

Abstract
Mutation rates vary across mammalian genomes, but little is known about the scale over which this variation occurs. Knowledge of the magnitude and scale of mutational variation is required to understand the processes that drive mutation, and is essential in formulating a robust null hypothesis for comparative genomics studies. Here we estimate the scale of mutational variation in the murid genome by calculating the spatial autocorrelation of nucleotide substitution rates in ancestral repeats. Such transposable elements are good candidates for neutrally evolving sequence and therefore well suited for the study of mutation rate variation. We find that the autocorrelation coefficient decays to a value close to zero by ∼15 Mb, with little apparent variation in mutation rate under 100 kb. We conclude that the primary scale over which mutation rates vary is subchromosomal. Furthermore, our analysis shows that within-chromosome mutational variability exceeds variation among chromosomes by approximately one order of magnitude. Thus, differences in mutation rate between different regions of the same chromosome frequently exceed differences both between whole autosomes and between autosomes and the X-chromosome. Our results indicate that factors other than the time spent in the male germ line are important in driving mutation rates. This raises questions about the biological mechanism(s) that produce new mutations and has implications for the study of male-driven evolution.
Much evidence now suggests that the point mutation rate varies considerably across the mammalian genome. Studies of nucleotide substitution rates at synonymous sites (Wolfe et al. 1989; Matassi et al. 1999; Malcom et al. 2003; Chuang and Li 2004), within long alignments of primate intergenic sequence (Chen et al. 2001; Ebersberger et al. 2002; Silva and Kondrashov 2002; Smith et al. 2002) and mammalian repetitive sequence (Waterston et al. 2002; Hardison et al. 2003), have revealed considerably more variation in the substitution rate than expected by chance. This is of interest because substantial mutational variability could seriously reduce the effectiveness of comparative methods to locate putatively functional regions within noncoding DNA. The efficiency of identification of such regions could be improved if we knew a priori which regions are expected to be evolving more slowly.
The regional mutation hypothesis proposes that different regions of the vertebrate genome are diverging at substantially different rates (Filipski 1988). Previous studies have provided evidence that mutation rates vary substantially between chromosomes (Wolfe et al. 1989; Malcom et al. 2003; Makova et al. 2004). Particularly notable is the apparent reduction in the rate of point (McVean and Hurst 1997; Ebersberger et al. 2002; Waterston et al. 2002) and indel substitution (Makova et al. 2004) on the X-chromosome. This reduction has been suggested to reflect the primarily male origin of most mutations, although the evidence on this point is inconsistent (McVean and Hurst 1997; Lercher et al. 2001). In addition, there is evidence that significant variation in the mutation rate also occurs along the length of a chromosome (Wolfe et al. 1989; Chuang and Li 2004). Although mutational variation has been studied at these two levels, an unresolved problem is the relative importance of chromosome number and position within a chromosome in determining the underlying mutation rate. Of particular relevance to this question is the scale of local similarity of mutation rates. If the domain or “unit” of mutational variation is considerably smaller than a chromosome and substantial interdomain variability exists, this would suggest that position within a chromosome is a more important factor in determining neutral mutation rate. This conclusion is reversed if local similarity extends across entire chromosomes.
One of the first studies to address the issue of local similarity of evolutionary rates compared estimates of the synonymous divergence (Ks) from human–mouse gene orthologs within 1 cM of each other, and concluded that there is evidence for the existence of “evolutionary rate units” between which substantial variation exists (Matassi et al. 1999). Lercher et al. (2001) extended this analysis to a larger data set and found that significant similarity of Ks extends from 1 cM to entire chromosomes in a human–rodent comparison. Although it may seem unexpected that mutation rates would remain approximately constant across entire chromosomes, this situation does appear to exist in yeast (Chin et al. 2005). Such a large scale of similarity would seem to reject a substantial role for within-chromosomal mutational heterogeneity and apparently suggests that the majority of mutational variation occurs between chromosomes. However, more recent work has suggested that synteny blocks (i.e., regions for which gene order has been conserved between species) may represent a more meaningful “unit” than whole chromosomes (Malcom et al. 2003; Webster et al. 2004). Malcom et al. (2003) found that although a weak effect of chromosomal number is evident in both human–mouse and mouse–rat comparisons, this is confounded by substantial within-chromosome variation. These authors indicate that differences between synteny blocks on the same chromosome outweigh those observed between chromosomes. Additional support for a subchromosomal mutational scale comes from Chuang and Li (2004), who use a human–mouse comparison to show that local similarity in mutation rates extends to ∼10 Mb. The relevance of a chromosome as an evolutionarily distinct entity is uncertain, however, particularly between highly diverged species such as human and mouse, for which genome sequencing projects have revealed many large-scale rearrangements (Nadeau and Taylor 1984; Hudson et al. 2001; Waterston et al. 2002).
Many of the above studies have used synonymous substitution rates to examine patterns of mutational variation. However, synonymous sites comprise a small fraction of most mammalian genomes and may misrepresent mutational processes outside of coding sequence. In addition, the importance of sequence context effects, in particular CpG hypermutability, is becoming increasingly apparent (Arndt et al. 2003). Given that the majority of sites both 5′ and 3′ of mammalian fourfold degenerate synonymous sites are under strong purifying selection, this may introduce bias in the estimation of Ks. For example, strong selective preservation of a C that is 5′ to a fourfold synonymous site may serve to elevate the observed substitution rate. Furthermore, there is now some evidence that selection, perhaps related to mRNA splice efficiency or mRNA stability, may be operating at some mammalian synonymous sites (Eyre-Walker 1999; Keightley and Gaffney 2003; Chamary and Hurst 2004; Willie and Majewski 2004; Keightley et al. 2005).
