation conditions on WRF metabolism, morphology, and lipid composition is necessary to optimize bioprocesses.
Systems biology encompasses a suite of techniques (e.g., transcriptomics, proteomics, metabolomics, lipidomics) that can be pieced together to understand the larger metabolic network within an organism21. Systems biology studies in WRF have predominantly centered on genomics, transcriptomics, and extracellular proteomics22. However, intracellular proteomics, metabolomics, and lipidomics should be also integrated into the analytical framework to provide further insights into the fate of the carbon utilized by these organisms. For example, the integration of these multi-omic datasets allowed metabolic pathway reconstruction and analysis of lignocellulosic carbon utilization in the yeast Rhodosporidioum toruloides23 and leveraged the foundation for understanding the catabolism of lignin-related compounds by WRF6. Therefore, systems biology is a powerful approach to discover pathways and enzymes for unlocking the full biotechnological potential of WRF.
In this study, we examine the effect of incubation mode (static and agitation) and AO presence on the metabolism of T. versicolor and G. subvermispora. We first elucidate the complete 4-HBA catabolic pathway in T. versicolor via in vivo enzyme functional analyses in a bacterium. Then, we integrate fungal phenotypes with targeted transcriptomics, proteomics, and metabolomics to understand 4-HBA catabolism in the various cultivation conditions. Lastly, extracellular metabolomics and lipidomics were used to identify fungal pathways and carbon fate, while microscopy provided mycelial structural information, offering valuable information from applied and fundamental biology perspectives.
Results
In vivo functional validation of oxidative decarboxylases in a bacterium solves the full hydroxyquinol pathway in T. versicolor and reveals functional redundancy in G. subvermispora
The catabolic pathway of 4-HBA towards central carbon metabolism has been recently elucidated in G. subvermispora8. However, the first biochemical reaction in the pathway (oxidative decarboxylation of 4-HBA to hydroquinone) remains undescribed in T. versicolor due to enzyme solubility issues during the heterologous expression of putative oxidative decarboxylases in Escherichia coli8. To fully analyze the 4-HBA catabolic pathway in T. versicolor, we utilized a different approach to validate enzyme function. We selected a putative oxidative decarboxylase from T. versicolor (TV_175239)6 and expressed it in vivo in an engineered Pseudomonas putida KT2440 bacterial strain (which lacks this catabolic activity). We also included a putative oxidative decarboxylase from G. subvermispora (GS_119636) in this study, which was also reported to be insoluble6. As positive control, we used an enzyme from G. subvermispora previously validated (GS_120062)8. As expected based on previous literature24, the wild type P. putida KT2440 fully utilized 4-HBA as carbon source and the engineered strain with pobAR knocked out hampered any 4-HBA conversion (Fig. 1). The positive control and strains containing TV_175239 and GS_119636 showed 4-HBA conversion to hydroquinone, which validates the function of these fungal enzymes (Fig. 1). Therefore, both enzymes were further included in this study.
Fig. 1. Functional validation of oxidative decarboxylases from WRF in the bacterium P. putida KT2440.
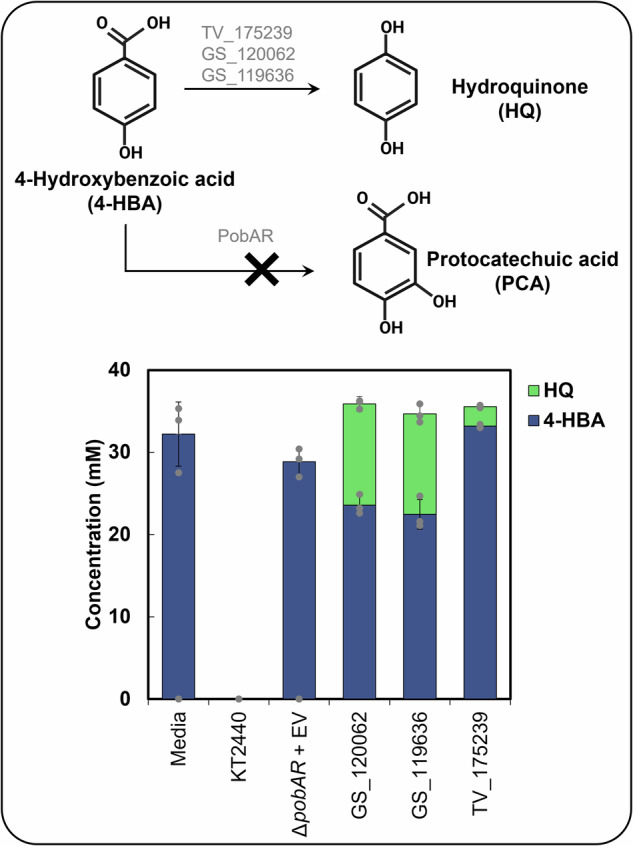
The figure shows 4-hydroxybenzoic acid (4-HBA) conversion to hydroquinone (HQ) by WRF and to protocatechuic acid by P. putida KT2440, as well as metabolite concentrations in the supernatant of cultivations conducted with engineered P. putida expressing oxidative decarboxylases from G. subvermispora (GS) and T. versicolor (TV). Bars represent the average concentration from biological triplicates and error bars show the standard deviation. Dot-plots depict individual data points. Media = abiotic media; KT2440= wild-type P. putida; pobAR + EV = P. putida KT2440 with pobAR knockout and empty vector.
Both incubation mode and AO supplementation impact 4-HBA and sugar conversion by G. subvermispora and Tversicolor
To better understand the catabolism of lignin-related aromatic compounds by G. subvermispora and T. versicolor, we evaluated the effect of cultivation conditions on 4-HBA conversion. Our recent study6 confirmed the catabolism of 4-HBA as a carbon source in both WRF in cultivations containing 4-HBA and cellobiose and incubated in static mode with AO (i.e., ascorbic acid and α-tocopherol). Here, we utilize a similar experimental setup, and include additional variables to understand the effect of incubation mode (static or agitation) and AO level (with or without AO). Specifically, the WRF were incubated in minimal media containing cellobiose as the sole carbon source in either static or agitation mode. After 6 and 10 days, 4-HBA was added to the cultivations of T. versicolor and G. subvermispora, respectively, with or without AO. Cellobiose and 4-HBA were monitored in the supernatant (Fig. 2) and, samples for systems biology analyses were harvested 24 h after the addition of 4-HBA.
Fig. 2. Conversion of cellobiose and 4-hydroxybenzoic acid (4-HBA) in the extracellular fractions in different cultivation conditions by WRF.

(a–c) G. subvermispora (GS) and (d–f) T. versicolor (TV) profiles. (a and d) Cellobiose concentration overtime, (b and e) glucose concentration overtime, (c and f) 4-HBA concentration after its addition in the cultivations and total conversion levels after 24 h. Data show averages from three biological replicates. Error bars represent standard deviation. Dot-plots depict individual data points. AO ascorbic acid and α-tocopherol antioxidants, NAO no antioxidants.
Cellobiose conversion by G. subvermispora was minimal when incubated in agitation while nearly 5 mM cellobiose was depleted from the media in static conditions after 11 days of incubation, regardless of AO presence (Fig. 2a). Most of that cellobiose was hydrolyzed to glucose and only a maximum concentration of 2 mM of glucose was utilized as a carbon source in static conditions (Fig. 2b). We also observed a maximum 4-HBA conversion (95%) in agitation without AO while the remaining conditions resulted in lower levels of conversion (ranging between 40–60%, Fig. 2c). In the case of T. versicolor, cellobiose conversion was nearly 100% in agitation conditions and approximately 50% in static conditions regardless of AO addition, at 7 days of incubation (Fig. 2d). Furthermore, glucose concentrations were close to zero during the cultivations (Fig. 2e), which indicates that T. versicolor is simultaneously consuming glucose from extracellular cellobiose hydrolysis or harbors cellobiose transporters similar to those identified in other fungi25. The conversion of 4-HBA was nearly 100% in agitation with and without AO (Fig. 2f). However, in static conditions the conversion was negligible in the presence of AO. These results show that both the incubation mode and level of AO impact 4-HBA conversion rates (Fig. 2).
The hydroxyquinol pathway is regulated differently in G. subvermispora and T. versicolor
Comparative analysis of WRF transcriptomes revealed significant differences in functional enrichments under different cultivation conditions (Supplementary Information, Supplementary Text 1, Supplementary Figs. 1 and 2 and Supplementary Data 1). In addition, as we demonstrated the impact of cultivation conditions on 4-HBA conversion, we conducted targeted gene expression and enzyme abundance analyses in the recently reported 4-HBA catabolic (hydroxyquinol) pathway8 (Fig. 3a). We also included potential pathways and putative enzymes for 4-HBA catabolism described in del Cerro et al. (2021)6 such as 4-HBA reduction to 4-hydroxybenzaldehyde (by carboxylic acid reductases, CARs26), hydroxylation to protocatechuate (by cytochrome P450s27), and methylation to 4-methoxybenzoate (by O-methyltransferases6,28) (Fig. 3a).
Fig. 3. Transcriptomic and proteomic profiles of G. subvermispora and T. versicolor of 4-HBA catabolic pathways, TCA cycle, and glyoxylate shunt under different cultivation conditions.