For these reasons, it is desirable to investigate mutational variation outside of coding sequence. Some authors have sought to address this by using long human–chimpanzee alignments of intergenic sequence (Ebersberger et al. 2002; Smith et al. 2002; Webster et al. 2004). Webster et al. (2004) estimated the extent of local similarity using substitution rates at ancestral repeat (AR), intronic, and intergenic sites from a human–chimp alignment of 14 Mb from human Chromosome 7. Their results indicate that the most significant local similarity of mutation rates occurs at a scale of 1–2 Mb. However, they did not investigate the rate of decay of this local similarity. Furthermore, it is becoming increasingly apparent that some of the noncoding nonrepetitive portion of the mammalian genome, assumed to be neutral in the above studies, may be under selection (Waterston et al. 2002; Thomas et al. 2003; Bejerano et al. 2004). Smith et al. (2002) and Webster et al. (2004) argue that such selected regions should have little influence on substitutional variation in closely related species. However, minimally diverged species are more susceptible to the influence of ancient polymorphism in the last common ancestor, and selection in noncoding DNA does become relevant when considering alternative, more distantly related taxa, such as mouse and rat. Thus, in these species pairs, long, intergenic alignments are not ideal for the study of mutational variation. One alternative is to focus on the remnants of repetitive elements that were inserted in the last common ancestor (e.g., Waterston et al. 2002; Hardison et al. 2003) The use of these ancestral repeats is appealing because, of all classes of noncoding DNA, they are the most likely candidates for neutrality (Ellegren et al. 2003). Additionally, the large quantities of repetitive sequence allow for investigation of mutational variation on much finer scales than is possible just using Ks.
We therefore collected a data set of repetitive elements present in the last common mouse–rat ancestor. Using these data, we sought to address the following questions: (1) What is the scale of local similarity of rodent mutation rates? (2) At this scale, what is the ratio of between-chromosome to within-chromosome mutation rate variation? Answers to these questions are important to accurately quantify mutational variation and improve our understanding of the processes that may cause point mutation. Furthermore, information on the scale of mutational variation is important in establishing a robust null hypothesis for comparative genomics methods.
Results
We extracted and aligned a total of 55 Mb of repetitive sequence. This can be broken down into the following contributions from various classes of repetitive elements: 17.5 Mb of SINE, 13.0 Mb of LINE, 21.0 Mb of LTR, and 3.7 Mb of DNA elements. The proportions of aligned sequence derived from each repeat family appears approximately consistent across autosomes (Fig. 1). However, LINE elements appear to be significantly more prevalent on the X-chromosome (P < 0.0001) than the autosomes. This would suggest either that LINE elements have been more active on the X-chromosome or that the rate of deletion of LINEs is less than on the autosomes. There is some evidence to suggest that the former scenario is more likely, as it seems that some retrotransposing sequences preferentially target the X-chromosome (Khil et al. 2005). It may also be that LINEs play a role in X-chromosome inactivation (Bailey et al. 2000; Waterston et al. 2002).
Figure 1.

Proportion of total sequence per mouse chromosome contributed by each repeat class.
Between-chromosome variation
We estimated the average chromosomal divergence at all sites and at sites not preceded by a C or followed by a G (non-CpG-prone sites) for each mouse chromosome (Fig. 2). Non-CpG-prone sites are the least likely to have been part of a hypermutable CpG dinucleotide, and therefore the least affected by potential covariation between nucleotide divergence and age of transposable element insertion (see Methods). We find that the X-chromosome is evolving more slowly at all sites than any of the autosomes, and we estimate a male-to-female mutation rate, α, of 1.5. This is slightly lower than previous estimates in rodents (Chang et al. 1994; Gibbs et al. 2004). Rates at non-CpG-prone sites are consistently lower than those estimated at all sites for all autosomes. This would suggest that rates at all sites are affected by the elevated mutation rates at CpG dinucleotides and the selection of non-CpG-prone sites goes some way to removing this effect. Interestingly, however, this situation is reversed on the X-chromosome, where substitution rates at non-CpG-prone sites are, in fact, marginally, although not significantly, higher than those estimated at all sites. This result appears to be roughly consistent within repeat families (Supplemental Table 1).
Figure 2.

Estimated average nucleotide substitution rates at all sites and non-CpG-prone sites for each mouse chromosome. Bars show the 95% bootstrap confidence intervals.
Scale of local similarity
We estimated the scale of local similarity of mutation rates using the autocorrelation of average substitution rates across a variety of block sizes. Figure 3 shows the autocorrelation of nucleotide substitution rates at all sites between blocks of 5 kb and 100 kb extending over intervals from 10 kb to 1 Mb and 200 kb to 20 Mb, respectively. Autocorrelation of rates across 5-kb blocks (Fig. 3A) remains highly significant compared to randomly permuted data across a distance of 1 Mb. There is minimal change in autocorrelation from 10 kb to 100 kb (Fig. 3A), suggesting that little variation in underlying mutation rate exists below 100 kb. The low magnitude of the correlation across 5-kb blocks reflects the relatively noisy estimates of substitution rates obtained from the small number of ancestral repeat sites (295 bp on average) within each block. In contrast, the number of sites within the average 100-kb block is approximately one order of magnitude larger than that in 5-kb blocks (2.3 kb on average), thus our estimate of the substitution rate is less noisy and the magnitude of autocorrelation is higher. Here, there is a slow decay of similarity in substitution rates extending to a distance of 10–15 Mb (Fig. 3B). It is important to note that autocorrelation in Figure 3A,B shows the same proportional change over the same distance. For example, autocorrelation across 5-kb blocks decays from ∼0.078 to ∼0.052 (a decrease of approximately one-third) over a distance of 1 Mb; autocorrelation across 100-kb blocks decays from ∼0.445 to ∼0.290 (again a decrease of approximately one-third) over the same distance.
Figure 3.

Autocorrelation of nucleotide substitution rates in ancestral repeats (A,B,C) and ancestral repeat flanking sequence (D) across 5-kb (A) and 100-kb (B,C,D) blocks. Substitution rates were estimated at all sites. Dotted lines show the upper and lower bounds of the 95% confidence interval of autocorrelation under the null hypothesis of no dependence of rates between blocks. Blocks were permuted randomly (A,B) and within common GC-content intervals (C,D).
The similarity of evolutionary rates between blocks within an interval of 0–15 Mb seems to be explained, in part, by the corresponding similarity of average GC content of adjacent blocks, since randomly permuting blocks within GC classes still produces a moderate signal of autocorrelation in the absence of local structure (Fig. 3C,D). This would suggest that local GC content, or one or more covariates of local GC content, influences neutral substitution rates in both repetitive and nonrepetitive DNA. However, this similarity does not seem to be a result of CpG hypermutability or compositional change, since our results were qualitatively similar when we estimated rates at non-CpG-prone sites or by counting A↔T and G↔C changes only (Supplemental Fig. 1).
We also estimated the partial autocorrelation of nucleotide substitution rates in both ancestral repeats and flanking sequence, averaged across 100-kb blocks (Fig. 4). Plots of partial autocorrelation coefficients suggest that all local similarity over distances >1 Mb can be explained by autocorrelations below 1 Mb. This suggests that the average “unit” of mutational variation is no larger than ∼1 Mb. The results are similar in both repetitive and nonrepetitive sequence (Fig. 4A,B, respectively).
Figure 4.

Partial autocorrelation of nucleotide substitution rates in ancestral repeats (A) and flanking sequences (B). Substitution rates are estimated for all sites. Dotted lines show the upper and lower bounds of the 95% confidence interval of partial autocorrelation under the null hypothesis of no dependence of rates between blocks.
Within- and between-chromosome mutational variation
The data were initially fitted to two linear models, one including terms for fixed chromosomal and random regional effects, and the other including a chromosomal effect only. We estimated the magnitude of within-chromosome mutational variation as the variation between levels of the random regional effect in the former model. Model fit was assessed using Akaike's Information Criterion (AIC). It appears that all models that included “regional variation” effects provide a substantially better fit to the data than those including chromosome means alone (Fig. 5). This is clearly seen from the decrease in AIC (models with a better fit have a lower AIC) for models including a random regional effect. The AIC for Model 1 was –844,146.7 for ancestral repeats and –933,598.5 for flanking sequence data. Including blocks of 1 Mb as a random effect in the model, for example, decreases the AIC to –854,057.2 for the ancestral repeat data (Fig. 5) and to –945,433.5 for the flanking sequence data. This is evidence that significant regional variation in neutral mutation rate does, indeed, occur along the length of a chromosome. The most parsimonious model (as adjudged by the AIC) in our analysis includes a block size of 1 Mb as a random effect. At this scale the variation between blocks is approximately one order of magnitude greater than that observed between chromosomes. The between-chromosome variance was 2.28 × 10–5 for ancestral repeats and 7.71 × 10–7 for flanking sequence, whereas the between-block variance in the most parsimonious model is 2.06 × 10–4 for ancestral repeats and 9.53 × 10–5 for flanking sequence. While the substitution rates in ancestral repeats appear more variable than flanking nonrepetitive sequence, the difference in between- and within-chromosome mutational variation is striking in both categories of sites. Our results are consistent whether we consider rates at non-CpG-prone sites or by counting only A↔T and G↔C changes (Supplemental Figs. 2 and 3).
Figure 5.

Between-block variation (σ2b) in substitution rates within ancestral repeats (A) and flanking sequence (B). Substitution rates are estimated at all sites. Between-block variances are estimated fitting the chromosome as a fixed effect and the block as a random effect across different block sizes, from 25 kb to 125 Mb. Block sizes are plotted on a log10 scale. The 95% confidence intervals of the between-block variance were as estimated by the lme routine of the nlme package in R. The Akaike Information Criterion (AIC) is shown for each fitted model.
We also determined whether there were significant chromosome effects by comparing the mixed model (Model 2) with a model that includes a term for random regional effects only (Model 3). Regional effects of 1 Mb were included in both models. We analyzed four different data sets, consisting of nucleotide substitution rates in ancestral repeats and flanking sequence, including and excluding the X-chromosome. Our results indicate that Model 2 describes the data most parsimoniously in all cases (Table 1). We note, however, that the difference in AIC between Model 2 and Model 3 is far smaller (approximately two orders of magnitude) than that observed between Model 1 and Model 2. This would support our conclusion that although there exist small but detectable chromosomal effects on nucleotide substitution rates, they are far outweighed by subchromosomal regional variation. Differences in AIC between Model 2 and Model 3 drop when the X-chromosome is excluded.
Table 1.
Akaike Information Criteria for Model 2 (chromosomal and regional effects) and Model 3 (regional only) when fitted to each of four data sets: nucleotide substitution rates in ancestral repeats and flanking, nonrepetitive sequence, including and excluding the X-chromosome
|
All chromosomes
|
Autosomes only
|
|||
|---|---|---|---|---|
| Ancestral repeat | Flank | Ancestral repeat | Flank | |
| Model 2 | -854,283.5 | -1,118,129 | -803,472 | -1,052,114 |
| Model 3 | -854,133 | -1,117,967 | -803,349.8 | -1,052,005 |
Both models included a term for a 1-Mb regional effect.
We investigated the efficiency of our approach by analyzing simulated data (Supplemental material). Results of this analysis indicate that when regional effects are absent, Model 1 (fixed chromosome effects only) explains the data more parsimoniously than Model 2 (fixed chromosome and random block effects), independent of the block size included in Model 2 (Supplemental Fig. 4). When regional effects of varying sizes are simulated, Model 2 provides a substantially better fit to the data, as is the case with our real data. In addition, the best-fitting mixed effects model (i.e., the model with the lowest AIC) is that which includes a block size closest to the true simulated block size (Supplemental Fig. 5).