Transcriptomic and proteomic profiles of enzymes involved in (a) 4-HBA conversion and (b) the TCA cycle, and the glyoxylate shunt in G. subvermispora and T. versicolor. Heatmaps associated to each biochemical step display log2fold changes of relative enzyme (top row) and transcripts (bottom row) abundances in four pairwise comparisons (from left to right): AO vs. without AO (NAO) in agitation, AO vs NAO in static conditions, static vs. agitation with AO, and static vs. agitation without AO. Green is indicative of a negative log2fold change (negative induction) while dark blue is indicative of a positive log2fold change (positive induction) in the corresponding pairwise comparison. Gray boxes indicate that the protein or gene did not return triplicate transcripts or peptides in upstream analyses. Asterisks (*) within the heatmap boxes indicate significant differences in those pairwise comparisons, determined via Tukey’s Honest Significant Difference test90. Discontinuous arrow lines are non-validated biochemical reactions but included based on systems biology observations in del Cerro et al. (2021)6. Protein IDs (in blue) are also shown for each biochemical step abbreviated as (in alphabetic order): 2-AKD 2-alpha ketoglutarate dehydrogenase, AH aconitate hydratase, CAR carboxylic acid reductase, CS citrate synthase, FH fumarate hydratase, HL hydroxylase, ID isocitrate dehydrogenase, IDD intradiol dioxygenase, IL isocitrate lyase, MAR maleylacetate reductase, MDH malate dehydrogenase, MS malate synthase, OD oxidative decarboxylase, PCS peroxisomal citrate synthase, P450 cytochrome P450, SD succinate dehydrogenase. Other abbreviations: AO ascorbic acid and α-tocopherol antioxidants, NAO no antioxidants. Omics results originate from three biological replicates for each cultivation condition.
Significant differences among cultivation conditions aligned in both transcriptomics and proteomics data in most of the targeted genes and enzymes (Fig. 3). In the hydroxyquinol pathway, 4-HBA is first oxidatively decarboxylated to hydroquinone8. Both oxidative decarboxylases in G. subvermispora were induced (hereafter, ‘induced’ means either statistically significant higher gene expression and/or relative protein abundance) in agitation conditions with no impact by AO levels (Fig. 3a), which suggests that agitation promotes conversion to hydroquinone. Differently, this biochemical step is induced in static conditions in T. versicolor (Fig. 3a). The second step in the hydroxyquinol pathway involves the hydroxylation of hydroquinone to hydroxyquinol. Hydroxylases responsible for this step were induced in both organisms in static conditions (Fig. 3a). For the subsequent reaction, ring cleavage dioxygenases and maleylacetate reductases exhibited similar trends than those observed for the oxidative decarboxylases in both WRF, suggesting potential similarities in metabolic regulation of this subgroup of enzymes. However, we note that genes encoding maleylacetate reductases were not universally detected across all T. versicolor cultivation conditions. This could be due to upstream metabolic bottlenecks (e.g., accumulation of 4-hydroxybenzaldehyde from potential pathways included in our analysis), and therefore a limited flux through the hydroxyquinol pathway and/or induction of enzymes in this pathway.
Regarding alternative 4-HBA conversion pathways, 4-HBA bioconversion to protocatechuate or 4-hydroxybenzaldehyde showed opposite trends to the oxidative decarboxylases in both WRF (induced in static or agitation in G. subvermispora and T. versicolor, respectively) at the transcript level. We note that CAR enzymes were not identified in G. subvermispora, which suggests the lack of 4-HBA reduction in this experimental setup or lack of sensitivity of the proteomic analysis. The formation of 4-hydroxybenzyl alcohol from 4-hydroxybenzaldehyde by a putative alcohol dehydrogenase6 was also investigated. At the transcript level, we found trends similar to those identified for CARs, with induction under static conditions in G. subvermispora and under agitation in T. versicolor, suggesting similar regulation patterns for both enzymes. We also examined the potential conversion of 4-HBA to 4-methoxybenzoate. The two enzymes previously proposed to be involved in this step in G. subvermispora were detected in the proteomics and transcriptomic datasets, but only showed significant differences at the transcript level and with similar trends as those identified for the oxidative decarboxylases. In T. versicolor, the putative O-methyltransferase was not detected, and no significant gene expression changes were observed among different cultivation conditions. We then compared these results with the total 4-HBA conversion levels (Fig. 2c, f). 4-HBA conversion was the highest in agitation without AO in G. subvermispora (Fig. 2c) which aligns with the induction of oxidative decarboxylases. In T. versicolor, the maximum 4-HBA conversion, also in agitation, aligns with the induction of the reductase and cytochrome P450 (Fig. 2f). These results propose preferred 4-HBA conversion pathways depending on the cultivation conditions.
The hydroxyquinol pathway, tricarboxylic acid cycle, and glyoxylate shunt are, in general, induced under similar cultivation conditions in T. versicolor but not in G. subvermispora
4-HBA may enter the tricarboxylic acid (TCA) cycle and glyoxylate shunt via acetyl-CoA and succinyl-CoA in WRF6. We reconstructed these pathways in both WRF to determine the induction of these pathways under the different cultivation conditions as well as any correlation with the hydroxyquinol pathway. Given that glucose also enters central carbon metabolism, we extended our analyses to the glycolysis pathway (Supplementary Fig. 3). The glycolysis pathway did not exhibit a clear induction trend among cultivation conditions, limiting any potential correlation among pathways and therefore, glycolysis is not further discussed.
In the TCA cycle of G. subvermispora, only the citrate synthase, which converts acetyl CoA to citrate, was significantly induced in agitation mode (Fig. 3b). The genes and/or enzymes leading to succinate from acetyl-CoA were induced in static conditions (Fig. 3b), which does not align with the trends observed in the hydroxyquinol pathway (Fig. 3a). Regarding AO, their presence also induced few biochemical steps in this WRF. In T. versicolor, in general, static conditions induced the TCA cycle (Fig. 3b), similar to the trends of the hydroxyquinol pathway (Fig. 3a). In the glyoxylate shunt of G. subvermispora and T. versicolor, we only identified significant differences at the transcript level. For G. subvermispora, both isocitrate lyase and malate synthase were either induced in agitation, while the same genes in T. versicolor were induced in static conditions (Fig. 3b). Because these trends align with the hydroxyquinol pathway in both WRF, we hypothesize preferential carbon flux from aromatics compounds to central metabolism via the glyoxylate pathway instead the TCA cycle. However, spatial 13C-isotopic labeling analyses is necessary to determine 4-HBA fate towards the peroxisome (glyoxylate shunt) or mitochondria (TCA cycle).
Targeted metabolomics provides insights into 4-HBA catabolism through different conversion pathways
To compare metabolite presence and/or relative abundance with cultivation conditions, we conducted targeted metabolomic analyses for the hydroxyquinol pathway and the putative alternative 4-HBA conversion pathways. With the goal of increasing the sensitivity of these analyses and detect transient metabolites, we modified the experimental setup and increased the concentration of 4-HBA from ~2 mM (Fig. 2) to 7 mM (Supplementary Fig. 4). Controls containing experimental media inoculated with inactive seed cultures or without fungal inoculation were also included. The sugar and 4-HBA conversion trends were highly similar for both 4-HBA concentrations by T. versicolor (Fig. 2 and Supplementary Fig. 4). However, in G. subvermispora, 4-HBA conversion increased in static conditions compared to the previous experiment, which indicates again the higher susceptibility of this WRF to varying cultivation conditions. Regardless, as intended, the total 4-HBA utilization was higher in this setup in both WRF. Overall targeted metabolomics results are included in Supplementary Data 2 and illustrated in Fig. 4 and Supplementary Figs. 5 and 6. All expected metabolites were detected, except hydroquinone and β-ketoadipate, which suggests either fast enzymatic conversion of these metabolites or instability during sample preparation and/or analysis.
Fig. 4. Intracellular metabolomic profiles for various 4-HBA conversion pathways in T. versicolor and G. subvermispora.

Heatmaps below each metabolite display log2fold changes of relative metabolite abundance in four pairwise comparisons (from left to right): AO vs. without AO (NAO) in agitation (Ag), AO vs NAO in static (St) conditions, St vs. Ag with AO, and St vs. Ag without AO. G. subvermispora (GS) is shown in the top row and T. versicolor (TV) in the bottom row. Green and dark blue colors are indicative of a negative and positive log2fold change, respectively, in each pairwise comparison. Asterisks (*) within heatmap boxes show significant differences in pairwise comparisons, confirmed by a Tukey’s honest significant difference test. A metabolite is considered as present if detected in at least three biological replicates. Gray boxes indicate the metabolite is not detected. Molecules without heatmaps do not have commercially available standards. Continuous and discontinuous black lines correspond to validated and proposed enzymatic steps, respectively. Metabolomics results originate from three biological replicates for each cultivation condition. AO ascorbic acid and α-tocopherol antioxidants, NAO no antioxidants, Ag agitation, St static, GS G. subvermispora, TV T. versicolor.
First, we focused on intracellular and extracellular 4-HBA abundances. Under static conditions, T. versicolor exhibited a significant increase of intracellular 4-HBA accumulation compared to agitation. Interestingly, 4-HBA conversion rates were slower in static conditions (quantified in the supernatant overtime, Supplementary Fig. 4). This suggests that the enzymes induced in static conditions (i.e., oxidative decarboxylases, Fig. 3a) may be also slower than those induced in agitation (e.g., CARs). G. subvermispora exhibited intracellular enrichment of 4-HBA in agitation (Fig. 4), which also correlated with the slower conversion rates (Supplementary Fig. 4). We note that recent in vitro results with enzymes from G. subvermispora showed that oxidative decarboxylation was the slowest step in the hydroxyquinol pathway8.
We then focused on intracellular downstream pathways and metabolites that exhibited significant differences among cultivation modes. G. subversmipora showed higher relative abundances of most intracellular metabolites in agitation conditions compared to static conditions (except for 4-methoxybenzoate) (Fig. 4) and T. versicolor showed higher abundances in static conditions (except for hydroxyquinol), which aligns with the induction of the hydroxyquinol, TCA, and glyoxylate pathways. In T. versicolor, 4-hydroxybenzaldehyde was also identified in the supernatant and its production was notable in all cultivation conditions (Supplementary Fig. 4). G. subvermispora did not accumulate 4-hydroxybenzaldehyde in the extracellular milieu (Supplementary Fig. 4). However, metabolomic analyses identified it in the intracellular fraction, with similar abundances among cultivation conditions (Fig. 4), indicating that it is not a bottleneck as in T. versicolor. Lastly, we observed that the presence of AO induced the accumulation of most catabolic intermediates in both WRF. Since proteomic and transcriptomic analyses showed no significant induction of enzymes in cultivations with AO, we attribute these results to the increased flux and stability of these compounds in the presence of AO.