It should be noted that the mixed model does not explain a large proportion of the variance in substitution rate (∼6%) when fitted to data consisting of observations on individual ancestral repeats, as we have presented above. However, it is likely that much of the residual variation is due to the considerable error involved in inferring substitution rates from such small sequences (on average ∼200 bp). This is supported by the observation that the proportion of variance explained by the mixed model when fitted to the slightly longer flanking sequences (on average ∼362 bp) is higher (∼9%). If we assume that there is minimal mutational variation below 50 kb and thus treat all ancestral repeats within a 50-kb window as a single sequence having a single mutation rate, the mixed model, including a term for a 1-Mb regional effect, explains ∼25% of the total variation. We consider this to be a reasonable estimate of the proportion of true mutational variation explained by the most parsimonious model in our analyses.
Discussion
Our study provides further evidence for, and clarification of, the regional mutation hypothesis. It appears that the primary scale over which mutation rates vary is subchromosomal and that within-chromosome effects are at least as important as male germ-line effects as a source of mutational variability, although the latter has received substantially more attention in the literature. The evidence for this conclusion is threefold. Firstly, partial autocorrelations suggest that all long-range (>1 Mb) similarity of mutation rates can be explained by “propagation” of similarity of mutation rates across distances of <1 Mb. Secondly, results of the mixed model analysis indicate that within-chromosome mutational variation greatly exceeds variation among chromosomes. Given that chromosomal location of X-linked sequence appears highly conserved between mouse and rat (Gibbs et al. 2004), it is unlikely that the within-chromosome variation we observe could be the result of differences in time spent within the male germ line. Thirdly, comparison of our Models 2 and 3 indicates that the effects of chromosome on mean nucleotide substitution rates are small.
We find little evidence in murids for significant similarity of substitution rates across scales as large as an entire chromosome, as a previous human–mouse study has indicated (Lercher et al. 2001). A possible explanation is that the mutation pattern has undergone a substantial shift in the lineage leading from the murid common ancestor to human, although how such an event might have occurred remains uncertain. Perhaps a more likely possibility is that the wide divergence between human and mouse simply affords greater power to detect such small effects. Notwithstanding, a recent large-scale study of the synonymous substitution rates at ∼15,000 human–mouse gene orthologs supports our conclusion of local similarity extending to 10–15-Mb intervals (Chuang and Li 2004).
It is interesting to note that while our estimates of between-chromosome variation are consistent with previous estimates from murid ancestral repeats (e.g., ∼3 × 10–5) (Makova et al. 2004), they are lower than the between-chromosome variation estimated at synonymous sites from a recent study (2.7 × 10–4) (Malcom et al. 2003). However, the average variance of the estimates of mean chromosomal Ks from Malcom et al. (2003) is also somewhat larger than the variance of chromosomal substitution rates we estimate from ancestral repeats (∼0.0069 vs. ∼0.0025). It seems, therefore, that substitution rates at synonymous sites are considerably more variable than rates within ancestral repeat sequences. This may be a result of selection on some synonymous sites, or interaction between the effects of strong selection on sites adjacent to synonymous sites and context-dependent mutational processes. It is likely, therefore, that the same pattern of variation (within-chromosome mutational variation exceeding variation among chromosomes) would also be evident if rates were estimated at synonymous sites.
Our results raise questions about the biological mechanisms that give rise to new mutations. We suggest that the pattern of variation that we observe could therefore be explained by two, nonmutually exclusive, processes. Firstly, the accuracy of DNA replication may vary regionally along the length of chromosomes. This could elevate or diminish the mutation rate in different regions of the same chromosome. We are, however, unaware of a specific biological mechanism that could produce regionally varying replication accuracy. Secondly, other factors, such as structural alterations and spontaneous degradation of nucleotide bases that are unaffected by DNA replication could contribute substantially to the production of single base-pair mutations. Such alterations could include processes such as the deamination of methylcytosine to thymine or oxidative base damage caused by oxygen free radicals. That the pattern of variation remains the same when considering substitution rates at non-CpG-prone sites (Supplemental Fig. 3) would suggest that CpG-derived mutation is not responsible for much of the regional variation we observe. It is unclear whether those mutations produced by oxidative base damage can be distinguished from mutations derived from other sources, however.
The magnitude of within-chromosomal mutational variation highlights the importance of accounting for regionally varying mutation rates in the identification of putatively functional regions of noncoding DNA. Although the coefficient of regional variation in nucleotide substitution rates we observe is not large (8.75%; 1-Mb regional effects), this still has an impact on the null expectation of conservation of a sequence between two species. As an example, assuming that mouse–rat divergence is normally distributed with a mean of 0.16 and a standard deviation of 0.014, 95% of divergence scores will be in the range 0.132–0.188. The probability of 95% sequence identity of a 100-bp sequence between two species at the lower 95% bound is more than two orders of magnitude larger than the probability of the same sequence at the upper 95% bound. This observation also emphasizes the importance of estimating neutral mutation rates locally. Additionally, our results illustrate that there is likely to be an effect of sampling when estimating average chromosomal substitution rates solely from genic regions. The majority of mammalian genes reside in GC-rich regions (Mouchiroud et al. 1991; Lander et al. 2001); thus even sampling all genes from a chromosome may return a regionally biased estimate of chromosomal evolutionary rate, and any subsamples thereof will potentially exaggerate this bias. Clearly, in order to accurately estimate an average chromosomal mutation rate, one must sample from all regions of a chromosome, not just genic regions, and this could explain some disparities between previous estimates of average X and autosomal substitution rates.
One implication of a subchromosomal mutational scale is that the major process or processes that drive point mutation could be expected to vary across similar scales. One candidate for such a driving process is recombination. Recombination rates have been previously shown to covary with neutral substitution rates in ancestral repeats (Hardison et al. 2003). It is also known that recombination rates in humans are significantly correlated with GC content, probably as a result of biased gene conversion (Kong et al. 2002; Meunier and Duret 2004). Recent results from the highly recombining human pseudoautosomal region provide further evidence that recombination may have an effect on the neutral mutation rate (Filatov 2004). In order to investigate the possibility that recombination rates are related to substitution rates, we collected mouse recombination rate data from a recent comparative study (Jensen-Seaman et al. 2004). These data consist of estimates of local recombination rate in 5-Mb windows across the mouse genome. We estimated average substitution rates for each of these windows from our data. However, we find little evidence for a relationship between mouse recombination rates and mouse–rat divergence; the slope of the regression line of substitution rates on recombination rates is approximately zero (Fig. 6). If recombination is driving mutation in murids, our data suggest that the relationship is not straightforward, on a genome-wide level at least. This conclusion is supported by recent work suggesting that the relationship between recombination rate and nucleotide substitution is at best moderate (Huang et al. 2005). Furthermore, some studies have suggested that the majority of recombinations in humans occur in a comparatively small proportion of the genome (Crawford et al. 2004; McVean et al. 2004). If such recombination “hotspots” also occur in murids, the lack of an observed relationship may be explained, in part, by this effect. For example, if recombination rates vary over scales of kilobases, as opposed to megabases, then any relationship between mutation and recombination may be obscured by averaging rates over large genomic distances. In addition, if recombination rates change rapidly over evolutionary time, this may cause problems in deciphering the true nature of any relationship between mutation and recombination, as the latter is measured over much shorter timescales than the former.
Figure 6.

The relationship between mouse–rat divergence and the mouse recombination rate average across 5-Mb windows. The equation of the regression line shown was estimated as y = 0.144 – 0.002x.
One problem to which our data are potentially susceptible is that of gene conversion in repetitive sequence. It has been shown recently that some gene conversion occurs in young Alu repeats (Roy et al. 2000). If gene conversion is biased in the direction of the ancestral state, then this will produce a negative correlation between nucleotide divergence and the rate of conversion. The distributions of repeat age within SINEs (results not shown) would suggest that Alu/B1 elements differ from the other families of SINEs in that there is a small proportion of Alu elements that are younger than other SINE elements. This would suggest either that we have retrieved more Alu elements from low-mutating regions or that biased gene conversion toward the ancestral repeat is occurring. If the latter is the case, then there is little we can do to remove this effect from our data, short of locating those elements that are ancestral in a more highly diverged species, for example, human, to minimize the proportion of young Alus in the data set. However, if gene conversion is occurring in some Alus in our data, it appears to have a small effect on our results. The pattern of autocorrelation is practically unchanged if we entirely remove Alus from our data set, as is the ratio of within- to between-chromosome substitutional variation. In addition, previous analyses have concluded that gene conversion in repetitive DNA appears to have small effects on neutral substitution rates at the genomic scale (Makova et al. 2004).
We have shown that the scale of mutational similarity in murids extends from 100 kb to 15 Mb and that the “unit” of mutational variation is no larger than 1 Mb. Our results indicate that, at this scale of regional effect, there exists approximately one order of magnitude more variation in mutation rates within chromosomes than among chromosomes. This has implications for the study of the processes driving mutation and identification of functional noncoding DNA using comparative genomic methods.
Methods
Data
Most mammalian transposable elements can be divided into four broad classes: Short INterspersed Elements (SINEs), Long INterspersed Elements (LINEs), Long Terminal Repeat (LTR) retroposons, and DNA transposons. We identified all SINE, LINE, LTR, and DNA repetitive elements in build 33.1 of the mouse genome using RepeatMasker (http://www.repeatmasker.org/). We identified those repetitive elements that were inserted prior to the mouse–rat divergence as follows. First, 250 bp of sequence upstream and downstream of the identified mouse repeat was extracted. Any repetitive sequence in these flanking sequences was masked, also using RepeatMasker. In order to ensure that matches were achieved using reasonable lengths of sequence, we excluded any element that did not contain at least 50 consecutive bases of unique, nonrepetitive sequence in both its adjacent flanking sequences. Following masking, the remaining unique sequence was compared to the rat chromosome(s) syntenic to the mouse chromosome on which the repeat originated using BLASTN (Altschul et al. 1997). Chromosomal synteny was as defined in Figure 4 of Gibbs et al. (2004). The following criteria were used to accept or reject BLAST hits of pairs of flanking sequence. (1) Hits with E-values of >10–5 were rejected. (2) Hits were only accepted if both flanks had a single unique match on the same rat contig. (3) To ensure returned BLAST hits were orthologous to the sequence immediately adjacent to the flanks of the original mouse repetitive element, matches of upstream (downstream) flanks were required to extend to within 50 bp of the flank end (start). Fulfilment of these criteria indicated that the sequence surrounding the mouse repeat in question was present in the last common murid ancestor. The region between the outer limits of the matched flanks was then extracted from the appropriate rat chromosome of NCBI build 3.1 of the rat genome and aligned to the original mouse flanks and repetitive element sequence using AVID (Bray et al. 2003). The presence of a clearly orthologous sequence in rat opposite the original mouse repeat in our alignment indicated that the transposable element in question was inserted prior to the mouse–rat divergence.
Estimation of substitution rates
Nucleotide substitution rates were estimated for each ancestral repeat and its flanking sequence, correcting for multiple hits using the Tamura-Nei method (Tamura and Nei 1993). Many transposable elements are GC and CpG rich, and this may affect nucleotide substitution rates, depending on the region of insertion of the element. In addition, analysis of the composition and age of large numbers of repetitive elements in the human genome indicated that element GC content tends to decay over the course of evolutionary time (Lander et al. 2001). This effect violates the assumption of stationarity, common to the majority of models used to estimate substitution rates. It is likely, however, that for moderately diverged species, such as mouse and rat, relatively little GC-content decay will have occurred since the two species split. Of greater concern is the fact that many mammalian repetitive consensus sequences contain hypermutable CpG dinucleotides at a substantially higher frequency than the genome at large. Hypermutability of CpG dinucleotides in vertebrates is well documented and poses a problem for the estimation of substitution rates using ancestral repeats. Following insertion, CpG dinucleotides within elements are by far the most likely sites to mutate. However, ancient elements will have experienced most CpG-related changes prior to mouse–rat divergence, whereas those more recent insertions may appear to be evolving at an inflated rate because of their comparatively higher CpG content. This effect could produce covariation between the age of insertion and overall divergence, with more CpG-rich recently inserted elements diverging proportionally more than their older counterparts. Although there have been recent advances in incorporating context dependency into models of sequence evolution (Arndt et al. 2003; Siepel and Haussler 2004), in this study we addressed these issues by estimating nucleotide substitution rates in three alternate ways: using all sites, at those sites not preceded by a C or followed by a G (non-CpG-prone sites), and by counting only A↔T and G↔C changes. The latter two categories are likely to be the least affected by CpG context effects and compositional change and allowed us to assess the impact, or otherwise, of these factors on our results.