Metabolomic analysis enables the identification of phenylpropanoid biosynthetic pathways
We further conducted metabolomic and lipidomic analyses in the intracellular (pellets) and extracellular (supernatant) fractions to provide insights into the overall metabolism of WRF and carbon fate in different cultivations conditions (Supplementary Fig. 4 and Supplementary Data 2). To explore active fungal pathways, we retrieved KEGG, HMDB, and PubChem IDs for the 171 metabolites identified in negative ionization mode and 155 in positive ionization mode in all the cultivation conditions and then mapped them onto the T. versicolor genome using KEGG mapper (Supplementary Data 3). Using this approach, we identified that phenylalanine, tyrosine, p-coumarate, and p-coumaroyl alcohol were mapped in the monolignol biosynthesis pathway and identified in all the cultivation conditions. Their higher abundance in the extracellular fraction compared to non-inoculated controls (Supplementary Data 2–3, Fig. 5a and Supplementary Figs. 6–7) prompted a deeper investigation into extracellular metabolites with similar mass features. We identified a clade of metabolites, including cinnamic, caffeic, and ferulic acids, that belong to the monolignol biosynthesis pathway29,30 but were not mapped in this pathway by KEGG Mapper. Phenylalanine and tyrosine have been reported to be precursors of these aromatic compounds in plants and some microbes (Fig. 5b)31–34 but information about this biosynthetic pathway is limited in WRF. Overall, based on these results, we hypothesize that these WRF produce and secrete aromatic compounds from aromatic amino acids. To validate this hypothesis, we conducted an additional experiment in media containing either cellobiose and 4-HBA or cellobiose and phenylalanine (10 mM) and quantified these metabolites in the supernatant after 5 days of incubation.
Fig. 5. Proposed phenylpropanoid biosynthetic pathway in T. versicolor.

a Metabolomic analysis in the extracellular milieu of T. versicolor for a group of mass features including cinnamic acid, p-coumaric acid, caffeic acid, ferulic acid, phenylalanine, and tyrosine. The heatmap, represented by the log10 of peak heights, shows relative metabolite abundances from cultivations conducted in different conditions. A metabolite is considered present if it is detected in at least three biological replicates. Data originated from three biological replicates. b Intracellular proteomic analysis showing log10 concentrations of putative enzymes of the proposed phenylpropanoid biosynthetic pathway. Reference enzymes for each reaction (from 1 to 10) are color-coded to highlight their origins:blue for plants, red for bacteria, and yellow for yeast. This heatmap represents the same conditions as in section A. Data originated from three biological replicates. c Proposed phenylpropanoid biosynthetic pathway in T. versicolor. The quantification of identified metabolites in the extracellular fraction is shown in bar graphs and the results derive from averages of biological duplicates. Error bars indicate the absolute difference between the biological duplicates. Dot-plots depict individual data points. The enzymes of each reaction are: Reaction 1:PAL L-phenylalanine ammonia-lyase, PTAL bifunctional L-phenylalanine/L-tyrosine ammonia-lyase, reaction 2:C4H cinnamate 4-hydroxylase, 3:C3H 4-coumarate 3-hydroxylase, reaction 4:Fcs feruloyl-CoA synthase, reaction 5:Ech enoyl-CoA hydratase/lyase, reaction 6:Vdh vanillin dehydrogenase, reaction 7:3-oxoacyl-(acyl-carrier protein) reductase, reaction 8: 3-oxoacyl CoA thiolase, reaction 9:Alpha/beta hydrolase family, reaction 10: COMT caffeate/5-hydroxyferulate 3-O-methyltransferase. Ag agitation, AO antioxidants, NAO no antioxidants, St static;controls:AOt0 = Time zero control (with seed media from T. versicolor), AOtn = 7-day control (with seed media from T. versicolor), NAOt0 = Time zero control (without seed media), NAOtn = 5-day control (without seed media), (no seed) NAOTn = 7-day control (without seed media and without antioxidants). Supplementary Data 6 includes source data underlying charts of Fig. 5c.
Targeted metabolites were detected exclusively in T. versicolor cultivations fed with phenylalanine and cellobiose, and in all cultivation conditions except for 4-hydroxybenzaldehyde, which was not detected under agitation without AO (Fig. 5 and Supplementary Fig. 7). This does not necessarily indicate absence of metabolites in the other cultivations, as metabolite levels may be below the detection limit. In T. versicolor, phenylalanine was partially consumed after 5 days of cultivation and the formation of cinnamic acid was observed in all tested cultivation conditions, up to 6 µM in agitation without AO (Fig. 5c). Interestingly, although we did not detect p-coumaric acid, caffeic acid, or ferulic acid following this setup, we observed the formation of 4-HBA and 4-hydroxybenzaldehyde (Fig. 5c). Based on the detection of 4-HBA and 4-hydroxybenzaldehyde, we hypothesize that T. versicolor holds a conversion pathway from cinnamic acid to p-coumaric acid and from p-coumaric acid to 4-HBA, as previously described and validated in bacteria and in yeasts (Fig. 5c)23,35.
To propose enzymatic steps for this pathway, we conducted protein sequence homology analyses with enzymes previously characterized in plants, bacteria, and yeasts (Supplementary Data 4 and 5) and the two WRF of this study. Employing cutoff of 30% identity and for IPR domain similarity, we down selected 33 enzymes in T. versicolor (Fig. 5b) and 18 in G. subvermispora (Supplementary Fig. 7). The initial step in the proposed pathway draws from the plant phenylpropanoid pathway31, involving the deamination of phenylalanine into cinnamic acid by phenylalanine ammonia-lyase (PAL). We identified a putative enzyme homologous to PAL in T. versicolor (TV_173713) exclusively produced in agitation (reaction 1, Fig. 5b, c). This finding could explain the increased catabolism of phenylalanine under this incubation mode (Fig. 5c). Putative enzymes for each of the subsequent multi-step conversion of p-coumaric acid to 4-HBA (reaction 2–9) were identified in T. versicolor. We note that p-coumaroyl-CoA has been reported to form two different intermediates, 4-hydroxybenzaldehyde in the bacterium P. putida and p-hydroxyphenyl-3-hydroxypropanoyl-CoA in yeast Rhodotorula toruloides (Fig. 5c)23,35, which could explain the presence of 4-hydroxybenzaldehyde and 4-HBA in all tested cultivation conditions (Fig. 5b, c). However, enzyme validations will be necessary to ascertain if both pathways are active in T. versicolor. Regarding G. subvermispora, homologous enzymes to PAL were also identified (GS_118180 and GS_116229 (Supplementary Fig. 7)). However, the absence of putative PAL in the proteomics datasets as well as homologs of other downstream enzymes (Supplementary Data 5), explains the lack of phenylalanine conversion to cinnamic acid and other downstream metabolites (Supplementary Fig. 7). Lastly, we note that p-coumarate could also be produced from tyrosine via a bifunctional PAL (PTAL) and ferulic acid from caffeic acid via caffeic acid 3-O-methyltransferase (COMT)31. We identified putative enzymes across various cultivation conditions for these reactions, except for the homolog of COMT which was exclusively identified in T. versicolor (TV_51611). These findings establish a foundation for future enzyme and pathway validation in WRF.
Lipid composition undergoes significant changes depending on both the incubation mode and the species of WRF
Lipids are key cellular components in several processes including structure (lipid bilayer) and cell signaling/shuttling (lipid rafts)36–39. Lipid groups have been reported to change as a response to cellular stress such as nutrient limitation, physical environmental stress, stress-induced ROS generation in filamentous fungi, yeast, algae, and bacteria40–43, but this has not been determined in WRF to our knowledge. Therefore, to add to the metabolic information of WRF, we extracted lipids from the same samples utilized for transcriptomics and proteomics (24 h after induction with 4-HBA) (Fig. 2), from the fungal pellets (including lipids from cell membranes and intracellular lipids) and extracellular fractions (Fig. 6a, b). The feature annotations, abbreviations, lipids category, main class, and sub class are detailed in Supplementary Table 1. At a high level, the most significant changes in the lipidome were identified between static and agitation incubation modes (Supplementary Figs. 8 and 9). These results are expected since the addition of AO occurred at the end of the cultivation, likely limiting their effect on lipid composition and structural changes.
Fig. 6. Intracellular and extracellular lipidomic profiles and visualization of G. subvermispora and T. versicolor mycelia under different cultivation conditions.

Summary of the (a) intracellular and (b) extracellular lipid classes detected in different cultivation conditions. The heatmaps associated with each lipid class are the results from pairwise comparisons, from left to right: are agitation with antioxidants (AO) vs. agitation without AO (NAO), static cultivation with AO vs. static without AO, static with AO vs. agitation with AO, and static without AO vs. agitation without AO. Green indicates that the feature is significantly higher, while blue indicates that the feature is significantly lower based on the pairwise analyses. Only statistically significant results are displayed in the heatmaps (at least 80% of the statistically significant features, evaluated by log2 fold change and confirmed by Tukey’s Honest Significant Difference test, are in one group or another). Gray boxes indicate the lipid class was not detected, and white boxes indicate no significant difference between the compared groups (< 80%). These -omic results originate from three biological replicates for each cultivation condition. c Microscopy images of G. subvermispora and T. versicolor mycelia stained with BODIPY-ceramide stain to highlight ceramide/sphingolipids patterns (top) and intensity of the staining (bottom). Scale bars= 50 µm (left) 10 µm91. Ag agitation, AO antioxidants, NAO no antioxidants, St static; and for the lipid features: Cer Ceramides, DG Diradylglycerols, PA Glycerophosphates, PC Glycerophosphocholines, PE Glycerophosphoethanolamines, PG Glycerophosphoglycerols, PI Glycerophosphoinositols, PS Glycerophosphoserines, Sphingolipids Phosphosphingolipids, TG Triradylglycerols.
In the G. subvermispora pellets, we identified glycerophosphocholines (PC), diradylglycerols (DGs), and glycerophosphoserines (PS) as positively induced in agitation cultivation conditions (Fig. 6a and Supplementary Fig. 8). Based on the study of Li et al. (2022) with the oleaginous fungus Cunninghamella echinulate44, we hypothesize that membrane lipids, including PC and PS, may be mobilized as part of the antioxidant response to safeguard cellular membranes against oxidative damage44. DGs could be employed in the synthesis of new lipids or serve as intermediaries for energy production45–47. Given that PC can also play crucial roles in cellular signaling48, the enrichment of these lipids during agitation could be associated with signaling events that coordinate adaptive responses to stress. Alternatively, this enrichment may relate to the structural composition of membranes in G. subvermispora. In static cultivation conditions in the presence of AO, we observed positive induction of glycerophosphates (PA) and triradylglycerols (TG), suggesting that these lipids are important in these conditions for the formation of the mycelial mat. Several ceramides and sphingolipids were also enriched in the static background. The enrichment in ceramides may be also linked to certain stress responses, as ceramides play a role in signaling pathways associated with adaptive responses47,49,50.
In T. versicolor, most of the detected features were positively induced in the AO background, including PC, DG, PS, and PA (Fig. 6a and Supplementary Fig. 9). Collectively, lipids including PC, PS, glycerophosphoethanolamines (PE), and glycerophosphoinositols (PI) exhibited positive induction under static conditions. Notably, ceramides exhibited increased levels during agitation, indicating their potential involvement in the cellular stress response and adaptive responses, or alternatively, reflecting changes in the structural composition of membranes. This differential induction suggests a complex interplay between lipid metabolism and environmental conditions. Overall, our results show that cultivation conditions impact both T. versicolor and G. subvermispora membrane and/or intracellular lipid composition.
Lipid secretion is promoted in cultivations incubated in static and agitation modes in T. versicolor and G. subvermispora, respectively
Recent studies have shown that T. versicolor secrete extracellular droplets rich in long-chain fatty acids in solid-state cultivation conditions51,52 whose composition varies depending on the feedstock used for growth and the age of the droplets. To assess whether T. versicolor and G. subvermispora secrete lipids in submerged cultivations, and if the secretion is influenced by cultivation conditions, we evaluated the extracellular lipidome in both WRF. In T. versicolor, we observed a significant increase of the features identified as TGs and DGs under static cultivation, both with and without AO (Fig. 6b and Supplementary Fig. 10. DGs were not detected intracellularly, which supports their uniqueness in the extracellular fraction. TG enrichments did not align between the extracellular and intracellular fraction either. There are no previous reports on the secretion of lipids or fatty acids from G. subvermispora and our findings suggest that lipid features such as PC, ceramides, and DGs are also enriched in the extracellular milieu, particularly when supplemented with AO in agitation conditions (Fig. 6b and Supplementary Fig. 10). Interestingly, ceramides in the fungal pellet were more abundant in static conditions than in agitation (Fig. 6a), which suggests that incubation in agitation favors their release (Fig. 6c).
Microscopy indicates that cultivation conditions do not impact mycelial structure but rather their composition
Lastly, we aimed to determine whether the cultivation conditions impact the mycelial composition and structure. At the macroscopic level, G. subvermispora and T. versicolor show different morphologies. Specifically, these WRF form large spheres of fungal pellets under agitation conditions, whereas they develop mat-like structures on the surface when incubated in static conditions (Supplementary Fig. 11). Considering this, we hypothesized that the distribution and content of cell wall components (e.g., carbohydrates and lipids) may also vary. Lipidomics data already showed significant differences in the content of several lipids, including ceramides, between cultivations conducted in static and agitation modes (Fig. 6a). To further test the hypothesis, we examined ceramide distribution in the mycelia. For this purpose, we conducted microscopy analysis with boron dipyrromethene difluoride (BODIPY-ceramide), which has been used as a stain for ceramides and sphingolipids. In addition, we used acriflavine, as an indicator of altered cell wall structure and chemistry (e.g., carbohydrates). In G. subvermispora, the enrichment of ceramides and sphingolipids in static cultivations is confirmed by microscopy, indicating that sphingolipid content is induced in those conditions and is evenly distributed throughout the hyphae (Fig. 6c), which is reflected by a single, uniform peak, in the chromatogram (Fig. 6c). In contrast, ceramides are concentrated in agitation conditions in specific regions of the hyphae, resulting in a bimodal frequency distribution (Fig. 6c). In T. versicolor, lipidomic analyses revealed ceramide enrichment in cultures conducted in agitation but microscopy with BODIPY stain does not show significant differences in intensity between cultivation conditions, shown as similar frequency in the chromatograms (Fig. 6c). Acriflavine staining revealed that the hyphae morphology remains similar under both cultivation conditions in the two WRF (Supplementary Fig. 12), suggesting that the observed morphological differences in the cultivations occur at the macroscopic scale. However, acriflavine tended to accumulate within the cells incubated in agitation, highlighting different chemistries, likely due to regions with varied carbohydrate content (Supplementary Fig. 13).
Discussion
This study demonstrates the significant role of cultivation conditions in the overall metabolism of two WRF and highlights the dependence of the metabolic responses on the strain used. G. subvermispora and T. versicolor present different lignocellulose degradation patterns in nature (preferential lignin degradation or simultaneous lignin and polysaccharide degradation, respectively)4,53,54. This study unravels that the regulation of the enzyme cascade involved in the catabolism of the lignin-related compound, 4-HBA, is also different when these WRF are cultivated under static or agitation conditions. We also hypothesized that AO would mitigate oxidative stress and/or enhance 4-HBA uptake by preventing the repolymerization of aromatic compounds in the extracellular milieu. While our 4-HBA analysis in the fungal supernatants does not directly support this hypothesis (Fig. 2), it does not contradict it either, as 4-HBA could still be modified extracellularly. In contrast, intracellular metabolomic analyses (Fig. 4) revealed elevated concentrations of catabolic intermediates in the presence of AO, likely indicating an increased influx of 4-HBA into the fungal cell. These insights are invaluable for future efforts to genetically manipulate these organisms rationally and redirect carbon flow from aromatic compounds in more productive ways. For instance, gene knockouts of competing pathways (e.g., CARs) during the catabolism of 4-HBA and tailored cultivation conditions could be used to induce certain enzymes for the accumulation of valuable products. One such product could be β-ketoadipate, a performance-advantaged replacement for adipic acid55, which is a catabolic intermediate in the hydroxyquinol pathway8. However, we note that, despite current advances56–58, genetic tools in WRF need further development.
Changes in the morphology and composition (e.g., glucans) of the fungal mycelia, as well as total biomass have been associated to different incubation conditions (static compared to agitation)59. In this study, we show that morphology mostly changes at the macroscopic level but does not significantly change at the microscopic level. However, the lipid composition varies significantly. Lipid concentration has direct implications for industries focused on producing fungal mycelia for food60 and biomaterials61. This study specifies the intracellular lipid composition and the cultivation conditions that enrich each type, which has additional implications in these and other applications62. In addition, we investigated lipid secretion by these WRF and identified the secretion of DGs and TGs by T. versicolor. These lipids can serve as biodiesel precursors63,64 and, importantly, their secretion would avoid costly downstream processes to extract lipids from cells. G. subvermispora also secreted DGs, along with PC and ceramides, which can be used in cosmetic formulations65,66. The quantification of these compounds in the supernatant as well as the understanding and optimization of their production, is warranted to either use WRF as production hosts or transfer pathways to other organisms for their production.
Our metabolomic analyses identified clusters of phenylpropanoids in T. versicolor and G. subvermipora. This suggested the existence of aromatic biosynthetic pathways in these WRF. Previous studies using the WRF Phanerochaete chrysosporium67 and Bjerkandera adusta68 confirmed the transformation of phenylalanine into various aromatic compounds, such as veratryl alcohol, whose function is to stabilize lignin peroxidases and promote the oxidation of various substrates during lignin decay67. In our study, we demonstrate that the addition of phenylalanine in the cultivations enhances the production of other phenylpropanoids (e.g., cinnamic acids) and phenolic acids (e.g., 4-HBA) and that T. versicolor can secrete them at the milligram per liter scale without any optimization. We also found that agitation without AO promotes that conversion. Validating and comparing the efficiency of the enzymes proposed for this conversion in this study with those from plants31, bacteria35, or other fungi23 would be critical for tailoring these pathways to increase the production of phenylpropanoids for a diversity of applications69.
This study has certain limitations that need to be acknowledged. The experiments were conducted using a single monomeric aromatic compound rather than a complex lignocellulosic substrate. Therefore, our findings cannot be directly translated to scenarios involving consolidated bioprocesses for lignin valorization. However, these results are relevant for lignin streams containing 4-HBA or any of the aromatic catabolic intermediates identified in this study. Additionally, from a biotechnological point of view, the rates of aromatic catabolism in these WRF and these experimental conditions are slower compared to bacteria70 or other yeasts71. Nevertheless, WRF excel when directly used on solid substrates compared to other microbes, which would avoid the need of conducting pretreatment and enzymatic hydrolysis of lignocellulose. This study also proposes pathway induction under certain cultivation conditions; however, it does not quantify differences in 4-HBA flux, as other tools (e.g.,13C-isotopic labeling) would be necessary to do so. Lastly, some of the enzymes identified in our study have not been experimentally validated, highlighting the need for further research to confirm their roles and activities.