Mean chromosomal divergence
We calculated the mean chromosomal divergence treating the entire chromosome as a single sequence and summing differences and sites across all elements. Estimates were also corrected for multiple hits using the method of Tamura and Nei (1993). In order to estimate confidence intervals for the average chromosomal substitution rate, we generated 1000 bootstrap data sets for each chromosome. Because adjacent substitution rates are autocorrelated, we bootstrap by 2-Mb blocks to minimize dependence between observations. We calculated the mean chromosomal divergence for each data set, and the bootstrap distribution of these was used to estimate 95% confidence intervals for each mouse chromosome. Bootstrap data sets were generated using the “boot” library in R (R Development Core Team 2004).
Local similarity
To investigate the scale of local similarity of substitution rates, we divided the mouse genome into 5-kb and 100-kb blocks and estimated an average block substitution rate by taking a weighted (by number of sites) average of the substitution rates of all elements found within a block. We then estimated the autocorrelation of substitution rates across blocks. The autocorrelation of substitution rate K in block i with block i + k, where k is the order or lag of the autocorrelation, is given by (Box et al. 1994):
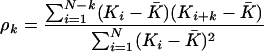 |
(1) |
where N is the total number of blocks. In order to provide confidence intervals for the distribution of ρ under the null hypothesis of no relationship between the evolutionary rates of adjacent blocks, we estimated ρ for 1000 data sets in which block order was randomized. Following Matassi et al. (1999) and Lercher et al. (2001), we assessed the impact of local GC content on the observed pattern of autocorrelation using data sets in which blocks were randomized according to their GC content. Because of the nonrandom pattern of insertion of transposable elements, in all cases elements were permuted while maintaining the structure of our original data set, for example, any empty blocks in the real data were maintained as empty blocks in all our randomized data sets. Local GC content was estimated as the average GC content of all masked mouse and rat flanking sequences within a block. Blocks were then assigned to one of several GC-content classes and randomly permuted only with blocks in the same GC-content class, where each GC-content class contained 5% of the data set.
To investigate the mean “unit” of mutational variation, we estimated the partial autocorrelation of substitution rates averaged across 100-kb blocks. Partial autocorrelation between the mean substitution rates in block xi and block xi+k, where k is the lag, is the amount of correlation that is not explained by the “propagation” of lower-order lags (k – 1, k – 2,...). In our case, partial autocorrelation becomes insignificant at the point beyond which all observed similarity of substitution rates can be explained by autocorrelation of rates across smaller distances. All partial autocorrelations were estimated in R. The significance of partial autocorrelations was again assessed using 1000 data sets in which block order was randomized. We estimated partial autocorrelation of substitution rates in both ancestral repeat and flanking sequence up to an interval distance of 5 Mb.
Between- and within-chromosome variation
We estimated a male-to-female mutation rate ratio, α, using the following formula:
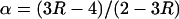 |
(2) |
(Miyata et al. 1987), where R = X/A, and X and A are the mean substitution rates at all sites on the X-chromosome and across all the autosomes, respectively.
In order to quantify between- and within-chromosome mutational variation, the data were fitted to a variety of linear models using the nlme library in R (R Development Core Team 2004). Substitution rates in ancestral repeats and flanking sequences were grouped by location into blocks of increasing size from 25 kb to average chromosome size (125 Mb). We then tested the significance of regional effects in explaining variation in the substitution rate by comparing two models:
- Model 1
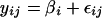
(3) - Model 2
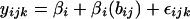
(4)
In Model 1, the substitution rate yij is described by an effect of Chromosome i, (βi), and a random error term (εij). In Model 2, the substitution rate yijk is again described by a mean chromosomal rate but also by a mean “regional” rate or effect of block j, bij, modeled as a normally distributed random effect, nested within the chromosome, that is, as a random variable representing the deviation from the chromosomal mean rate. If substantial regional effects exist, then Model 2 will provide a significantly better fit to the data than Model 1. Both models were fitted to the data using restricted maximum likelihood.
We also tested for significant chromosomal effects by comparing the fit of Model 2 to the data with the following model (Model 3), which includes a term for a random regional effect only:
Model 3
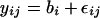 |
(5) |
If there are significant chromosomal effects, Model 2 will provide a better fit to the data than Model 3. Model 2 and Model 3 were fitted to data both including and excluding the X-chromosome, which is a chromosomal outlier. In this case the data were fitted using “full” maximum likelihood as Model 2 and Model 3 differ in their fixed effects specification and their log-restricted likelihoods cannot be compared (Pinheiro and Bates 2000).
For all comparisons we used the Akaike Information Criterion (AIC) to assess the fit of the model to the data. The AIC is a model selection criterion that incorporates information about the fit of the model to the data and the model complexity:
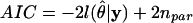 |
(6) |
where 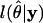 is the log-likelihood of the model
is the log-likelihood of the model 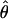 , given the data y, and npar is the number of parameters in the model (Pinheiro and Bates 2000).
, given the data y, and npar is the number of parameters in the model (Pinheiro and Bates 2000).