This study paves the way for future research on a variety of areas. Enzyme validation for the various 4-HBA catabolic pathways and phenylpropanoid synthesis is warranted. Additionally, pathway discovery to enhance the production of extracellular fatty acids using these WRF would be a promising avenue. Alternatively, these pathways could be transferred to other hosts. As demonstrated in this study, the bacterium P. putida—a robust host for the utilization of aromatic compounds, production of value-added molecules, and scalability72—is likely promising for generating functional WRF enzymes. Moreover, analyzing where specific reactions occur in WRF (e.g., peroxisome vs. mitochondria), as described above, would be beneficial to understand energy outputs (which differ between organelles)58 and for the future successful engineering of these organisms. Overall, our study reveals key factors impacting WRF metabolism and provides additional insights into the dual catabolism of carbohydrates and lignin related compounds as well as potential routes for carbon flux and sequestration in nature.
Materials and methods
Functional validation of fungal enzymes in Pseudomonas putida KT2440
Gene structure
Genomes for T. versicolor v1.03 and Ceriporiopsis (Gelatoporia) subvermispora B73 were downloaded from Joint Genome Institute (JGI) Mycocosm database74. Integrated Genomics Viewer75 was used to visualize the exon junction maps generated by Tophat2 in down selected genes.
Gene synthesis
Candidate genes for functional analysis were codon optimized using the OPTIMIZER codon optimization software76 using the guided random method based on the codon usage of the highly expressed genes (HEG) in P. putida KT2440. The De Novo DNA software’s RBS Calculator77 was used to generate RBS sequences for each gene with a target translation initiation rate of 10,000. Candidate genes were synthesized in pBTL-2 by TWIST Biosciences. Sanger sequencing data was provided by Genewiz. All sequences can be found in Supplementary Table 2 in the Supplementary Information.
Strain construction
Each candidate enzyme was expressed individually from Plac on a pBTL-2 backbone. Plasmids were electroporated into the appropriate P. putida strain. Briefly, an overnight culture of P. putida was grown at 30 °C and 225 rpm. 2.5 mL of culture per transformation was spun down at 8000 rcf for 1 min. Supernatant was decanted and cells were resuspended in 750 μL sterile sucrose (300 mM) two times. After washing, cell pellets were resuspended in 50 μL. Plasmids were added to the resuspended cells at approximately 30 ng and pipetted up and down gently to mix. Cells and plasmid were transferred to 1 mm electro cuvettes and electroporated at 1.6 kV, 25 μF, and 200 Ω. Cells were recovered in 950 μL of S.O.C. (Super Optimal broth with Catabolite repression) medium at 30 °C for 1 h, shaking at 225 rpm. Approximately 20 μL of each recovery was spotted onto a LB+kanamycin (50 µg/mL) plate and streaked out. Plates were incubated at 30 °C overnight. Glycerol stocks were made of single colonies.
Cultivations with wild-type and engineered P. putida
The bacterial strains were grown overnight in LB, with antibiotics (kanamycin, 50 µg/mL) as necessary, at 30 °C and 225 rpm. Cultures were centrifuged at 8000 rcf for 1 min and washed twice with M9 media containing no carbon source. M9 media consisted of 6.78 g/L Na2HPO4, 3.00 g/L K2HPO4, 0.50 g/L NaCl, 1.66 g/L NH4Cl, 0.24 g/L MgSO4, 0.01 g/L CaCl2, and 0.002 g/L FeSO4. The optical density at 600 nm (OD600) of the washed cells was measured using a spectrophotometer and each strain was inoculated at an initial OD600 of 0.1 into 125 mL baffled flasks to a volume of 25 mL. Cultivations were conducted as biological triplicates. M9 medium was supplemented with 20 mM of 4-HBA and 20 mM of glucose. Flasks were incubated at 30 °C and 225 rpm and samples were collected at 24 h of incubation. Supernatant was filtered through a 0.22 μm nylon syringe filter. A volume of 100 μL of supernatant was mixed with 900 μL of analytical grade DMSO in an amber glass HPLC vial and sealed with an aluminum cap with a rubber septum to analyze 4-HBA and hydroquinone.
Fungal cultivations
Fungal strains
Monokaryon strains of Ceriporiopsis (Gelatoporia) subvermispora FP-105752 and T. versicolor FP-101664 were used in this study6. Yeast-maltose-peptone-glucose (YMPG)-agar medium was used to maintain the strains at 4 °C. Additional details on media composition and preparation can be found in Supplementary Methods in the Supplementary Information.
Seed culture preparation
An agar plug with fungal mycelia was taken from agar plates maintained at 4 °C, deposited in the center of an YMPG agar plate, and incubated for 7 days at 28 °C. Then, 4–5 agar plugs with mycelia were inoculated in 50 mL of liquid YMPG and incubated at 220 rpm and 28 °C for 7 days (pre-seed). Fungal pellets were then homogenized (10 s, speed 6) in sterile conditions using an Omni Mixer 115 V with a 50 mL SS Chamber PTFE (Omni International, GA, USA). The fungal suspension (2 mL) was re-inoculated in YMPG under the same conditions than the pre-seed culture and incubated for 5 days. A ratio of 2:25 (v:v) of further homogenized fungal suspension per media volume was used to inoculate the cultivations.
Cultivations for multi-omics experiments (transcriptomics, proteomics, and lipidomics)
T. versicolor and G. subvermispora cultivations were conducted in 250 mL flasks containing 25 mL of Czapek-Dox medium (CDM) with cellobiose (15 mM). Details on media composition and preparation can be found in Supplementary Methods in the Supplementary Information. Cultivations were incubated at 28 °C in either agitation (220 rpm) or static conditions using a humidity-controlled incubator (> 50% humidity). After 6 and 10 days of incubation with T. versicolor and G. subvermispora, respectively, we induced all the cultivations with 4-HBA (~2 mM) from a 4-HBA stock solution of 724 mM. Antioxidants (5 mM ascorbic acid and 1 mM α-tocopherol), and prepared as previously described6, were also added in the media 30 min before the induction in half of the cultivations incubated in either static or agitation conditions. Cultivations were harvested 24 h after the induction, corresponding to 7 days and 11 days for T. versicolor and G. subvermispora, respectively. The controls for this experiment were treated in the same conditions as the test cases and included media not inoculated with fungi. Samples (1 mL) were taken periodically to analyze extracellular metabolites via HPLC. Fungal cultivations were performed as biological triplicates.
Cultivations for metabolomic experiments
T. versicolor and G. subvermispora cultivations were conducted as detailed above with some modifications. Specifically, CDM was supplemented with both cellobiose (3.75 mM) and 4-HBA (7 mM) before inoculation. Antioxidants were also added to half of the samples before inoculation. Cultivations were harvested at days 5 and 7 for T. versicolor and G. subvermispora, respectively. Controls included (1) media not inoculated with fungi and (2) media inoculated with inactivated seed cultures. To inactivate seed cultures, broth was filter-sterilized using 0.22 µm PES Steriflip filter units (Millipore, MA, USA) followed by incubation at 98 °C for 10 min. These controls were used for the analysis of the extracellular metabolomics. Samples (1 mL) were also taken periodically to quantify extracellular metabolites via HPLC. Fungal cultivations were performed as four biological replicates.
Cultivations for the exploration of the phenylpropanoid pathway
Cultivations were conducted as detailed above with some modifications. Specifically, for the 4-HBA cultivations CDM was supplemented with both cellobiose (3.75 mM) and 4-HBA (7.5 mM) before inoculation, and for the phenylalanine cultivations CDM was supplemented with both cellobiose (3.75 mM) and phenylalanine (10 mM) before inoculation. Antioxidants were also added to the samples before inoculation when AO was indicated. Cultivations were harvested at days 5 and 7 for T. versicolor and G. subvermispora, respectively. Controls included (1) media not inoculated with fungi and (2) media inoculated with inactivated seed cultures. To inactivate seed cultures, broth was filter-sterilized using 0.22 µm PES Steriflip filter units (Millipore, MA, USA) followed by incubation at 98 °C for 10 min. Samples (1 mL) were quantified via HPLC for the specific extracellular metabolites of interest. Fungal cultivations were performed with two biological replicates and errors for the different metabolites are shown as the absolute difference between the biological duplicates.
Quantification of extracellular metabolites via HPLC
Metabolites from P. putida and WRF cultivations were quantified as follows.
Cellobiose and glucose analysis
Quantification of cellobiose and glucose was performed on an Agilent Technologies 1260 series high-performance liquid chromatography instrument equipped with an Agilent evaporative light scattering detector (ELSD). Chromatographic separation of analytes was achieved using a Shodex SUGAR SZ5532 (6 µm, 6.0 × 150 mm) column and compatible guard. The column was held at a constant temperature of 60 °C and a mobile phases (A) 0.1% formic acid in water and (B) 0.1% formic acid in acetonitrile held at a constant flow rate of 1.0 mL/min was used with the following gradient conditions; initial to t = 5 min (A) = 20% and (B) = 80%; t = 12 min (A) = 35% and (B) = 65%; then immediately returning to initial conditions at t = 12.01 min and held for 2.9 min for a total runtime of 15 min. The ELSD was configured with an evaporator temperature of 80 °C, a nebulizer temperature of 60 °C, and nitrogen flow rate of 1.60 L/min. Samples and standards were injected at volume of 5.0 µL. A minimum of 7 calibration levels were used for each compound and quantitation ranges of 100 µg/mL to 3000 µg/mL. A quadradic fit was utilized with an r2 coefficient ≥ 0.995. A calibration verification standard (CVS) was analyzed every 10–20 samples to monitor detector and instrument drift.
Analysis of aromatic compounds
Aromatic compounds were analyzed by ultra-high pressure liquid chromatography UHPLC) as previously described78. Briefly, samples were analyzed by UHPLC with diode array detection by reverse phase chromatography. The protocol utilizes a BEH C18 column from Waters Corporation and a mobile phase gradient of 0.2% formic acid in water and acetonitrile to achieve separation. Commercial standards were for quantitation of each analyte of interest.
Multi-omic analyses
Sample preparation and analyses via lipidomics, proteomics, metabolomics, and transcriptomics analyses are detailed in supplementary information (Supplementary Methods in the Supplementary Information).
Bioinformatic analyses
Pathway construction for central carbon metabolism and biosynthesis of amino acids
To propose pathways and enzymes for central carbon metabolism and the biosynthesis of amino acids in T. versicolor and G. subvermispora, we utilized the pathway map information for T. versicolor FP-101664 SS1 shown at the Kyoto Encyclopedia of Genes and Genomes (KEGG)79,80 for T. versicolor. Then, we conducted sequence homology searches to identify the homologs (best hit) in G. subvermispora.
Pathway construction for the biosynthesis of phenylpropanoids and monomeric aromatic compounds
For in silico pathway discovery efforts, blastP searches81 were performed using biochemically characterized proteins (plants, bacteria, and yeast, Supplementary Data 4 and 5) and the two WRF proteomes. Due to the phylogenetic distance between some of the model enzymes and the WRF enzyme sequences, all hits achieving a minimum identity score of 20% (Fsc and COMT) or 30% (all the additional protein sequences), a maximum e-value of 1e−10 were considered as putative homologous candidate proteins in the proposed pathways. Putative homologous candidate proteins were additionally, researched for conserved functional domains (PFAM/ InterPro)82.
Transcriptomic bioinformatic analyses
Differential expression analysis was performed using DESeq2 (PMID: 25516281). We conducted non-targeted analyses in overall transcriptomic changes in pairwise comparisons of each treatment: AO vs. without AO (NAO) in static conditions, AO vs NAO in agitation, agitation vs. static in AO, and agitation vs. static without AO. Results were presented as volcano plots and constructed in R (v 2.2.1) and the following packages: ggplot2, tidyverse, stringr (within tidyverse), and gridExtra. To determine enriched functions within transcriptomic profiles, we utilized eukaryotic clusters of orthogroups (KOG) functional classification and gene ontology83 terms as two annotation properties for enrichment analyses as previously described84. Briefly, to generate the two annotation properties, we used the protein model sequences of T. versicolor and G. subvermipsora for gene annotation through eggNOG mapper 4.585. We then extracted GO terms and KOG class attributes. We performed functional enrichment analyses for each pairwise comparison. For KOG classification, we performed a hypergeometric test (Fisher’s exact test) using a customized R script. For GO terms, we performed the analyses through GOseq package83. All p-values from the analyses were adjusted using a false discovery rate correction method.
Proteomic bioinformatic analyses
Biological triplicates were averaged, requiring the protein to be present in at least two biological replicates. For the pairwise studies (enrichments between two groups) we performed Fold Change analysis and t-tests (0.05). For the proteins that passed the t-test (0.05), meaning that a significant difference in the two compared groups was identified, we calculated the log2FC for data visualization in bar plots or heatmaps using R (v 4.1.1).
Lipidomics and metabolomics bioinformatic analyses
For the data processing and normalization, we used R (v 4.1.1) package MetaboAnalyst6.086. We performed a Log10 normalization and data scaling by the mean-centered and divided by standard deviation of each variable (z-score). We then performed analysis of One-way ANOVA (for four conditions) to ascertain whether the overall comparison is significant or not, followed by post-hoc analyses to identify which two levels are different (Fisher’s LSD). We used hierarchical clustering for heatmaps visualizations. For the pairwise studies (enrichments between two groups) we performed Fold Change (FC) analysis and t-tests (0.05). For the features/metabolites that passed the t-test (0.05), meaning that a significant difference in the two compared groups was identified, we calculated the log2FC for data visualization in bar plots or heatmaps.
To explore active fungal pathways, we retrieved KEGG, HMDB, and PubChem IDs for the 171 metabolites identified in negative ionization mode and 155 in positive ionization mode across various cultivation conditions. We then mapped these metabolites onto the T. versicolor (Tvs) genome using KEGG Mapper87,88, generating a list of pathways and modules that they matched. We note that some metabolites may have more than one entry as KEGG IDs (e.g., enantiomers).
Microscopy
Sample preparation and staining
Fungal samples from cultivations conducted in static and agitation mode were fixed in two exchanges of 1% paraformaldehyde and 2.5% glutaraldehyde in 0.2 M cacodylate buffer (EMS, Hatfield, PA) and stored at 4 °C overnight. The fixed samples were placed on microscope slides and manually manipulated to achieve close to a monolayer of mycelium. The samples were prone to clumping. The stain was applied under the coverslip. 0.1% acriflavine (Sigma-Aldrich, St. Louis, MO) was used to stain cell wall components, and BODIPY-ceramide (ThermoFisher) was integrated with and localized to sphingolipids.
Confocal scanning laser microscopy (CLSM)
Stained samples were imaged with a 60×, 1.4 NA Plan Apo objective lens on a Nikon C1 Plus microscope (Nikon, Tokyo, Japan) equipped with the Nikon C1 confocal system with four lasers (403 nm, 561 nm, 643 nm, and tunable 458/477/488/515 nm), and operated via Nikon’s EZ-C1 software. Acriflavine-stained samples were excited using a 488 nm laser, and the emission fluorescence was captured through a 30 nm bandpass centered on 515 nm. BODIPY-ceramide-stained samples were excited with a 561 nm laser, and the emission fluorescence was captured through a 60 nm bandpass centered on 590 nm. The laser power was fixed at 75%, and detection gains were set at 6 for all images captured. Images were captured as eighteen-slice Z-stacks at 100 nm thickness per slice. The result is that each fluorescent image is a series of images from different focal planes through the entire sample that is then compacted into a single two-dimensional image.
Image processing and analysis
The raw image stacks were opened in Fiji (fiji.com) with split channels. Image registration was performed using the Rigid Registration plugin. The registered image stacks were recombined using the Z-project tool, median projection for the brightfield channel, and maximum projection for the fluorescent channel. The registered z-projection of the fluorescent channel was used for analysis. To visualize and quantify an observed change in the intensity and distribution of acriflavine-stained cell wall components, the acriflavine-stained images were analyzed by line scan. A line region of interest was drawn perpendicular to the long axes of the hyphae in an area away from septae or nuclei, and the fluorescence intensity was plotted on an 8-bit (0–255) scale. Twelve-line scans were measured per image. To quantify the intensity of the BODIPY-ceremide detection 150, pixel diameter circular ROIs were drawn around individual fungal hyphae identified as single-layer, scattered across the micrographs. These ROIs effectively isolated the hyphae from the background. Twelve ROIs were selected from each image, and all pixel intensity values were plotted as frequency histograms of 8-bit intensity values.
Statistics and reproducibility
The data were analyzed in R, version 4.1.1. Statistical significance was established at a p value of less than 0.05. For the pairwise studies (enrichments between two groups) we performed Fold Change analysis and p value of less than 0.05. Statistics were conducted in data points derived from tree or four biological replicates, as described in methodology.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
This material is based on work supported by the US Department of Energy (DOE) Office of Science within the Biological and Environmental Research program and under the Early Career Research program. This work was authored, in part, by the National Renewable Energy Laboratory, operated by Alliance for Sustainable Energy, LLC, for the US DOE under contract no. DE-AC36-08GO28308. A portion of the research has been performed using the Environmental Molecular Sciences Laboratory (grid.436923.9), a DOE Office of Science user facility sponsored by the Office of Biological and Environmental Research. A portion of the work was conducted by the U.S. Department of Energy Joint Genome Institute (https://ror.org/04xm1d337), a DOE Office of Science User Facility supported by the Office of Science of the U.S. Department of Energy operated under Contract No. DE-AC02-05CH11231. The views expressed in the article do not necessarily represent the views of the DOE or the US government. The US government retains and the publisher, by accepting the article for publication, acknowledges that the US government retains a nonexclusive, paid up, irrevocable, worldwide license to publish or reproduce the published form of this work, or allow others to do so, for US government purposes. G. subvermispora FP105752 RP-B was kindly provided by Daniel Cullen from the US Department of Agriculture CFMR. T. versicolor (L.) Lloyd FP-101664-Sp was also received from CFMR.
Author contributions
Lummy Monteiro: Data curation, formal analysis, investigation, methodology, validation, visualization, writing- original draft, writing- review & editing. Carlos del Cerro: Investigation, methodology, writing- review & editing. Teeratas Kijpornyongpan: Investigation, methodology, writing- review & editing. Allison Yaguchi: Investigation, methodology, writing- review & editing. Anna Bennett: Investigation, methodology, writing- review & editing. Bryon S. Donohoe: Investigation, methodology, visualization, writing- review & editing. Kelsey J. Ramirez: data curation, methodology, writing- review & editing. Alex F. Benson: data curation, methodology, writing- review & editing. Hugh D. Mitchell: data curation, methodology, writing- review & editing. Samuel O. Purvine: data curation, formal analysis, methodology, writing- review & editing. Lye M Markillie: data curation, methodology, writing- review & editing. Meagan C. Burnet: methodology, writing- review & editing. Kent Bloodsworth: data curation, methodology, writing- review & editing. Benjamin P. Bowen: data curation, methodology, writing- review & editing. Thomas V. Hardwood: data curation, methodology, writing- review & editing. Katherine Louie: data curation, methodology, writing- review & editing. Trent Northen: data curation, methodology, writing- review & editing. Davinia Salvachúa: Conceptualization, funding acquisition, investigation, methodology, project administration, supervision, writing- original draft, writing- review & editing.