Acknowledgments
We thank Daniel Halligan, Ian White, Toby Johnson, Bill Hill, Gabriel Marais, Alex Kondrashov, and two anonymous referees for helpful comments and discussion. We also thank the Blaxter Lab for the use of their Linux cluster. D.J.G. is funded by a University of Edinburgh postgraduate scholarship.
[Supplemental material is available online at www.genome.org.]
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.3895005. Article published online before print in July 2005.
References
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J.H., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arndt, P.F., Burge, C.B., and Hwa, T. 2003. DNA sequence evolution with neighbor-dependent mutation. J. Comput. Biol. 10: 313–322. [DOI] [PubMed] [Google Scholar]
- Bailey, J.A., Carrel, L., Chakravarti, A., and Eichler, E.E. 2000. Molecular evidence for a relationship between LINE-1 elements and X chromosome inactivation: The Lyon repeat hypothesis. Proc. Natl. Acad. Sci. 97: 6634–6639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejerano, G., Pheasant, M., Makunin, I., Stephen, S., Kent, W.J., Mattick, J.S., and Haussler, D. 2004. Ultraconserved elements in the human genome. Science 304: 1321–1325. [DOI] [PubMed] [Google Scholar]
- Box, G.E.P., Jenkins, G.M., and Reinsel, G.C. 1994. Time series analysis: Forecasting and control, 3rd ed. Prentice-Hall, Upper Saddle River, NJ.
- Bray, N., Dubchak, I., and Pachter, L. 2003. AVID: A global alignment program. Genome Res. 13: 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamary, J.V. and Hurst, L.D. 2004. Similar rates but different modes of sequence evolution in introns and at exonic silent sites in rodents: Evidence for selectively driven codon usage. Mol. Biol. Evol. 21: 1014–1023. [DOI] [PubMed] [Google Scholar]
- Chang, B.H.J., Shimmin, L.C., Shyue, S.K., Hewettemmett, D., and Li, W.H. 1994. Weak male-driven molecular evolution in rodents. Proc. Natl. Acad. Sci. 91: 827–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, F.C., Vallender, E.J., Wang, H., Tzeng, C.S., and Li, W.H. 2001. Genomic divergence between human and chimpanzee estimated from large-scale alignments of genomic sequences. J. Hered. 92: 481–489. [DOI] [PubMed] [Google Scholar]
- Chin, C., Chuang, J.H., and Li, H. 2005. Genome-wide regulatory complexity in yeast promoters: Separation of functionally conserved and neutral sequence. Genome Res. 15: 205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang, J.H. and Li, H. 2004. Functional bias and spatial organization of genes in mutational hot and cold regions in the human genome. PLoS Biol. 2: 253–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford, D.C., Bhangale, T., Li, N., Hellenthal, G., Rieder, M.J., Nickerson, D.A., and Stephens, M. 2004. Evidence for substantial fine-scale variation in recombination rates across the human genome. Nat. Genet. 36: 700–706. [DOI] [PubMed] [Google Scholar]
- Ebersberger, I., Metzler, D., Schwarz, C., and Paabo, S. 2002. Genomewide comparison of DNA sequences between humans and chimpanzees. Am. J. Hum. Genet. 70: 1490–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren, H., Smith, N.G.C., and Webster, M.T. 2003. Mutation rate variation in the mammalian genome. Curr. Opin. Genet. Dev. 13: 562–568. [DOI] [PubMed] [Google Scholar]
- Eyre-Walker, A. 1999. Evidence of selection on silent site base composition in mammals: Potential implications for the evolution of isochores and junk DNA. Genetics 152: 675–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filatov, D.A. 2004. A gradient of silent substitution rate in the human pseudoautosomal region. Mol. Biol. Evol. 21: 410–417. [DOI] [PubMed] [Google Scholar]
- Filipski, J. 1988. Why the rate of silent codon substitutions in variable within a vertebrate genome. J. Theor. Biol. 134: 159–164. [DOI] [PubMed] [Google Scholar]
- Gibbs, R.A., Weinstock, G.M., Metzker, M.L., Muzny, D.M., Sodergren, E.J., Scherer, S., Scott, G., Steffen, D., Worley, K.C., Burch, P.E., et al. 2004. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature 428: 493–521. [DOI] [PubMed] [Google Scholar]
- Hardison, R.C., Roskin, K.M., Yang, S., Diekhans, M., Kent, W.J., Weber, R., Elnitski, L., Li, J., O'Connor, M., Kolbe, D., et al. 2003. Covariation in frequencies of substitution, deletion, transposition, and recombination during eutherian evolution. Genome Res. 13: 13–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, S.-W., Friedman, R., Yu, N., Yu, A., and Li, W.-H. 2005. How strong is the mutagenicity of recombination in mammals? Mol. Biol. Evol. 22: 426–431. [DOI] [PubMed] [Google Scholar]
- Hudson, T.J., Church, D.M., Greenaway, S., Nguyen, H., Cook, A., Steen, R.G., Van Etten, W.J., Castle, A.B., Strivens, M.A., Trickett, P., et al. 2001. A radiation hybrid map of mouse genes. Nat. Genet. 29: 201–205. [DOI] [PubMed] [Google Scholar]
- Jensen-Seaman, M.I., Furey, T.S., Payseur, B.A., Lu, Y.T., Roskin, K.M., Chen, C.F., Thomas, M.A., Haussler, D., and Jacob, H.J. 2004. Comparative recombination rates in the rat, mouse, and human genomes. Genome Res. 14: 528–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley, P.D. and Gaffney, D.J. 2003. Functional constraints and frequency of deleterious mutations in noncoding DNA of rodents. Proc. Natl. Acad. Sci. 100: 13402–13406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley, P.D., Lercher, M.J., and Eyre-Walker, A. 2005. Evidence for widespread degradation of gene control regions in hominid genomes. PLoS Biol. 3: e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khil, P.P., Oliver, B., and Camerini-Otero, R.D. 2005. X for intersection: Retrotransposition both on and off the X chromosome is more frequent. Trends Genet. 21: 3–7. [DOI] [PubMed] [Google Scholar]