Peer review
Peer review information
Communications Biology thanks Mao Peng, Marie-Noelle Rosso, and the other, anonymous, reviewer for their contribution to the peer review of this work. Primary Handling Editor: Ophelia Bu.
Data availability
All data are available in the manuscript, Supplementary Data 1–5(excel format), and deposited in public repositories for (1) transcriptomics raw data (public database GEO, study number GSE272920), (2) mass spectrometry proteomics raw data have been deposited in MassIVE under accession ID: MSV000095519 and accessible at https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=feaf0bfbf966466684bfe23c7d0c886f and to the ProteomeXchange89 consortium with the dataset identifier PXD054613, (3) lipidomics data are deposited at the following 10.25582/data.2024-08.3200044/2426957, (4) Metabolomics raw data are deposited in MassIVE under accession ID: MSV000094781 and 10.25345/C5H41JZ41. Source data corresponding to Figs. 1, 2, 5c can be obtained in Supplementary Data 6.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s42003-025-07640-9.
References
- 1.Boerjan, W., Ralph, J. & Baucher, M. Lignin biosynthesis. Annu. Rev. Plant Biol.54, 519–546 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Zakzeski, J., Bruijnincx, P. C., Jongerius, A. L. & Weckhuysen, B. M. The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev.110, 3552–3599 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Floudas, D. et al. The paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science336, 1715–1719 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Martínez, A. T. et al. Biodegradation of lignocellulosics: microbial, chemical, and enzymatic aspects of the fungal attack of lignin. Int. Microbiol.8, 195–204 (2005). [PubMed] [Google Scholar]
- 5.Erickson, E. et al. Critical enzyme reactions in aromatic catabolism for microbial lignin conversion. Nat. Catal.5, 86–98 (2022). [Google Scholar]
- 6.del Cerro, C. et al. Intracellular pathways for lignin catabolism in white-rot fungi. Proc. Natl. Acad. Sci. USA118, e2017381118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fuchs, G., Boll, M. & Heider, J. Microbial degradation of aromatic compounds — from one strategy to four. Nat. Rev. Microbiol.9, 803–816 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Kuatsjah, E. et al. Biochemical and structural characterization of enzymes in the 4-hydroxybenzoate catabolic pathway of lignin-degrading white-rot fungi. Cell Reports, in print (2024). [DOI] [PubMed]
- 9.Beckham, G. T., Johnson, C. W., Karp, E. M., Salvachúa, D. & Vardon, D. R. Opportunities and challenges in biological lignin valorization. Curr. Opin. Biotechnol.42, 40–53 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Ragauskas, A. J. et al. Lignin valorization: improving lignin processing in the biorefinery. Science344, 1246843 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Li, Q., Harvey, L. M. & McNeil, B. Oxidative stress in industrial fungi. Crit. Rev. Biotechnol.29, 199–213 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Bai, Z., Harvey, L. M. & McNeil, B. Oxidative stress in submerged cultures of fungi. Crit. Rev. Biotechnol.23, 267–302 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Klis, F. M., Boorsma, A. & De Groot, P. W. J. Cell wall construction in saccharomyces cerevisiae. Yeast23, 185–202 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Furuno, S. et al. Mycelia promote active transport and spatial dispersion of polycyclic aromatic hydrocarbons. Environ. Sci. Technol.46, 5463–5470 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Salvachúa, D. et al. Versatile peroxidase as a valuable tool for generating new biomolecules by homogeneous and heterogeneous cross-linking. Enzym. Microb. Technol.52, 303–311 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Marzullo, L., Cannio, R., Giardina, P., Santini, M. T. & Sannia, G. Veratryl alcohol oxidase from pleurotus ostreatus participates in lignin biodegradation and prevents polymerization of laccase-oxidized substrates. J. Biol. Chem.270, 3823–3827 (1995). [DOI] [PubMed] [Google Scholar]
- 17.Salvachúa, D. et al. Lignin depolymerization by fungal secretomes and a microbial sink. Green. Chem.18, 6046–6062 (2016). [Google Scholar]
- 18.Shao, H. B., Chu, L. Y., Lu, Z. H. & Kang, C. M. Primary antioxidant free radical scavenging and redox signaling pathways in higher plant cells. Int J. Biol. Sci.4, 8–14 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang, H. et al. Structural mechanism of ergosterol regulation by fungal sterol transcription factor upc2. Nat. Commun.6, 6129 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Van Hauwenhuyse, F., Fiori, A. & Van Dijck, P. Ascorbic acid inhibition of candida albicans hsp90-mediated morphogenesis occurs via the transcriptional regulator upc2. Eukaryot. Cell13, 1278–1289 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Veenstra, T. D. Omics in systems biology: current progress and future outlook. PROTEOMICS21, 2000235 (2021). [DOI] [PubMed] [Google Scholar]
- 22.Kijpornyongpan, T., Schwartz, A., Yaguchi, A. & Salvachúa, D. Systems biology-guided understanding of white-rot fungi for biotechnological applications: a review. iScience25, 104640 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim, J. et al. Multi-omics driven metabolic network reconstruction and analysis of lignocellulosic carbon utilization in rhodosporidium toruloides. Front. Bioeng. Biotechnol.8, 612832 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu, S., Plan, M. R., Winter, G. & Krömer, J. O. Metabolic engineering of pseudomonas putida kt2440 for the production of para-hydroxy benzoic acid. Front. Bioeng. Biotechnol.4, 90 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cai, P. et al. Evidence of a critical role for cellodextrin transporte 2 (cdt-2) in both cellulose and hemicellulose degradation and utilization in neurospora crassa. PLoS ONE9, e89330 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qu, G., Guo, J., Yang, D. & Sun, Z. Biocatalysis of carboxylic acid reductases: Phylogenesis, catalytic mechanism and potential applications. Green. Chem.20, 777–792 (2018). [Google Scholar]
- 27.Lah, L. et al. The versatility of the fungal cytochrome p450 monooxygenase system is instrumental in xenobiotic detoxification. Mol. Microbiol.81, 1374–1389 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Jeffers, M. R., McRoberts, W. C. & Harper, D. B. Identification of a phenolic 3-o-methyltransferase in the lignin-degrading fungus phanerochaete chrysosporium. Microbiol. (Read.)143, 1975–1981 (1997). [DOI] [PubMed] [Google Scholar]
- 29.Shi, R. et al. Towards a systems approach for lignin biosynthesis in populus trichocarpa: transcript abundance and specificity of the monolignol biosynthetic genes. Plant Cell Physiol.51, 144–163 (2009). [DOI] [PubMed] [Google Scholar]
- 30.Amthor, J. S. Efficiency of lignin biosynthesis: a quantitative analysis. Ann. Bot.91, 673–695 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barros, J. et al. 4-coumarate 3-hydroxylase in the lignin biosynthesis pathway is a cytosolic ascorbate peroxidase. Nat. Commun.10, 1994 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barros, J. & Dixon, R. A. Plant phenylalanine/tyrosine ammonia-lyases. Trends Plant Sci.25, 66–79 (2020). [DOI] [PubMed] [Google Scholar]
- 33.Hyun, M. W., Yun, Y. H., Kim, J. Y. & Kim, S. H. Fungal and plant phenylalanine ammonia-lyase. Mycobiology39, 257–265 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Emiliani, G., Fondi, M., Fani, R. & Gribaldo, S. A horizontal gene transfer at the origin of phenylpropanoid metabolism: a key adaptation of plants to land. Biol. Direct4, 7 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ravi, K., García-Hidalgo, J., Gorwa-Grauslund, M. F. & Lidén, G. Conversion of lignin model compounds by pseudomonas putida kt2440 and isolates from compost. Appl. Microbiol. Biotechnol.101, 5059–5070 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jackson, CatherineL., Walch, L. & Verbavatz, J.-M. Lipids and their trafficking: an integral part of cellular organization. Dev.l Cell39, 139–153 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Yao, Y., Ding, L. & Huang, X. Diverse functions of lipids and lipid metabolism in development. Small Methods4, 1900564 (2020). [Google Scholar]
- 38.Bloom, M., Evans, E. & Mouritsen, O. G. Physical properties of the fluid lipid-bilayer component of cell membranes: a perspective. Q. Rev. Biophys.24, 293–397 (1991). [DOI] [PubMed] [Google Scholar]
- 39.Walther, T. C. & Farese, R. V. Lipid droplets and cellular lipid metabolism. Annu. Rev. Biochem.81, 687–714 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang, X. et al. Proteomics analysis of high lipid-producing strain mucor circinelloides wj11: an explanation for the mechanism of lipid accumulation at the proteomic level. Microb. Cell Fact.15, 35 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pedroso, N. et al. Modulation of plasma membrane lipid profile and microdomains by h2o2 in saccharomyces cerevisiae. Free Radic. Biol. Med.46, 289–298 (2009). [DOI] [PubMed] [Google Scholar]
- 42.Mandotra, S. K., Kumar, P., Suseela, M. R., Nayaka, S. & Ramteke, P. W. Evaluation of fatty acid profile and biodiesel properties of microalga scenedesmus abundans under the influence of phosphorus, ph and light intensities. Bioresour. Technol.201, 222–229 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Semchyshyn, H., Bagnyukova, T., Storey, K. & Lushchak, V. Hydrogen peroxide increases the activities of soxrs regulon enzymes and the levels of oxidized proteins and lipids in escherichia coli. Cell Biol. Int.29, 898–902 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Li, S., Feng, X., Zhang, X., Xie, S. & Ma, F. Phospholipid and antioxidant responses of oleaginous fungus cunninghamella echinulata against hydrogen peroxide stress. Arch. Biochem. Biophys.731, 109447 (2022). [DOI] [PubMed] [Google Scholar]
- 45.Chandel, N. S. Lipid metabolism. Cold Spring Harb. Perspect. Biol. 13, 1–20 (2021). [DOI] [PMC free article] [PubMed]
- 46.Barbosa, A. D. & Siniossoglou, S. Membranes that make fat: roles of membrane lipids as acyl donors for triglyceride synthesis and organelle function. FEBS Lett.598, 1226–1234 (2024). [DOI] [PubMed]
- 47.Hou, Q., Ufer, G. & Bartels, D. Lipid signalling in plant responses to abiotic stress. Plant, Cell Environ.39, 1029–1048 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Cassilly, C. D. & Reynolds, T. B. Ps, it’s complicated: the roles of phosphatidylserine and phosphatidylethanolamine in the pathogenesis of candida albicans and other microbial pathogens. J. Fungi4, 1–14 (2018). [DOI] [PMC free article] [PubMed]
- 49.Fabri, J. H. T. M., de Sá, N. P., Malavazi, I. & Del Poeta, M. The dynamics and role of sphingolipids in eukaryotic organisms upon thermal adaptation. Prog. Lipid Res.80, 101063 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ali, U., Li, H., Wang, X. & Guo, L. Emerging roles of sphingolipid signaling in plant response to biotic and abiotic stresses. Mol. Plant11, 1328–1343 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Hao, G. & Barker, G. C. Fatty acid secretion by the white-rot fungus, trametes versicolor. J. Ind. Microbiol. Biotechnol.49, kuab083 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rizzo, J., Rodrigues, M. L. & Janbon, G. Extracellular vesicles in fungi: past, present, and future perspectives. Front. Cell. Infect. Microbiol.10, 346 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hori, C. et al. Temporal alterations in the secretome of the selective ligninolytic fungus ceriporiopsis subvermispora during growth on aspen wood reveal this organism’s strategy for degrading lignocellulose. Appl Environ. Microbiol80, 2062–2070 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Salvachúa, D. et al. Fungal pretreatment: an alternative in second-generation ethanol from wheat straw. Bioresour. Technol.102, 7500–7506 (2011). [DOI] [PubMed] [Google Scholar]
- 55.Werner, A. Z. et al. Lignin conversion to β-ketoadipic acid by pseudomonas putida via metabolic engineering and bioprocess development. Sci. Adv.9, eadj0053 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Okuda, N. et al. Overexpressing pleurotus ostreatus rho1b results in transcriptional upregulation of the putative cellulolytic enzyme-encoding genes observed in ccl1 disruptants. Environ. Microbiol23, 7009–7027 (2021). [DOI] [PubMed] [Google Scholar]
- 57.Wu, H. et al. Transcriptional shifts in delignification-defective mutants of the white-rot fungus pleurotus ostreatus. FEBS Lett.594, 3182–3199 (2020). [DOI] [PubMed] [Google Scholar]
- 58.Demarquoy, J. & Le Borgne, F. Crosstalk between mitochondria and peroxisomes. World J. Biol. Chem.6, 301–309 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ibrahim, D., Weloosamy, H. & Lim, S. H. Effect of agitation speed on the morphology of aspergillus niger hfd5a-1 hyphae and its pectinase production in submerged fermentation. World J. Biol. Chem.6, 265–271 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Strong, P. J. et al. Filamentous fungi for future functional food and feed. Curr. Opin. Biotechnol.76, 102729 (2022). [DOI] [PubMed] [Google Scholar]
- 61.Haneef, M. et al. Advanced materials from fungal mycelium: fabrication and tuning of physical properties. Sci. Rep.7, 41292 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Passoth, V. In Biotechnology of yeasts and filamentous fungi (ed. Sibirny, A. A.). 1, 149–204 (Springer International Publishing, 2017).
- 63.Zendejas, F. J., Benke, P. I., Lane, P. D., Simmons, B. A. & Lane, T. W. Characterization of the acylglycerols and resulting biodiesel derived from vegetable oil and microalgae (thalassiosira pseudonana and phaeodactylum tricornutum). Biotechnol. Bioeng.109, 1146–1154 (2012). [DOI] [PubMed] [Google Scholar]
- 64.Monteiro, R. R. C. et al. Biosynthesis of alkanes/alkenes from fatty acids or derivatives (triacylglycerols or fatty aldehydes). Biotechnol. Adv.61, 108045 (2022). [DOI] [PubMed] [Google Scholar]
- 65.Schorsch, C., Boles, E. & Schaffer, S. Biotechnological production of sphingoid bases and their applications. Appl. Microbiol. Biotechnol.97, 4301–4308 (2013). [DOI] [PubMed] [Google Scholar]
- 66.Allegretti, C., Denuccio, F., Rossato, L. & D’Arrigo, P. Polar head modified phospholipids by phospholipase d-catalyzed transformations of natural phosphatidylcholine for targeted applications: an overview. Catalysts10, 997 (2020). [Google Scholar]
- 67.Jensen, K. A., Evans, K. M., Kirk, T. K. & Hammel, K. E. Biosynthetic pathway for veratryl alcohol in the ligninolytic fungus phanerochaete chrysosporium. Appl. Environ. Microbiol60, 709–714 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lapadatescu, C., Giniès, C., Le Quéré, J.-L. & Bonnarme, P. Novel scheme for biosynthesis of aryl metabolites from l-phenylalanine in the fungusbjerkandera adusta. Appl. Environ. Microbiol.66, 1517–1522 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cesarino, I., Eudes, A., Urbanowicz, B. & Xie, M. Editorial: phenylpropanoid systems biology and biotechnology. Front. Plant Sci.13, 866164 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kuatsjah, E. et al. Debottlenecking 4-hydroxybenzoate hydroxylation in pseudomonas putida kt2440 improves muconate productivity from p-coumarate. Metab. Eng.70, 31–42 (2022). [DOI] [PubMed] [Google Scholar]
- 71.Rodriguez, A. et al. Base-catalyzed depolymerization of solid lignin-rich streams enables microbial conversion. ACS Sustain. Chem. Eng.5, 8171–8180 (2017). [Google Scholar]
- 72.Weimer, A., Kohlstedt, M., Volke, D. C., Nikel, P. I. & Wittmann, C. Industrial biotechnology of pseudomonas putida: advances and prospects. Appl Microbiol. Biotechnol.104, 7745–7766 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fernández-Fueyo, E. et al. Comparative genomics of ceriporiopisis subvermispora and phanerochaete chrysosporium provide insight into selective ligninolysis. Proc. Natl. Acad. Sci. USA.109, 5458–5463 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Grigoriev, I. V. et al. Mycocosm portal: Gearing up for 1000 fungal genomes. Nucleic Acids Res.42, D699–D704 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol.29, 24–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Puigbò, P., Guzmán, E., Romeu, A. & Garcia-Vallvé, S. Optimizer: a web server for optimizing the codon usage of DNA sequences. Nucleic Acids Res.35, W126–W131 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Salis, H. M., Mirsky, E. A. & Voigt, C. A. Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol.27, 946–950 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Woodworth, S. P, Haugen, S. J, Michener, W. E, Ramirez, K. J, Beckham, G. T Muconic acid isomers and aromatic compounds analyzed by uhplc-dad. Protocols.io https://www.protocols.io/view/muconic-acid-isomers-and-aromatic-compounds-analyz-36wgqjjxyvk5/v3 (2024).
- 79.Kanehisa, M. & Goto, S. Kegg: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res28, 27–30 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kanehisa, M., Sato, Y., Furumichi, M., Morishima, K. & Tanabe, M. New approach for understanding genome variations in kegg. Nucleic Acids Res.47, D590–D595 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Altschul, S. F. et al. Gapped blast and psi-blast: a new generation of protein database search programs. Nucleic Acids Res.25, 3389–3402 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Paysan-Lafosse, T. et al. Interpro in 2022. Nucleic Acids Res.51, D418–D427 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Young, M., Wakefield, M., Smyth, G. & Oshlack, A. Goseq: gene ontology testing for rna-seq datasets. R Bioconductor.8, 1–25 (2012).
- 84.McQuilton, P., & Gene Ontology Consortium. Expansion of the gene ontology knowledgebase and resources. Nucleic Acids Res.45, 331–338 (2016).
- 85.Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. Eggnog-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol.38, 5825–5829 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chong, J. & Xia, J. Metaboanalystr: an r package for flexible and reproducible analysis of metabolomics data. Bioinformatics34, 4313–4314 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kanehisa, M. & Sato, Y. Kegg mapper for inferring cellular functions from protein sequences. Protein Sci.29, 28–35 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kanehisa, M., Sato, Y. & Kawashima, M. Kegg mapping tools for uncovering hidden features in biological data. Protein Sci.31, 47–53 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Deutsch, E. W. et al. The proteomexchange consortium in 2020: enabling ‘big data’ approaches in proteomics. Nucleic Acids Res.48, D1145–D1152 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Abdi, H. & Williams, L. J. Tukey’s honestly significant difference (hsd) test. Encycl. Res. Des.3, 1–5 (2010). [Google Scholar]
- 91.Swift, C. L. et al. Anaerobic gut fungi are an untapped reservoir of natural products. Proc. Natl. Acad. Sci.118, e2019855118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
All data are available in the manuscript, Supplementary Data 1–5(excel format), and deposited in public repositories for (1) transcriptomics raw data (public database GEO, study number GSE272920), (2) mass spectrometry proteomics raw data have been deposited in MassIVE under accession ID: MSV000095519 and accessible at https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=feaf0bfbf966466684bfe23c7d0c886f and to the ProteomeXchange89 consortium with the dataset identifier PXD054613, (3) lipidomics data are deposited at the following 10.25582/data.2024-08.3200044/2426957, (4) Metabolomics raw data are deposited in MassIVE under accession ID: MSV000094781 and 10.25345/C5H41JZ41. Source data corresponding to Figs. 1, 2, 5c can be obtained in Supplementary Data 6.