- Kong, A., Gudbjartsson, D.F., Sainz, J., Jonsdottir, G.M., Gudjonsson, S.A., Richardsson, B., Sigurdardottir, S., Barnard, J., Hallbeck, B., Masson, G., et al. 2002. A high-resolution recombination map of the human genome. Nat. Genet. 31: 241–247. [DOI] [PubMed] [Google Scholar]
- Lander, E.S., Linton, L.M., Birren, B., Nusbaum, C., Zody, M.C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh, W., et al. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860–921. [DOI] [PubMed] [Google Scholar]
- Lercher, M.J., Williams, E.J.B., and Hurst, L.D. 2001. Local similarity in evolutionary rates extends over whole chromosomes in human–rodent and mouse–rat comparisons: Implications for understanding the mechanistic basis of the male mutation bias. Mol. Biol. Evol. 18: 2032–2039. [DOI] [PubMed] [Google Scholar]
- Makova, K.D., Yang, S., and Chiaromonte, F. 2004. Insertions and deletions are male biased too: A whole-genome analysis in rodents. Genome Res. 14: 567–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malcom, C.M., Wyckoff, G.J., and Lahn, B.T. 2003. Genic mutation rates in mammals: Local similarity, chromosomal heterogeneity, and X-versus-autosome disparity. Mol. Biol. Evol. 20: 1633–1641. [DOI] [PubMed] [Google Scholar]
- Matassi, G., Sharp, P.M., and Gautier, C. 1999. Chromosomal location effects on gene sequence evolution in mammals. Curr. Biol. 9: 786–791. [DOI] [PubMed] [Google Scholar]
- McVean, G.T. and Hurst, L.D. 1997. Evidence for a selectively favourable reduction in the mutation rate of the X chromosome. Nature 386: 388–392. [DOI] [PubMed] [Google Scholar]
- McVean, G.A.T., Myers, S.R., Hunt, S., Deloukas, P., Bentley, D.R., and Donnelly, P. 2004. The fine-scale structure of recombination rate variation in the human genome. Science 304: 581–584. [DOI] [PubMed] [Google Scholar]
- Meunier, J. and Duret, L. 2004. Recombination drives the evolution of GC-content in the human genome. Mol. Biol. Evol. 21: 984–990. [DOI] [PubMed] [Google Scholar]
- Miyata, T., Hayashida, H., Kuma, K., Mitsuyasu, K., and Yasunaga, T. 1987. Male-driven molecular evolution—A model and nucleotide sequence analysis. Cold Spring Harbor Symp. Quant. Biol. 52: 863–867. [DOI] [PubMed] [Google Scholar]
- Mouchiroud, D., Donofrio, G., Aissani, B., MacAya, G., Gautier, C., and Bernardi, G. 1991. The distribution of genes in the human genome. Gene 100: 181–187. [DOI] [PubMed] [Google Scholar]
- Nadeau, J.H. and Taylor, B.A. 1984. Lengths of chromosomal segments conserved since divergence of man and mouse. Proc. Natl. Acad. Sci. 81: 814–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro, J.C. and Bates, D.M. 2000. Mixed-effects models in S and S-PLUS.. Springer-Verlag, New York.
- R Development Core Team. 2004. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. 3-900051-07-0.
- Roy, A.M., Carroll, M.L., Nguyen, S.V., Salem, A.H., Oldridge, M., Wilkie, A.O.M., Batzer, M.A., and Deininger, P.L. 2000. Potential gene conversion and source genes for recently integrated Alu elements. Genome Res. 10: 1485–1495. [DOI] [PubMed] [Google Scholar]
- Siepel, A. and Haussler, D. 2004. Phylogenetic estimation of context-dependent substitution rates by maximum likelihood. Mol. Biol. Evol. 21: 468–488. [DOI] [PubMed] [Google Scholar]
- Silva, J.C. and Kondrashov, A.S. 2002. Patterns in spontaneous mutation revealed by human–baboon sequence comparison. Trends Genet. 18: 544–547. [DOI] [PubMed] [Google Scholar]
- Smith, N.G.C., Webster, M.T., and Ellegren, H. 2002. Deterministic mutation rate variation in the human genome. Genome Res. 12: 1350–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura, K. and Nei, M. 1993. Estimation of the number of nucleotide substitutions in the control region of mitochondrial-DNA in humans and chimpanzees. Mol. Biol. Evol. 10: 512–526. [DOI] [PubMed] [Google Scholar]
- Thomas, J.W., Touchman, J.W., Blakesley, R.W., Bouffard, G.G., Beckstrom-Sternberg, S.M., Margulies, E.H., Blanchette, M., Siepel, A.C., Thomas, P.J., McDowell, J.C., et al. 2003. Comparative analyses of multi-species sequences from targeted genomic regions. Nature 424: 788–793. [DOI] [PubMed] [Google Scholar]
- Waterston, R.H., Lindblad-Toh, K., Birney, E., Rogers, J., Abril, J.F., Agarwal, P., Agarwala, R., Ainscough, R., Alexandersson, M., An, P., et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562. [DOI] [PubMed] [Google Scholar]
- Webster, M., Smith, N., Lercher, M., and Ellegren, H. 2004. Gene expression, synteny, and local similarity in human noncoding mutation rates. Mol. Biol. Evol. 21: 1820–1830. [DOI] [PubMed] [Google Scholar]
- Willie, E. and Majewski, J. 2004. Evidence for codon bias selection at the pre-mRNA level in eukaryotes. Trends Genet. 20: 534–538. [DOI] [PubMed] [Google Scholar]
- Wolfe, K.H., Sharp, P.M., and Li, W.H. 1989. Mutation rates differ among regions of the mammalian genome. Nature 337: 283–285. [DOI] [PubMed] [Google Scholar]
Web site references
- http://www.repeatmasker.org/; the program RepeatMasker is available for download from this site.